
Green Synthesis and Anticancer Potential of 1,4-Dihydropyridines-Based Triazole Derivatives: In Silico and In Vitro Study

 , ,
, ,  , , ,
, , ,  , , and
, , and
Abstract
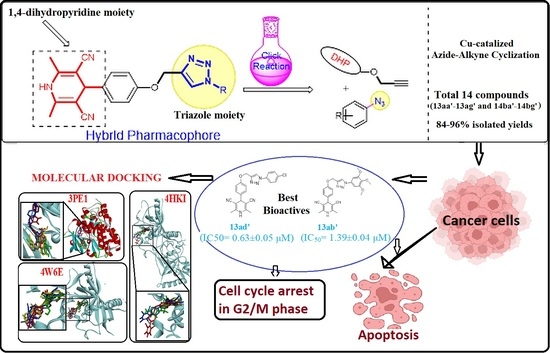
:
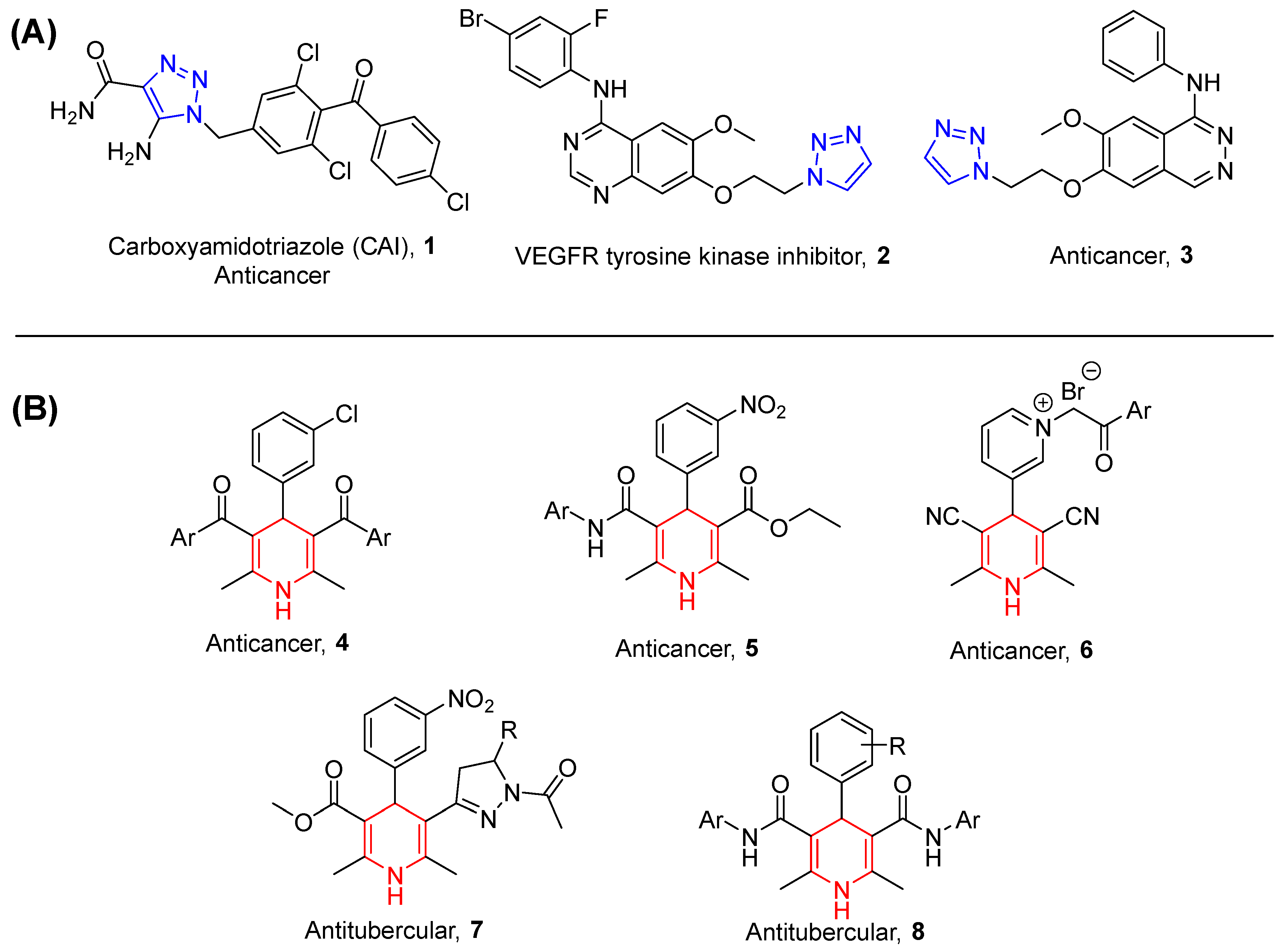
1. Introduction
2. Methodology
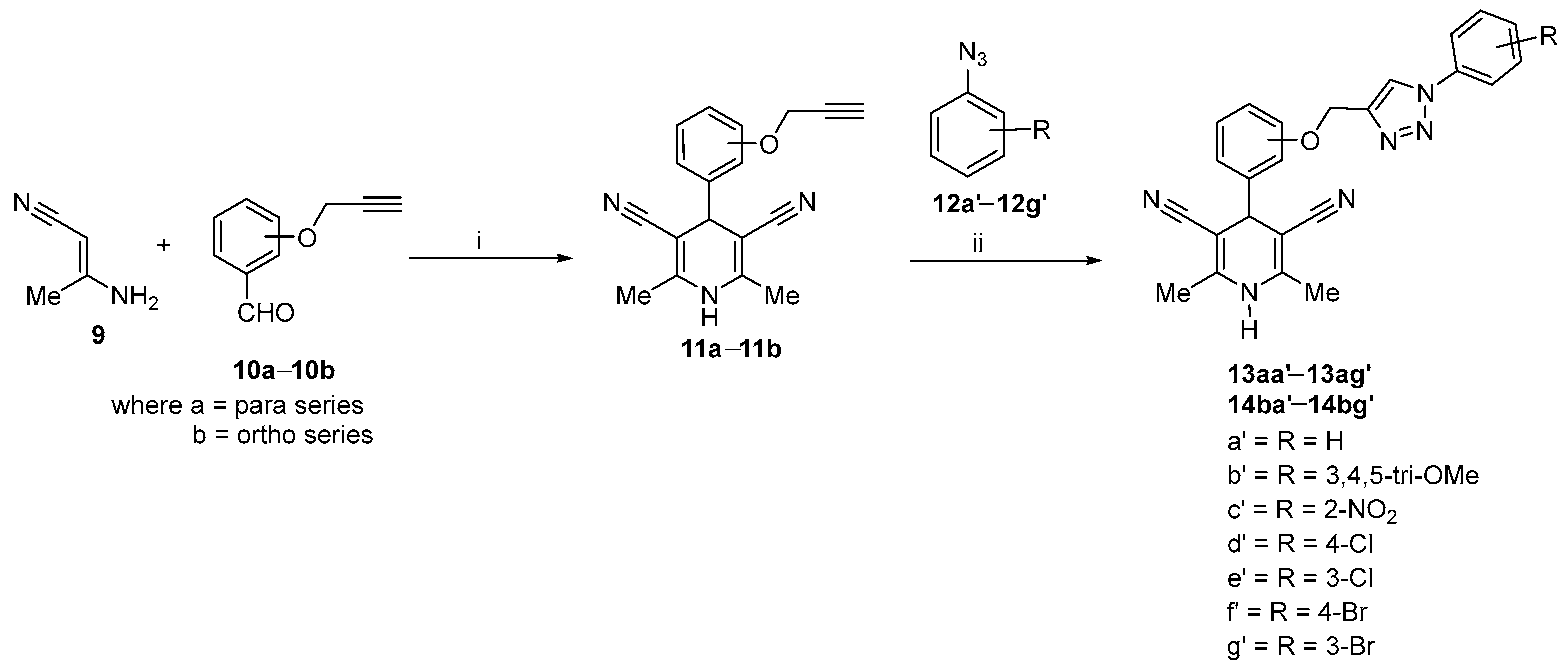
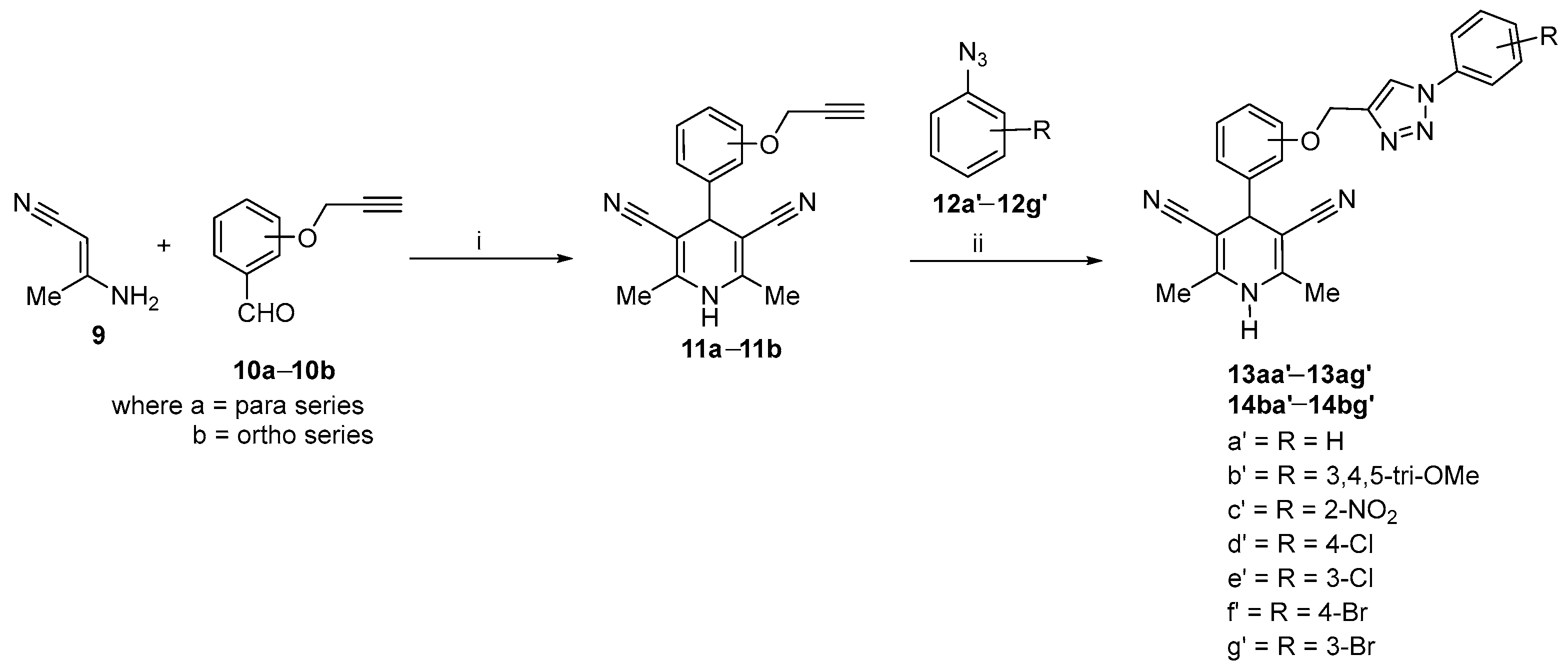
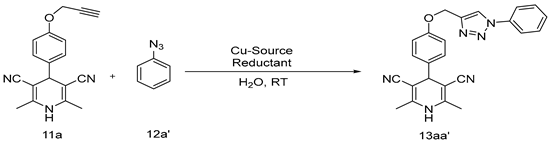
2.1. Synthetic Procedure for 1,4-Dihydropyridines-Based 1,2,3–Triazole Derivatives
2.2. Anticancer Evaluation
2.2.1. Cell Culture and Cell Viability Assay
2.2.2. Apoptosis/Cell Death Assay
2.2.3. Cell Cycle Assay
2.2.4. Statistical Analysis
2.3. Cheminformatics Molecular Interaction Study
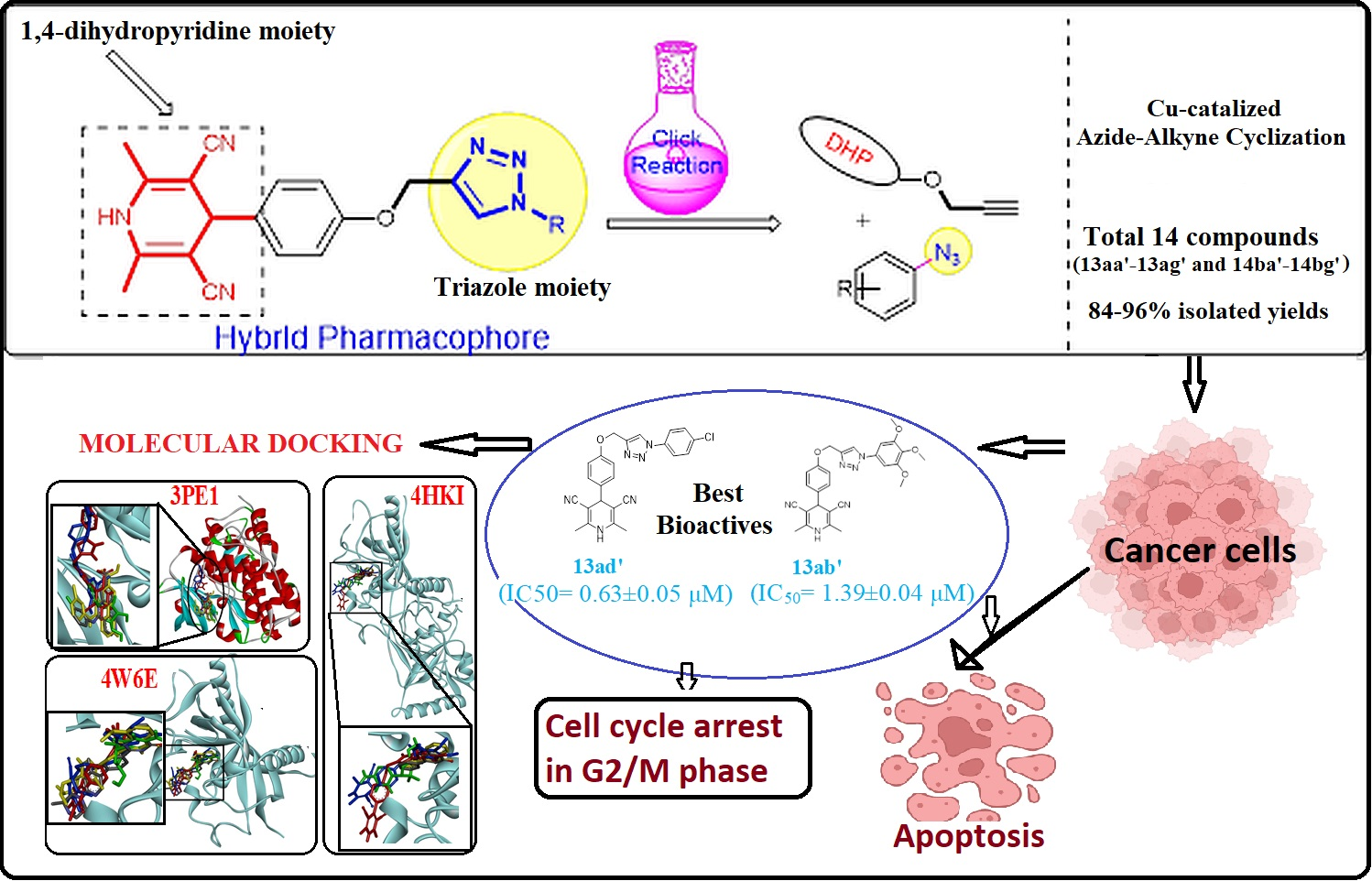
3. Results
3.1. Chemistry
Optimization and Synthesis of Compounds
3.2. Biological Anticancer Activity
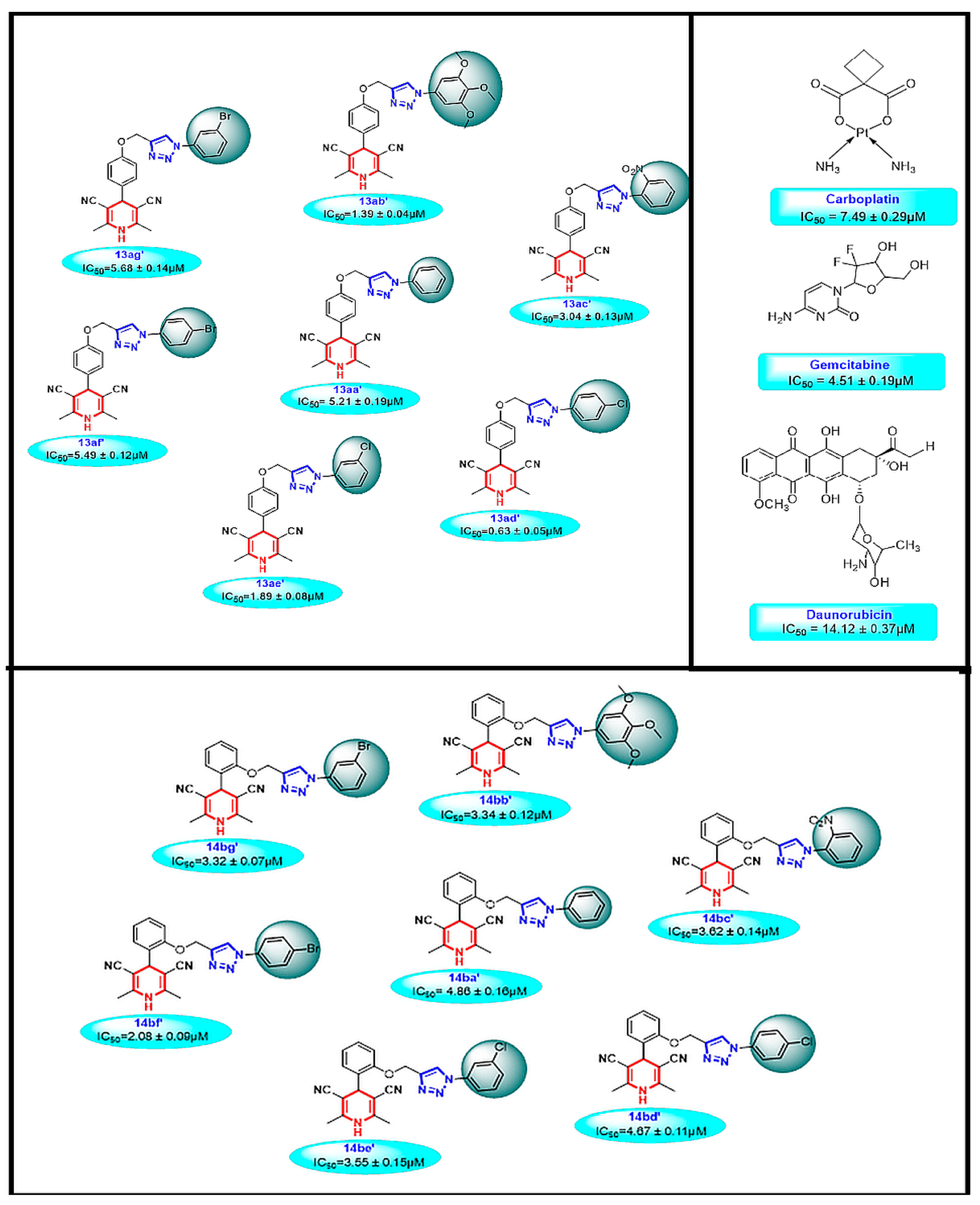
3.2.1. In Vitro Cytotoxicity
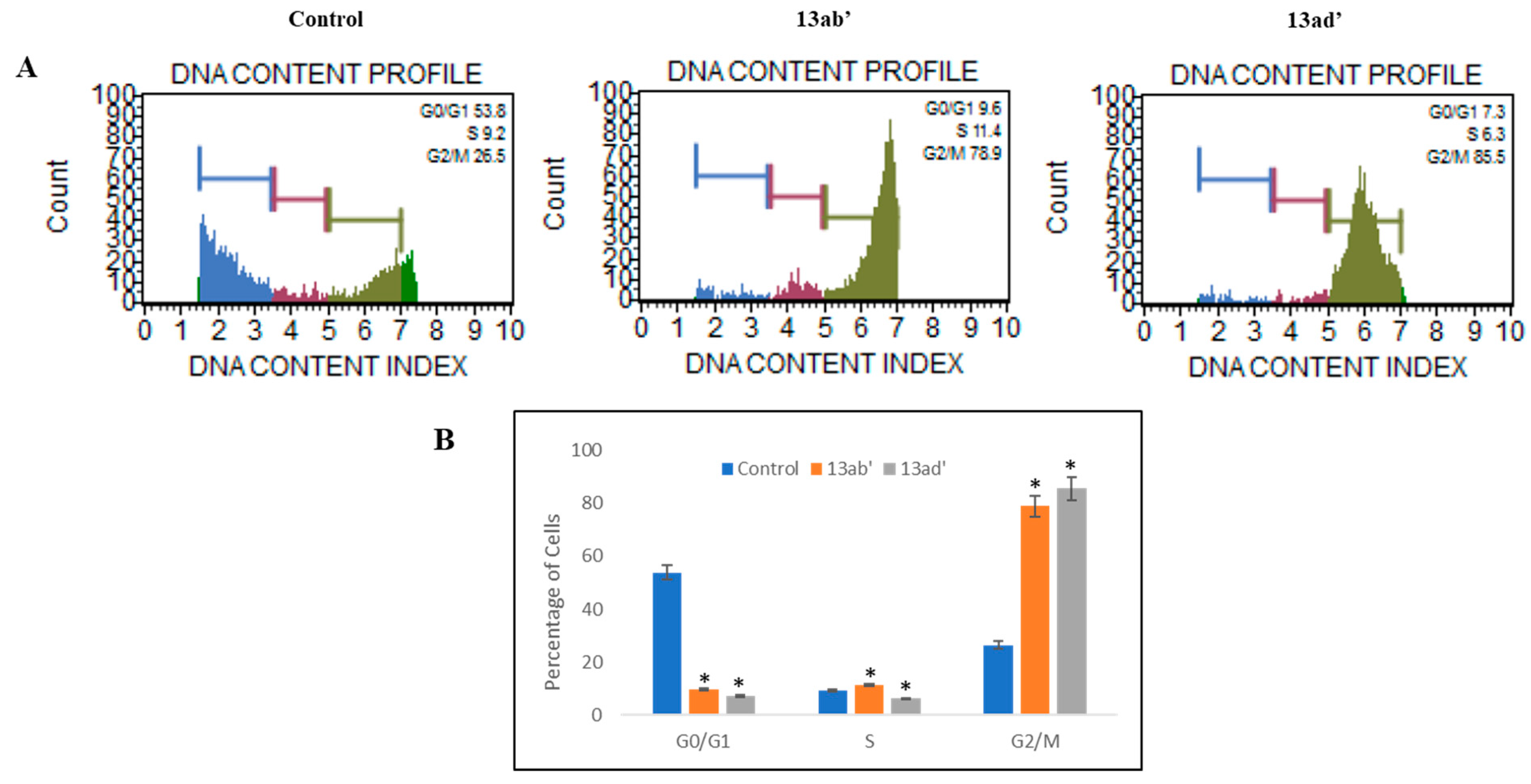
3.2.2. Cell Cycle Inhibition
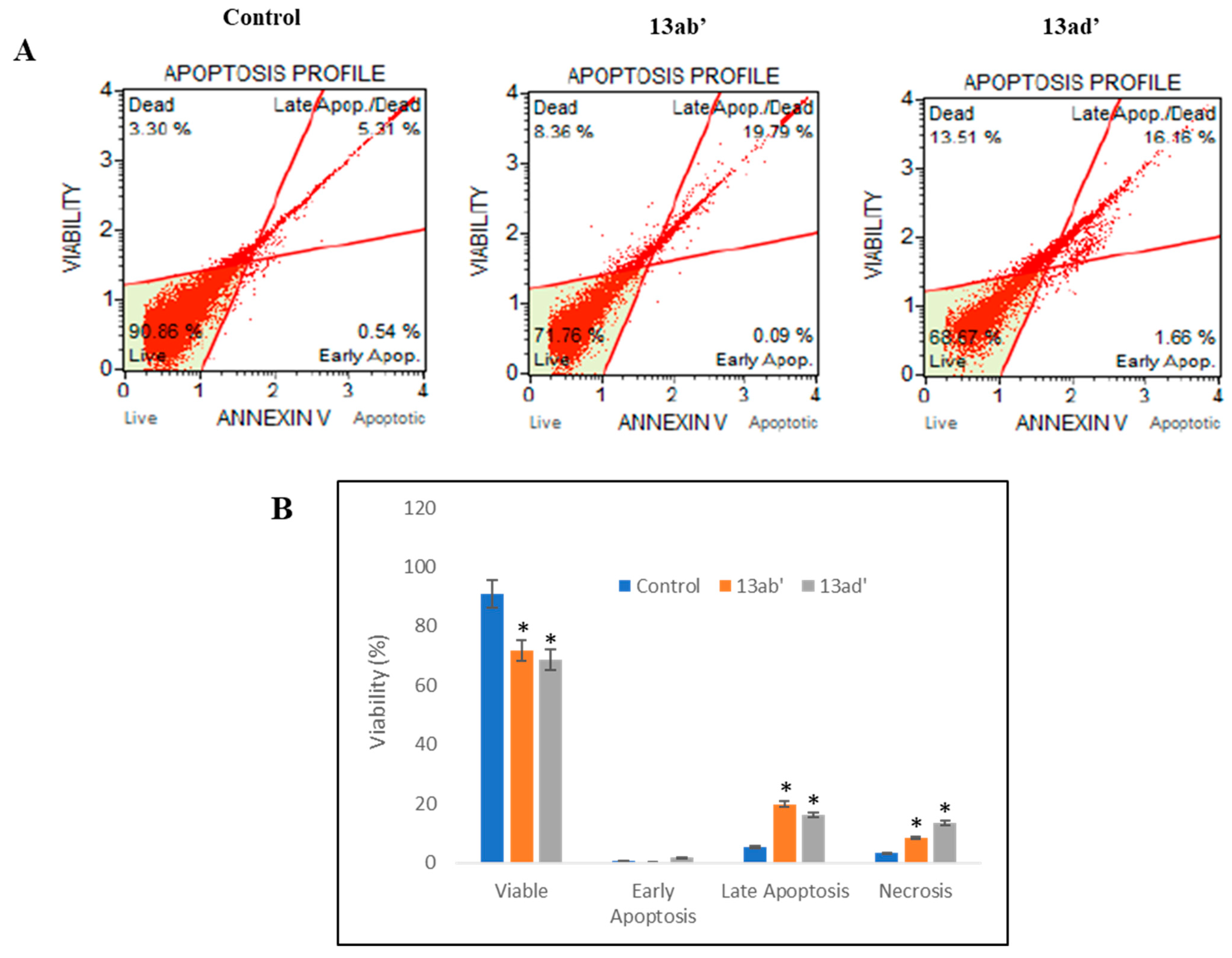
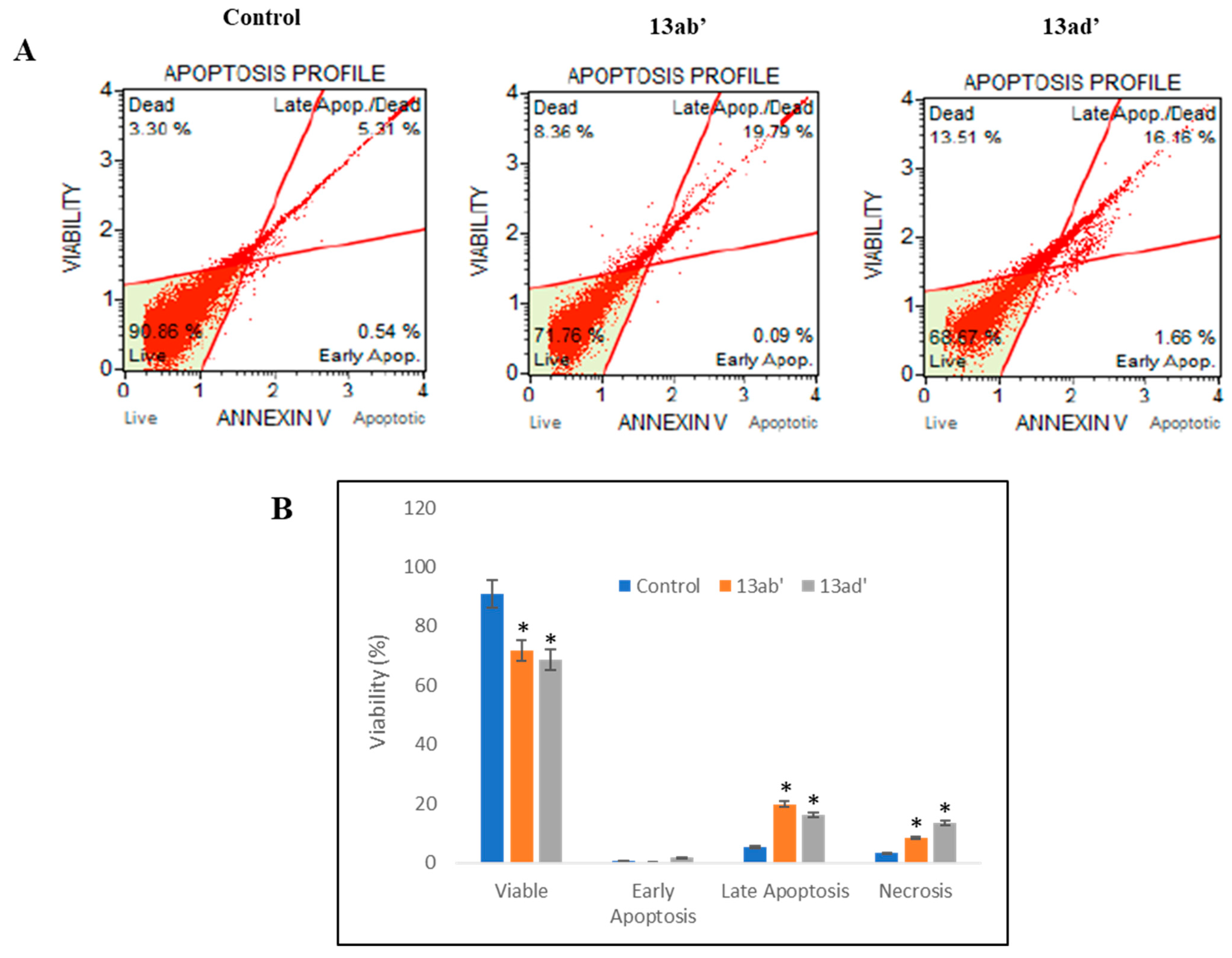
3.2.3. Induction of Apoptosis
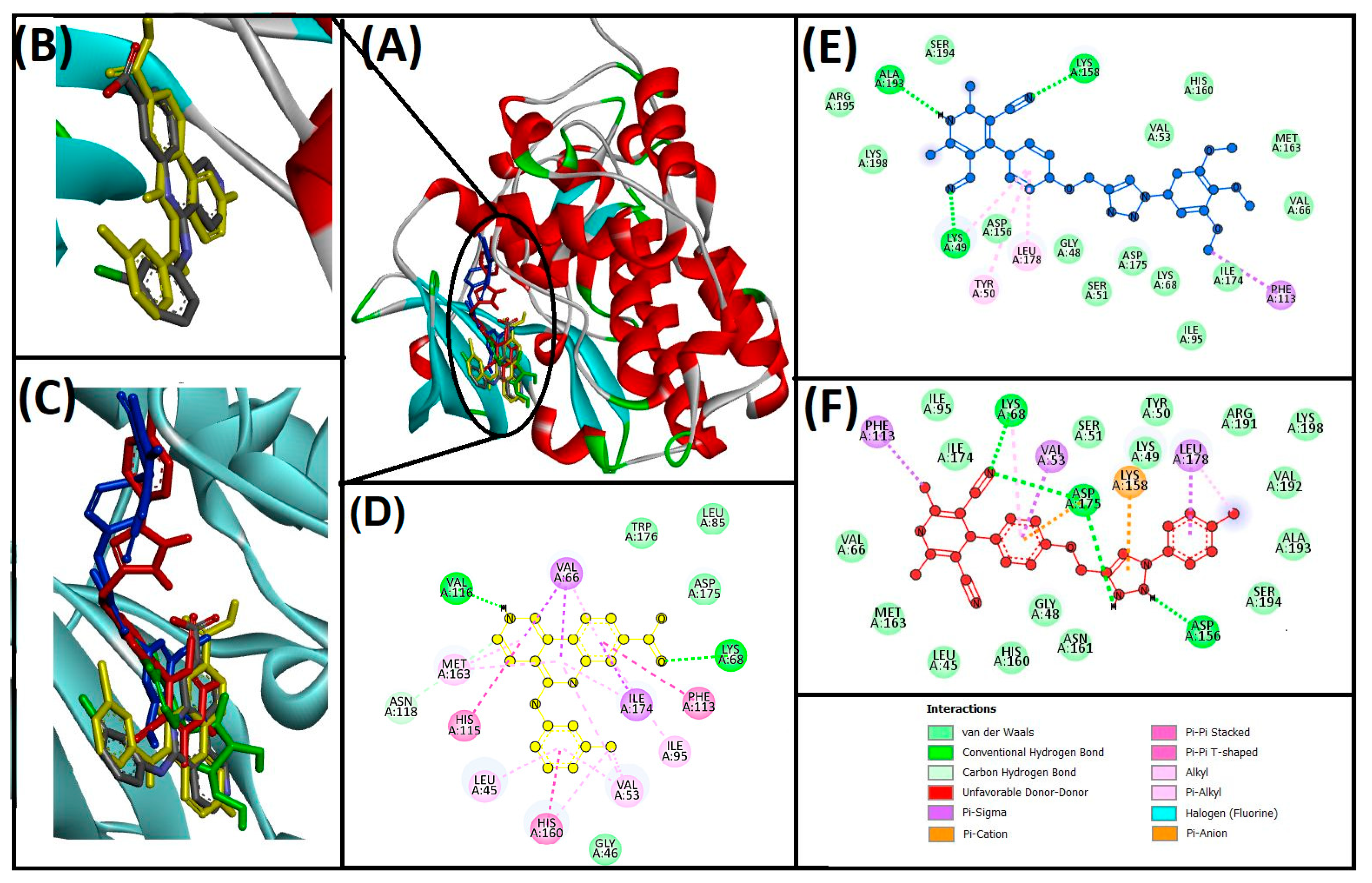
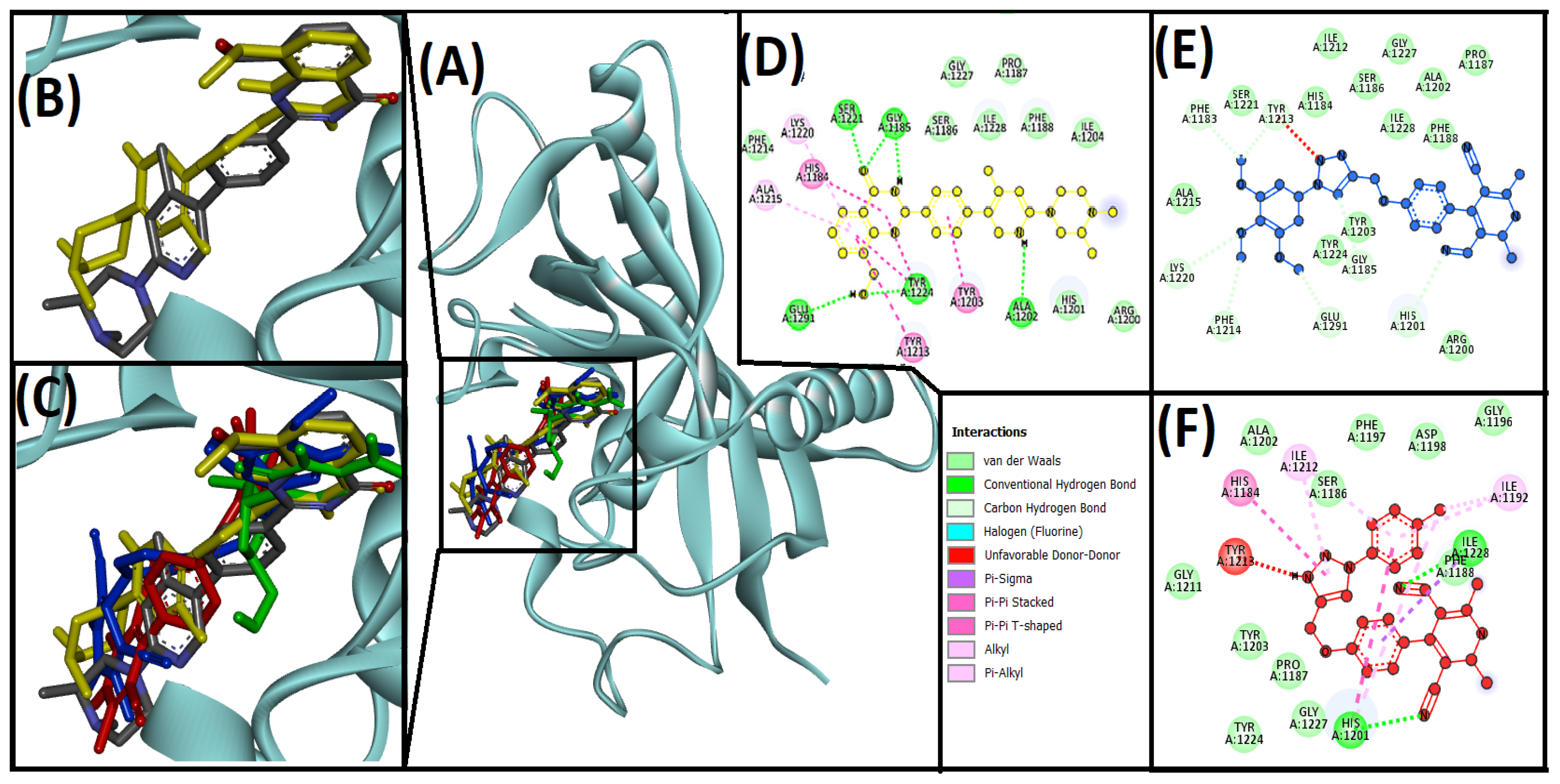
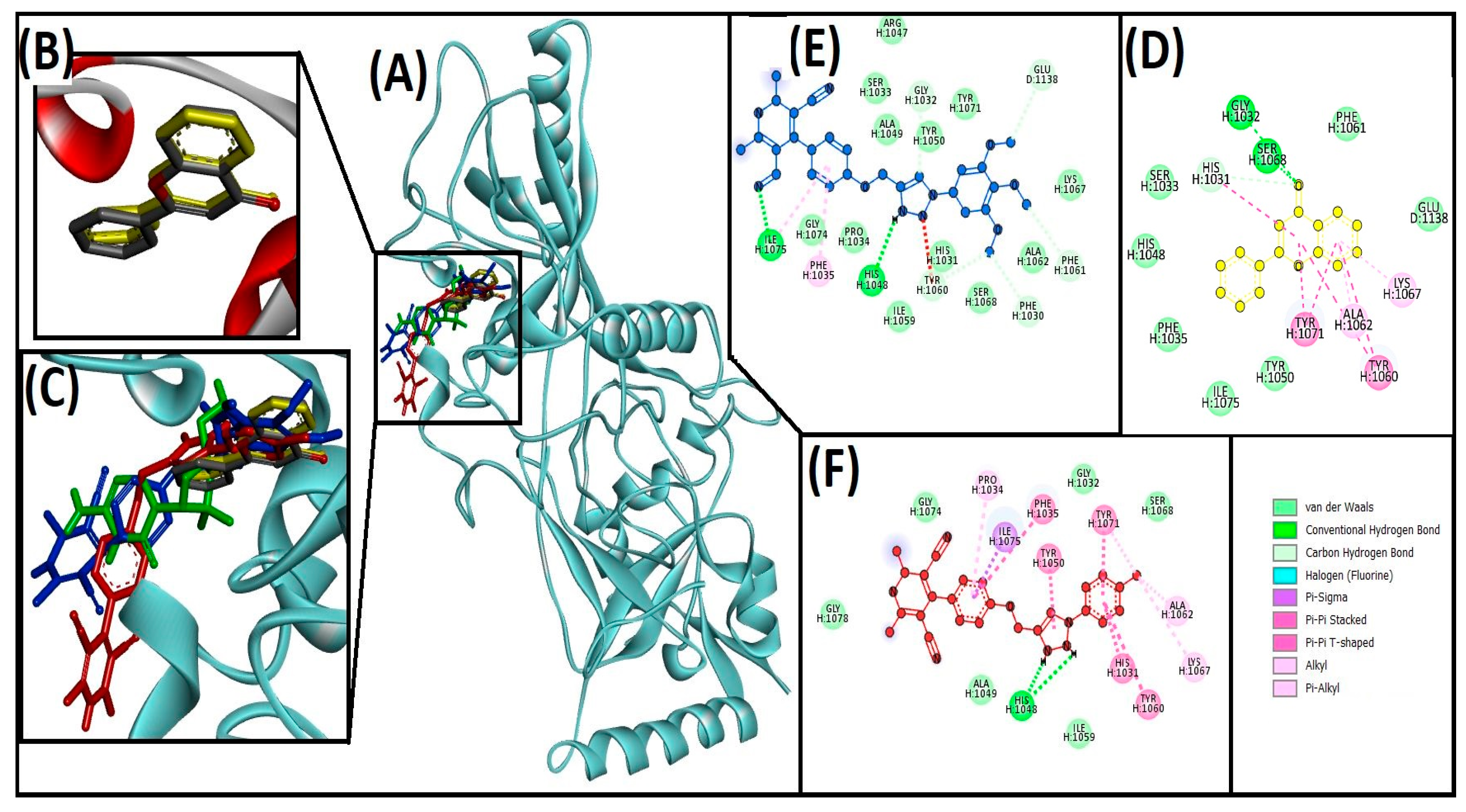
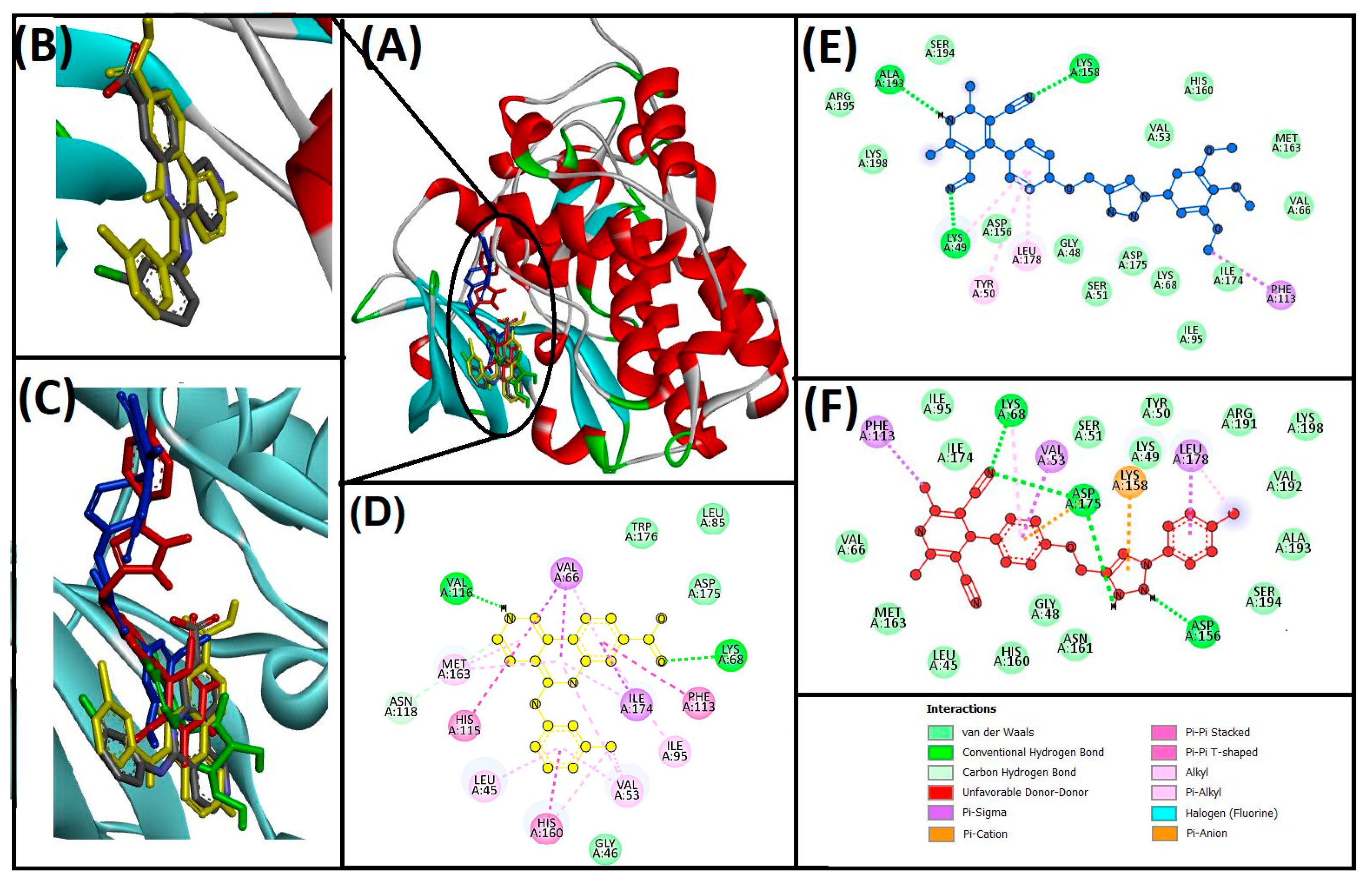
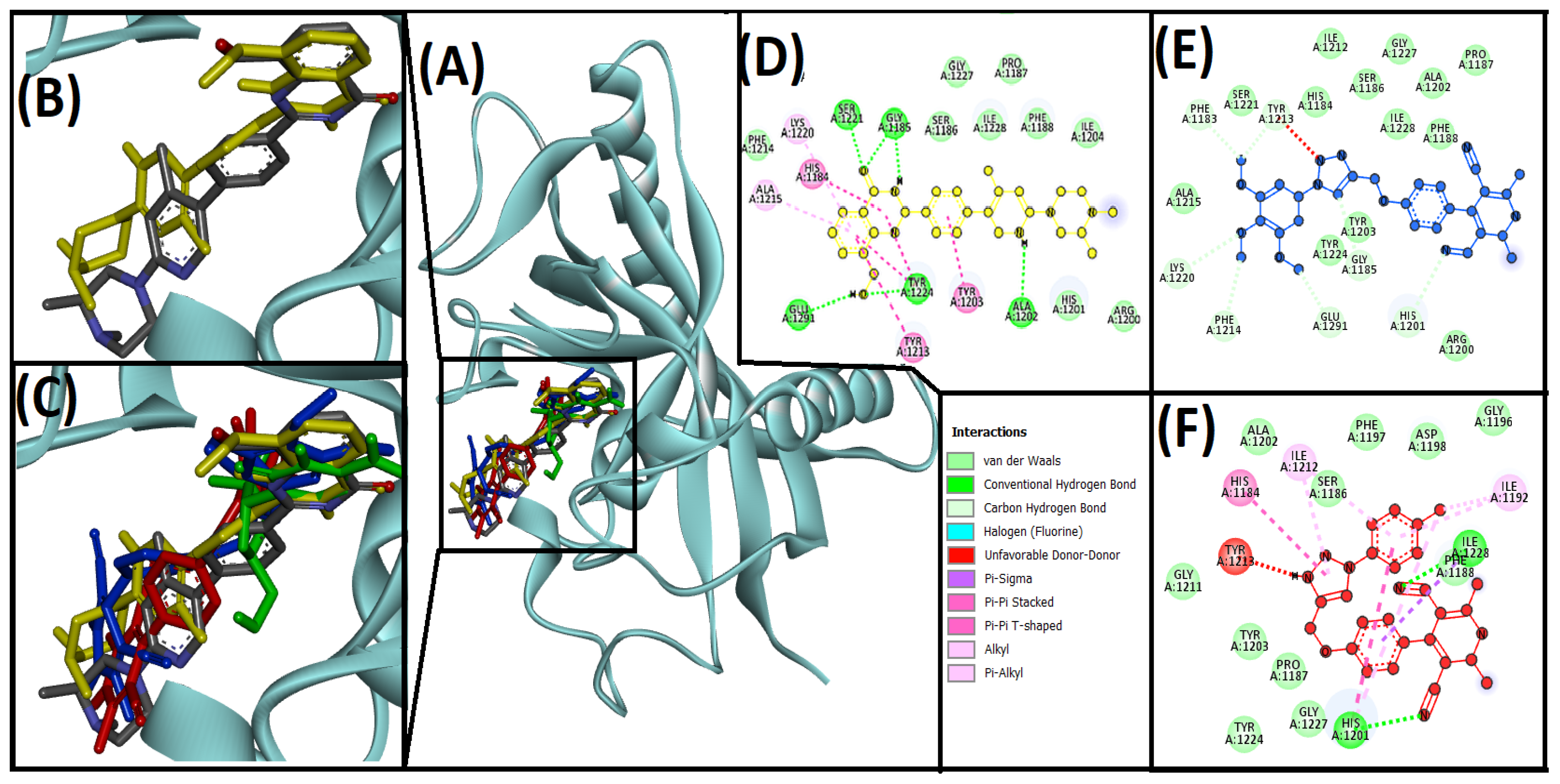
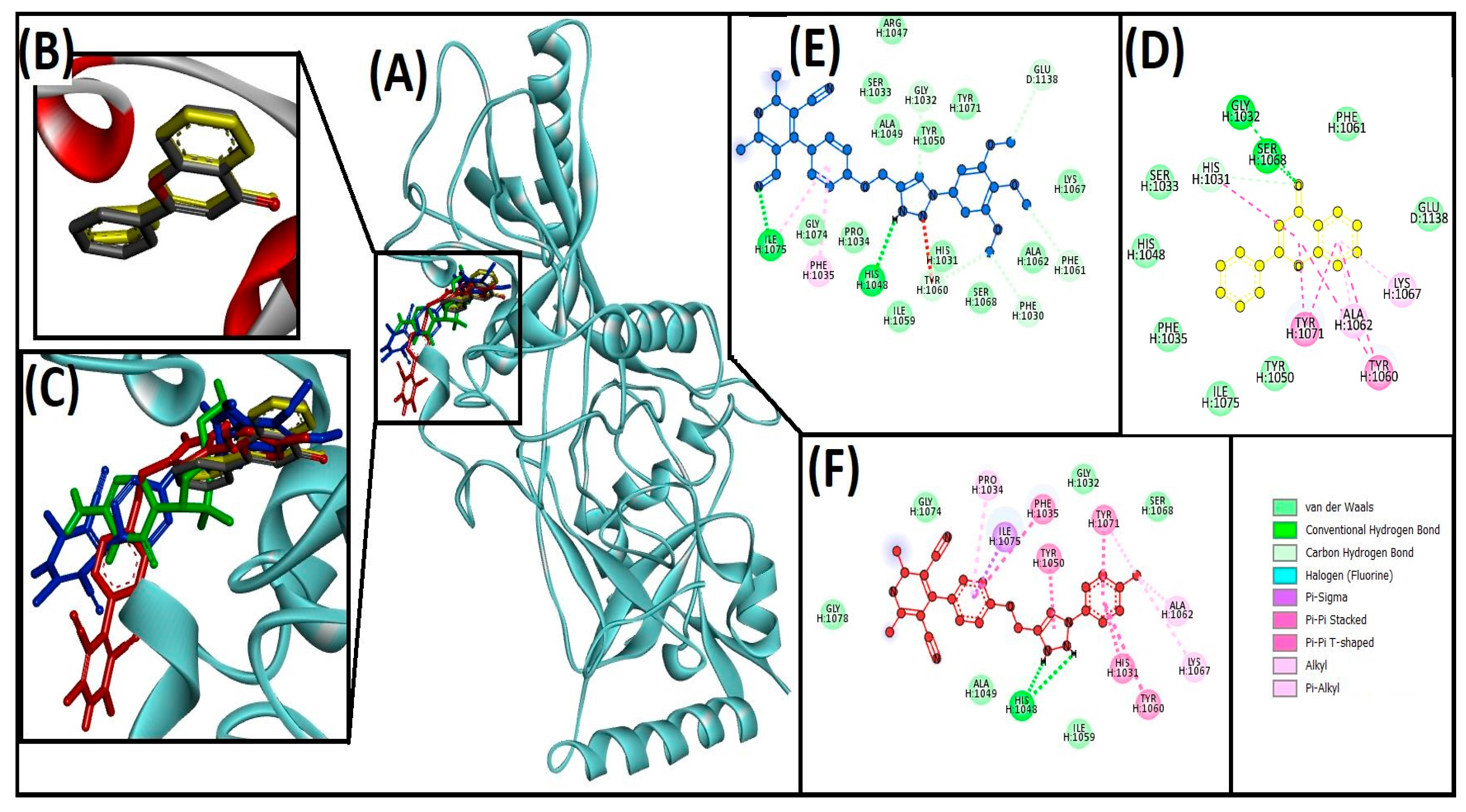
3.3. Cheminformatics Molecular Interaction Analysis
4. Discussion
4.1. Chemistry
4.2. Anticancer Activity
4.3. Cheminformatics Molecular Interaction Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; D’amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Lehtiö, L.; Chi, N.-W.; Krauss, S. Tankyrases as drug targets. FEBS J. 2013, 280, 3576–3593. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Borgo, C.; Pinna, L.A.; Ruzzene, M. Targeting CK2 in cancer: A valuable strategy or a waste of time? Cell Death Discov. 2021, 7, 325. [Google Scholar] [CrossRef]
