
Tree Reconciliation Methods for Host-Symbiont Cophylogenetic Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
I can understand how a flower and a bee might slowly become, either simultaneously or one after the other, modified and adapted in the most perfect manner to each other, by the continued preservation of individuals presenting mutual and slightly favorable deviations of structure [1].
2. The Phylogenetic Reconciliation Problem
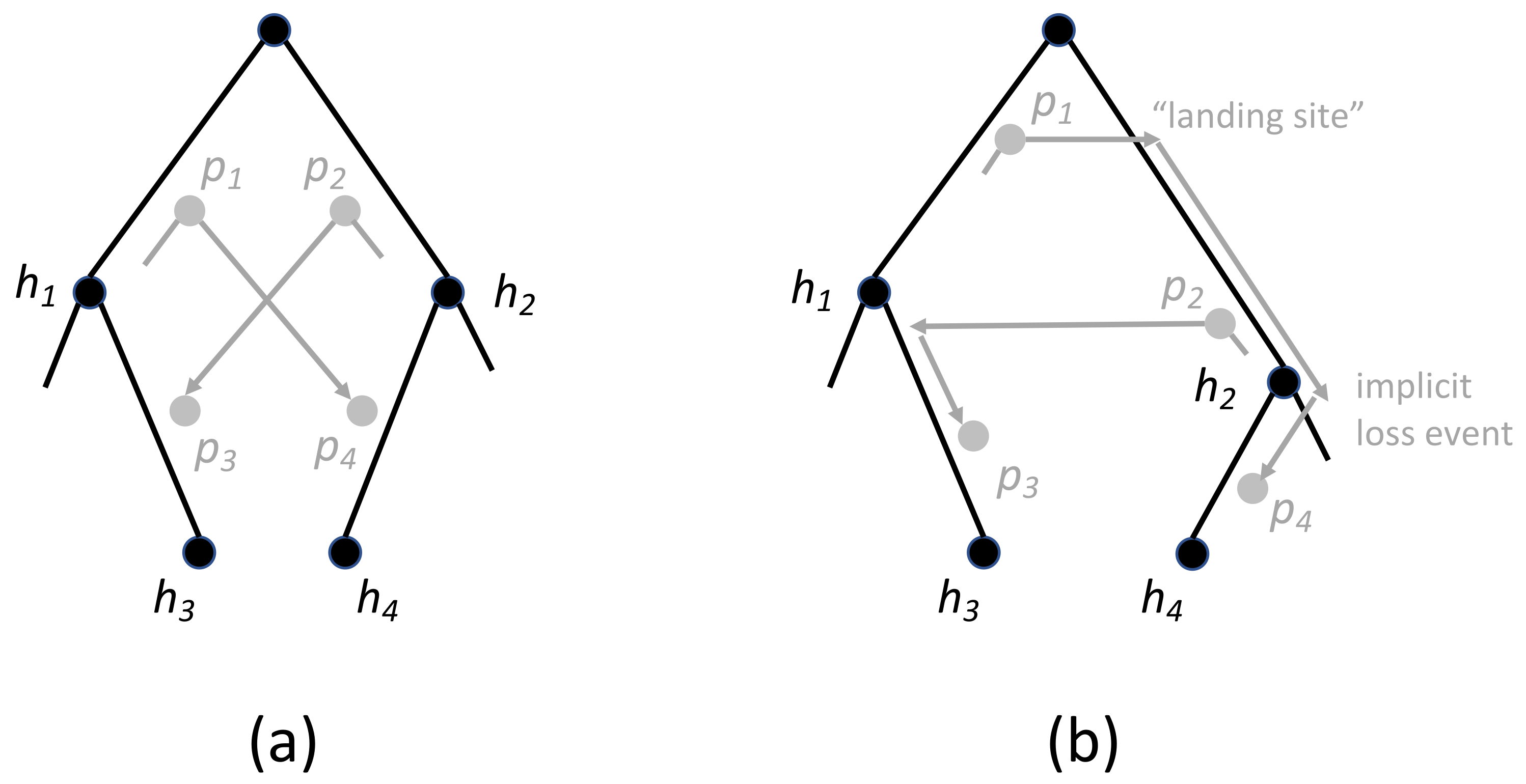
2.1. Reconciliations
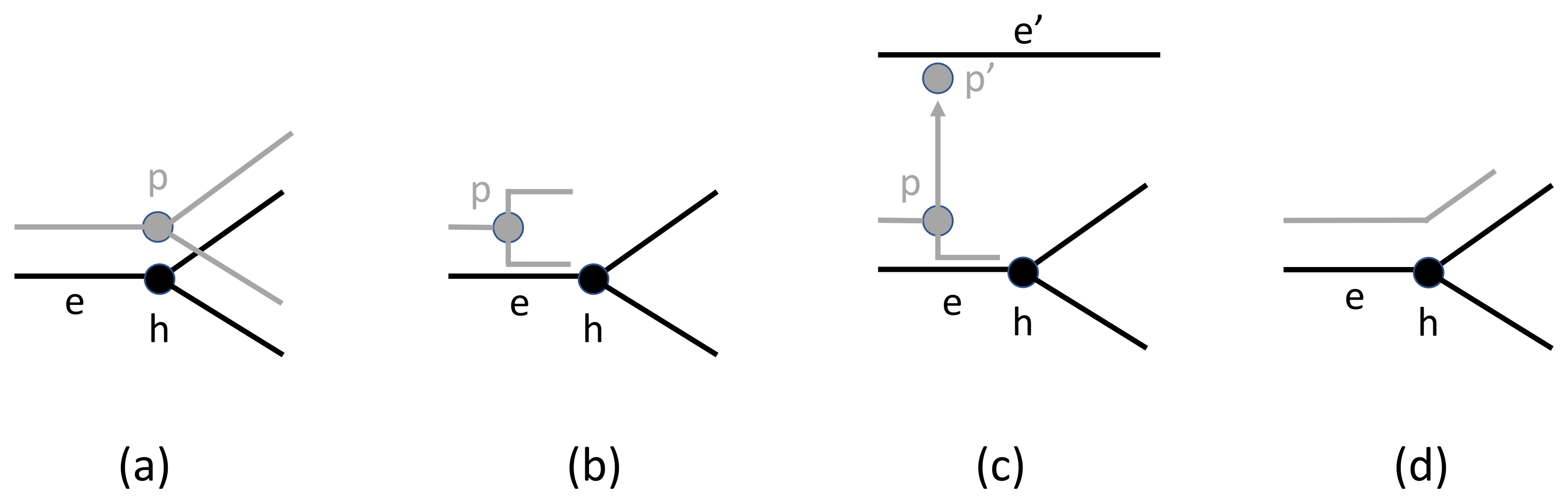
2.2. Events
2.3. Maximum Parsimony and Optimal Reconciliations
3. The Challenges of Reconciliation
3.1. Event Costs
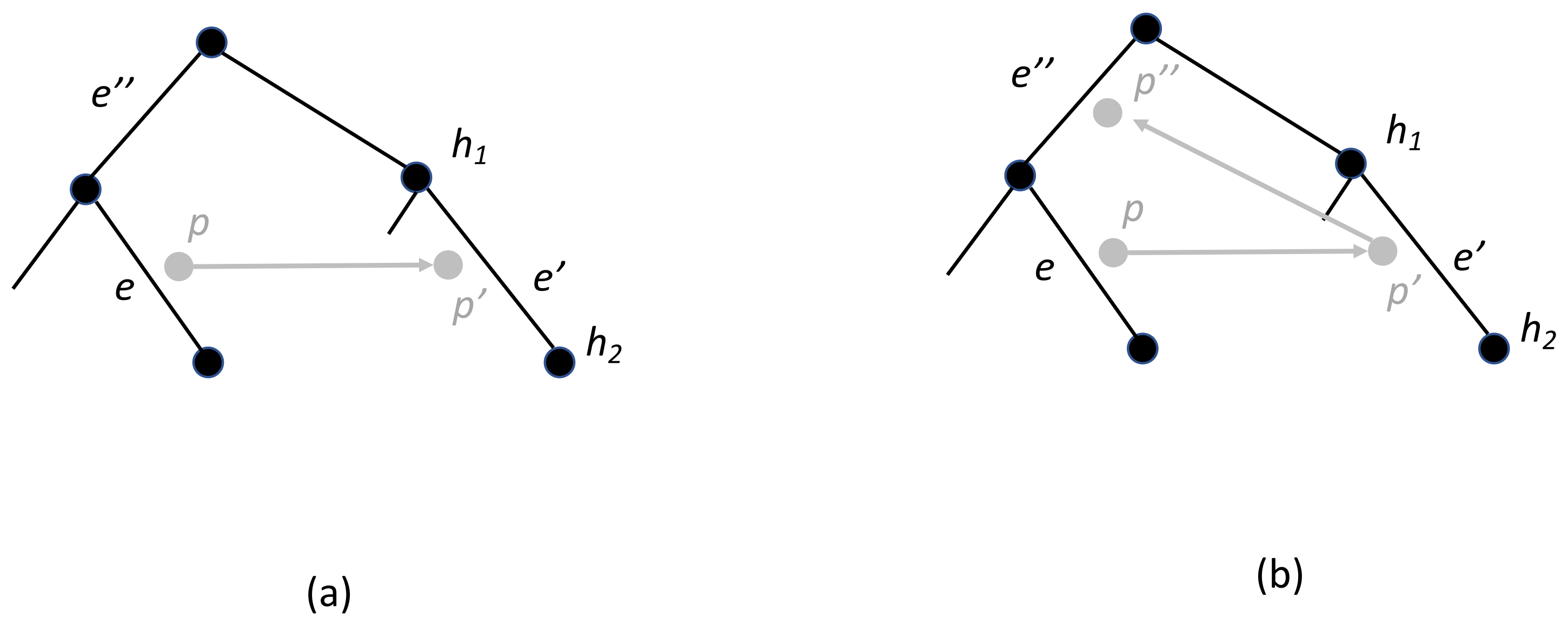
3.2. Dating and Time-Consistency