- Verma, A.; Kumar, A.; Chugh, A.; Kumar, S.; Kumar, P. Tankyrase inhibitors: Emerging and promising therapeutics for cancer treatment. Med. Chem. Res. 2021, 30, 50–73. [Google Scholar] [CrossRef]
- Zamudio-Martinez, E.; Herrera-Campos, A.B.; Muñoz, A.; Rodríguez-Vargas, J.M.; Oliver, F.J. Tankyrases as modulators of pro-tumoral functions: Molecular insights and therapeutic opportunities. J. Exp. Clin. Cancer Res. 2021, 40, 144. [Google Scholar] [CrossRef]
- Zou, J.; Luo, H.; Zeng, Q.; Dong, Z.; Wu, D.; Liu, L. Protein kinase CK2α is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. J. Transl. Med. 2011, 9, 97. [Google Scholar] [CrossRef] [Green Version]
- Bridges, A.J.; Mehta, R.K.; Shukla, S.; Schipper, M.J.; Lawrence, T.S.; Nyati, M.K. Drug-development, dose-selection, rational combinations from bench-to-bedside: Are there any lessons worth revisiting? Oncotarget 2021, 12, 1032–1036. [Google Scholar] [CrossRef]
- Jahan, S.; Mukherjee, S.; Ali, S.; Bhardwaj, U.; Choudhary, R.K.; Balakrishnan, S.; Naseem, A.; Mir, S.A.; Banawas, S.; Alaidarous, M.; et al. Pioneer Role of Extracellular Vesicles as Modulators of Cancer Initiation in Progression, Drug Therapy, and Vaccine Prospects. Cells 2022, 11, 490. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Gupta, R.; Jain, A. Microwave-assisted synthesis of nitrogen-containing heterocycles. Green Chem. Lett. Rev. 2013, 6, 151–182. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.M. 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Pharm. 2021, 355, e2100158. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, J.; Kadhim, H.; Makki, K.; Ali, B. Review on Antioxidant Evaluation of 1,2,3-Triazole Derivatives Synthesized by Click Chemistry. Ann. Rom. Soc. Cell Biol. 2021, 25, 2765–2796. [Google Scholar]
- Sahu, A.; Agrawal, R. A Recent Review on Drug Modification Using 1,2,3-triazole. Curr. Chem. Biol. 2020, 14, 71–87. [Google Scholar] [CrossRef]
- Varala, R.; Bollikolla, H.B.; Kurmarayuni, C.M. Synthesis of Pharmacological Relevant 1,2,3-Triazole and its Analogues-A Review. Curr. Org. Synth. 2021, 18, 101–124. [Google Scholar] [CrossRef]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click Chemistry: 1,2,3-Triazoles as Pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorganic Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Mishra, A.P.; Bajpai, A.; Rai, A.K. 1,4-Dihydropyridine: A Dependable Heterocyclic Ring with the Promising and the Most Anticipable Therapeutic Effects. Mini-Rev. Med. Chem. 2019, 19, 1219–1254. [Google Scholar] [CrossRef]
- Gaudio, A.C.; Korolkovas, A.; Takahata, Y. Quantitative Structure-Activity Relationships for 1,4-Dihydropyridine Calcium Channel Antagonists (Nifedipine Analogues): A Quantum ChemicalKlassical Approach. J. Pharm. Sci. 1994, 83, 1110–1115. [Google Scholar] [CrossRef]
- Mannhold, R.; Jablonka, B.; Voigt, W.; Schönafinger, K.; Schraven, E. Calcium- and calmodulin-antagonism of elnadipine derivatives: Comparative SAR. Eur. J. Med. Chem. 1992, 27, 229–235. [Google Scholar] [CrossRef]
- Reid, J.L.; Meredith, P.A.; Pasanisi, F. Clinical Pharmacological Aspects of Calcium Antagonists and Their Therapeutic Role in Hypertension. J. Cardiovasc. Pharmacol. 1985, 7, S18–S20. [Google Scholar] [CrossRef]
- Safak, C.; Simsek, R. Fused 1,4-dihydropyridines as potential calcium modulatory compounds. Mini-Rev. Med. Chem. 2006, 6, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.; Sureja, D.; Naliapara, Y.; Shah, A.; Saxena, A.K. Synthesis and QSAR Studies of 4-Substituted phenyl-2,6-dimethyl-3, 5-bis-N-(substituted phenyl)carbamoyl-1,4-dihydropyridines as potential antitubercular agents. Bioorganic Med. Chem. 2001, 9, 1993–1998. [Google Scholar] [CrossRef]
- Mahendra, M.; Doreswamy, B.H.; Sridhar, M.A.; Prasad, J.S.; Kinnari, D.; Dinesh, M.; Anamik, S. N,N′-Bis(2-chlorophenyl)-4-(4-chlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide. Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, o2567–o2569. [Google Scholar] [CrossRef] [Green Version]
- Radadiya, A.; Khedkar, V.; Bavishi, A.; Vala, H.; Thakrar, S.; Bhavsar, D.; Shah, A.; Coutinho, E. Synthesis and 3D-QSAR study of 1,4-dihydropyridine derivatives as MDR cancer reverters. Eur. J. Med. Chem. 2014, 74, 375–387. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorganic Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Khwaza, V.; Mlala, S.; Oyedeji, O.; Aderibigbe, B. Pentacyclic Triterpenoids with Nitrogen-Containing Heterocyclic Moiety, Privileged Hybrids in Anticancer Drug Discovery. Molecules 2021, 26, 2401. [Google Scholar] [CrossRef]
- Thakrar, S.; Bavishi, A.; Bhavsar, D.; Parekh, S.; Vala, H.; Radadiya, A.; Parmar, M.; Savant, M.; Shah, A. Efficient and Rapid Synthesis of Highly Functionalized Novel Symmetric 1,4-Dihydropyridines Using Glacial Acetic Acid as Solvent. Synth. Commun. 2012, 42, 3269–3278. [Google Scholar] [CrossRef]
- Jiang, Y.; Kong, D.; Zhao, J.; Qi, Q.; Li, W.; Xu, G. Cu(OAc)2·H2O/NH2NH2·H2O: An efficient catalyst system that in situ generates Cu2O nanoparticles and HOAc for Huisgen click reactions. RSC Adv. 2013, 4, 1010–1014. [Google Scholar] [CrossRef]
- Denish, V.; Sheefa, M.; Faraz, S.; Rajesh, K.; Anand, R.; Nayan, J.; Rakesh, R.; Anamik, S. Design and Synthesis of 1,4-Dihydropyridine Derivatives as Anti-Cancer Agent. Anti-Cancer Agents Med. Chem. 2017, 17, 1003–1013. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Dziedzic, A.; Kubina, R.; Kabała-Dzik, A.; Tanasiewicz, M. Induction of Cell Cycle Arrest and Apoptotic Response of Head and Neck Squamous Carcinoma Cells (Detroit 562) by Caffeic Acid and Caffeic Acid Phenethyl Ester Derivative. Evid.-Based Complement. Altern. Med. 2017, 2017, 6793456. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Wani, M.Y.; Al-Bogami, A.S.; Ahmad, A. Piperidine based 1,2,3-triazolylacetamide derivatives induce cell cycle arrest and apoptotic cell death in Candida auris. J. Adv. Res. 2021, 29, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, D.; Rehman, T.; Bin Dukhyil, A.; Rizvi, S.M.D.; Al Ajmi, M.F.; Alshehri, B.M.; Banawas, S.; Khan, M.S.; Alturaiki, W.; Alsaweed, M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals 2021, 14, 937. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA Discovery Studio-BIOVIA-Dassault Systèmes®. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 1 October 2021).
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented Selectivity and Structural Determinants of a New Class of Protein Kinase CK2 Inhibitors in Clinical Trials for the Treatment of Cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Narwal, M.; Haikarainen, T.; Fallarero, A.; Vuorela, P.M.; Lehtiö, L. Screening and Structural Analysis of Flavones Inhibiting Tankyrases. J. Med. Chem. 2013, 56, 3507–3517. [Google Scholar] [CrossRef]
- Iqbal, D.; Khan, M.S.; Waiz, M.; Rehman, T.; Alaidarous, M.; Jamal, A.; Alothaim, A.S.; AlAjmi, M.F.; Alshehri, B.M.; Banawas, S.; et al. Exploring the Binding Pattern of Geraniol with Acetylcholinesterase through In Silico Docking, Molecular Dynamics Simulation, and In Vitro Enzyme Inhibition Kinetics Studies. Cells 2021, 10, 3533. [Google Scholar] [CrossRef]
- Bijani, S.; Jain, V.; Padmanabhan, D.; Pandey, B.; Shah, A. Mixed Pd/C and Pt/C as efficient catalysts for deuteration of Mesalamine. Tetrahedron Lett. 2015, 56, 1211–1214. [Google Scholar] [CrossRef]
- Jain, V.; Bijani, S.; Ambasana, P.; Bhoya, U.; Shah, A. Development of Nitromethane Catalyzed C-H Activation for the Preparation of Chromeno[3,4-d]imidazol-4-ones as Hybrid Scaffolds. Chem. Biol. Interface 2015, 5, 347–364. [Google Scholar]
- Jain, V.; Bijani, S.; Ambasana, P.; Mehariya, K.; Bhoya, U.; Pandey, B.; Shah, A. Diversity-oriented expedient route for the synthesis of 3-tetrahydropyrimidinyl-coumarins via MCR. Synth. Commun. 2015, 46, 63–72. [Google Scholar] [CrossRef]
- Morshed, S.R.M.D.; Hashimoto, K.; Murotani, Y.; Kawase, M.; Shah, A.; Satoh, K.; Kikuchi, H.; Nishikawa, H.; Maki, J.; Sakagami, H. Tumor-specific cytotoxicity of 3,5-dibenzoyl-1,4-dihydropyridines. Anticancer Res. 2005, 25, 2033–2038. [Google Scholar]
- Ben El Ayouchia, H.; Bahsis, L.; Anane, H.; Domingo, L.R.; Stiriba, S.-E. Understanding the mechanism and regioselectivity of the copper(i) catalyzed [3 + 2] cycloaddition reaction between azide and alkyne: A systematic DFT study. RSC Adv. 2018, 8, 7670–7678. [Google Scholar] [CrossRef] [Green Version]
- Bock, V.D.; Hiemstra, H.; van Maarseveen, J.H. Cu I-Catalyzed Alkyne–Azide “Click” Cycloadditions from a Mechanistic and Synthetic Perspective. Eur. J. Org. Chem. 2006, 2006, 51–68. [Google Scholar] [CrossRef]
- Chassaing, S.; Kumarraja, M.; Sido, A.S.S.; Pale, P.; Sommer, J. Click Chemistry in CuI-zeolites: The Huisgen [3 + 2]-Cycloaddition. Org. Lett. 2007, 9, 883–886. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, Y.; Meng, X.; Chen, Y. Pyridinyl-triazole ligand systems for highly efficient CuI-catalyzed azide-alkyne cycloaddition. Catal. Commun. 2021, 148, 106165. [Google Scholar] [CrossRef]
- Jiang, Y.; Kong, D.; Zhao, J.; Zhang, W.; Xu, W.; Li, W.; Xu, G. A simple, efficient thermally promoted protocol for Huisgen-click reaction catalyzed by CuSO4·5H2O in water. Tetrahedron Lett. 2014, 55, 2410–2414. [Google Scholar] [CrossRef]
- Kapadiya, K.; Jadeja, Y.; Khunt, R. Synthesis of Purine-based Triazoles by Copper (I)-catalyzed Huisgen Azide-Alkyne Cycloaddition Reaction. J. Heterocycl. Chem. 2018, 55, 199–208. [Google Scholar] [CrossRef]
- Cushman, M.; Georg, G.I.; Holzgrabe, U.; Wang, S. Absolute Quantitative 1H NMR Spectroscopy for Compound Purity Determination. J. Med. Chem. 2014, 57, 9219. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.F.; Chen, S.-N.; Simmler, C.; Lankin, D.C.; Gödecke, T.; Jaki, B.U.; Friesen, J.B.; McAlpine, J.B.; Napolitano, J.G. Importance of Purity Evaluation and the Potential of Quantitative 1H NMR as a Purity Assay. Available online: https://pubs.acs.org/doi/10.1021/jm500734a (accessed on 9 January 2022).
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2021, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A.M. Targeting cell cycle and apoptosis for the treatment of human malignancies. Curr. Opin. Cell Biol. 2004, 16, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Bak, D.-H.; Chung, B.Y.; Bai, H.-W. Centipedegrass extract enhances radiosensitivity in melanoma cells by inducing G2/M cell cycle phase arrest. Mol. Biol. Rep. 2021, 48, 1081–1091. [Google Scholar] [CrossRef]
- Bhosale, P.-B.; Vetrivel, P.; Ha, S.-E.; Kim, H.-H.; Heo, J.-D.; Won, C.-K.; Kim, S.-M.; Kim, G.-S. Iridin Induces G2/M Phase Cell Cycle Arrest and Extrinsic Apoptotic Cell Death through PI3K/AKT Signaling Pathway in AGS Gastric Cancer Cells. Molecules 2021, 26, 2802. [Google Scholar] [CrossRef]
- Reddy, D.; Kumavath, R.; Ghosh, P.; Barh, D. Lanatoside C Induces G2/M Cell Cycle Arrest and Suppresses Cancer Cell Growth by Attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR Signaling Pathways. Biomolecules 2019, 9, 792. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Zhang, W.; Cheng, C.; Mo, F.; Chen, L.; Peng, G.; Cai, X.; Wang, J.; Yang, S.; Liu, X. Dioscin inhibits the growth of human osteosarcoma by inducing G2/M-phase arrest, apoptosis, and GSDME-dependent cell death in vitro and in vivo. J. Cell. Physiol. 2020, 235, 2911–2924. [Google Scholar] [CrossRef]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy—Potential clinical relevance. Cell. Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef]
- Dubach, V.R.A.; Guskov, A. The Resolution in X-ray Crystallography and Single-Particle Cryogenic Electron Microscopy. Crystals 2020, 10, 580. [Google Scholar] [CrossRef]
- Martín-Acosta, P.; Amesty, Á.; Guerra-Rodríguez, M.; Guerra, B.; Fernández-Pérez, L.; Estévez-Braun, A. Modular Synthesis and Antiproliferative Activity of New Dihydro-1H-pyrazolo[1,3-b]pyridine Embelin Derivatives. Pharmaceuticals 2021, 14, 1026. [Google Scholar] [CrossRef]
- Neagu, G.; Stefaniu, A.; Albulescu, A.; Pintilie, L.; Pirvu, L.C. Antiproliferative Activity of Stokesia laevis Ethanolic Extract in Combination with Several Food-Related Bioactive Compounds; In Vitro (Caco-2) and In Silico Docking (TNKS1 and TNKS2) Studies. Appl. Sci. 2021, 11, 9944. [Google Scholar] [CrossRef]
- Brear, P.; Ball, D.; Stott, K.; D’Arcy, S.; Hyvönen, M. Proposed Allosteric Inhibitors Bind to the ATP Site of CK2α. J. Med. Chem. 2020, 63, 12786–12798. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Hirota, T. Pharmacological Interventions to Circadian Clocks and Their Molecular Bases. J. Mol. Biol. 2020, 432, 3498–3514. [Google Scholar] [CrossRef]
- Oramas-Royo, S.; Haidar, S.; Amesty, Á.; Martín-Acosta, P.; Feresin, G.; Tapia, A.; Aichele, D.; Jose, J.; Estévez-Braun, A. Design, synthesis and biological evaluation of new embelin derivatives as CK2 inhibitors. Bioorganic Chem. 2020, 95, 103520. [Google Scholar] [CrossRef]
- Damale, M.G.; Pathan, S.K.; Shinde, D.B.; Patil, R.H.; Arote, R.B.; Sangshetti, J.N. Insights of tankyrases: A novel target for drug discovery. Eur. J. Med. Chem. 2020, 207, 112712. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lian, H.; Shen, N.; Korm, S.; Lam, A.K.P.; Layton, O.; Huiting, L.N.; Li, D.; Miao, K.; Zeng, A.; et al. The multifaceted role of protein kinase CK2 in high-risk acute lymphoblastic leukemia. Haematologica 2021, 106, 1461–1465. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Cu Sources | Reductants | Time (Hours) | % Yield b |
| 1 | CuSO4·5H2O | - | 18 | Traces |
| 2 | CuSO4·5H2O | Sodium Ascorbate | 16 | 84 |
| 3 | CuSO4·5H2O | Hydrazine Hydrate | 3 | 69 |
| 4 | Cu(OAc)2 | - | 24 | Traces |
| 5 | Cu(OAc)2 | Sodium Ascorbate | 24 | 82 |
| 6 | Cu(OAc)2 | Hydrazine Hydrate | 0.5 | 96 |
| 7 | CuI | - | 36 | Traces |
| 8 | CuI | Hydrazine Hydrate | 24 | 72 |
| 9 | CuBr | - | 22 | Traces |
| 10 | CuBr | Hydrazine Hydrate | 18 | 76 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Sr. No. | Compound | Position | R1 | R2 | R3 | R4 | % Yield |
| 1 | 13aa′ | para | H | H | H | H | 96 |
| 2 | 13ab′ | para | H | OCH3 | OCH3 | OCH3 | 87 |
| 3 | 13ac′ | para | NO2 | H | H | H | 89 |
| 4 | 13ad′ | para | H | H | Cl | H | 84 |
| 5 | 13ae′ | para | H | Cl | H | H | 88 |
| 6 | 13af′ | para | H | H | Br | H | 92 |
| 7 | 13ag′ | para | H | Br | H | H | 89 |
| 8 | 14ba′ | ortho | H | H | H | H | 87 |
| 9 | 14bb′ | ortho | H | OCH3 | OCH3 | OCH3 | 89 |
| 10 | 14bc′ | ortho | NO2 | H | H | H | 85 |
| 11 | 14bd′ | ortho | H | H | Cl | H | 91 |
| 12 | 14be′ | ortho | H | Cl | H | H | 83 |
| 13 | 14bf′ | ortho | H | H | Br | H | 89 |
| 14 | 14bg′ | ortho | H | Br | H | H | 84 |
| 3PE1 | |||||
|---|---|---|---|---|---|
| Compounds | Hydrogen Bond | Hydrophobic | Unfavorable | ΔG (kcal/mol) | Kd (M−1) |
| 13ab′ | LYS49, LYS158, ALA193 | PHE113, LYS49, LEU178, TYR50 | −9.3 | 6.56 × 106 | |
| 13ad′ | LYS68, ASP175, ASP156, LYS158 | VAL53, LEU178, PHE113, LYS68 | −11 | 1.16 × 108 | |
| CX−4945 (native ligand) | LYS68, VAL116, ASN118 | LEU45, VAL53, VAL66, ILE95, PHE113, HIS115, HIS160, MET163, ILE174 | −10.8 | 8.3 × 107 | |
| 4HKI | |||||
| Compounds | Hydrogen Bond | Hydrophobic | Unfavorable | ΔG (kcal/mol) | Kd (M−1) |
| 13ab′ | ILE1075, HIS1048, GLY1032, GLU1138, PHE1061, PHE1030, TYR1060 | ILE1075, PHE1035 | −9 | 3.9 × 106 | |
| 13ad′ | HIS1048 | ILE1075, HIS1031, TYR1060, TYR1071, TYR1050, PHE1035, ALA1062, LYS1067, TYR1071, PRO1034 | −10.4 | 4.20 × 107 | |
| Flavone (native ligand) | GLY1032, SER1068, and HIS1031 | HIS1031, ALA1062, LYS1067, TYR1060, TYR1071 | −9.7 | 1.3 × 107 | |
| 4W6E | |||||
| Compounds | Hydrogen Bond | Hydrophobic | Unfavorable | ΔG (kcal/mol) | Kd (M−1) |
| 13ab′ | HIS1201, LYS1220, GLY1185, GLU1291, PHE1214, PHE1183, TYR1213 | −8.6 | 2.01 × 106 | ||
| 13ad′ | HIS1201, ILE1228 | ILE1228, HIS1201, HIS1184, ILE1192, HIS1201, ILE1212, ILE1198 | TYR1213 | −10 | 2.14 × 107 |
| AZ6102 (native ligand) | GLY1185, GLY1185, ALA1202, SER1221 | HIS1184, TYR1203, TYR1213, ALA1215, LYS1220 | TYR1224 | −10.9 | 9.8 × 107 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bijani, S.; Iqbal, D.; Mirza, S.; Jain, V.; Jahan, S.; Alsaweed, M.; Madkhali, Y.; Alsagaby, S.A.; Banawas, S.; Algarni, A.; et al. Green Synthesis and Anticancer Potential of 1,4-Dihydropyridines-Based Triazole Derivatives: In Silico and In Vitro Study. Life 2022, 12, 519. https://doi.org/10.3390/life12040519
Bijani S, Iqbal D, Mirza S, Jain V, Jahan S, Alsaweed M, Madkhali Y, Alsagaby SA, Banawas S, Algarni A, et al. Green Synthesis and Anticancer Potential of 1,4-Dihydropyridines-Based Triazole Derivatives: In Silico and In Vitro Study. Life. 2022; 12(4):519. https://doi.org/10.3390/life12040519
Chicago/Turabian StyleBijani, Sabera, Danish Iqbal, Sheefa Mirza, Vicky Jain, Sadaf Jahan, Mohammed Alsaweed, Yahya Madkhali, Suliman A. Alsagaby, Saeed Banawas, Abdulrahman Algarni, and et al. 2022. "Green Synthesis and Anticancer Potential of 1,4-Dihydropyridines-Based Triazole Derivatives: In Silico and In Vitro Study" Life 12, no. 4: 519. https://doi.org/10.3390/life12040519
APA StyleBijani, S., Iqbal, D., Mirza, S., Jain, V., Jahan, S., Alsaweed, M., Madkhali, Y., Alsagaby, S. A., Banawas, S., Algarni, A., Alrumaihi, F., Rawal, R. M., Alturaiki, W., & Shah, A. (2022). Green Synthesis and Anticancer Potential of 1,4-Dihydropyridines-Based Triazole Derivatives: In Silico and In Vitro Study. Life, 12(4), 519. https://doi.org/10.3390/life12040519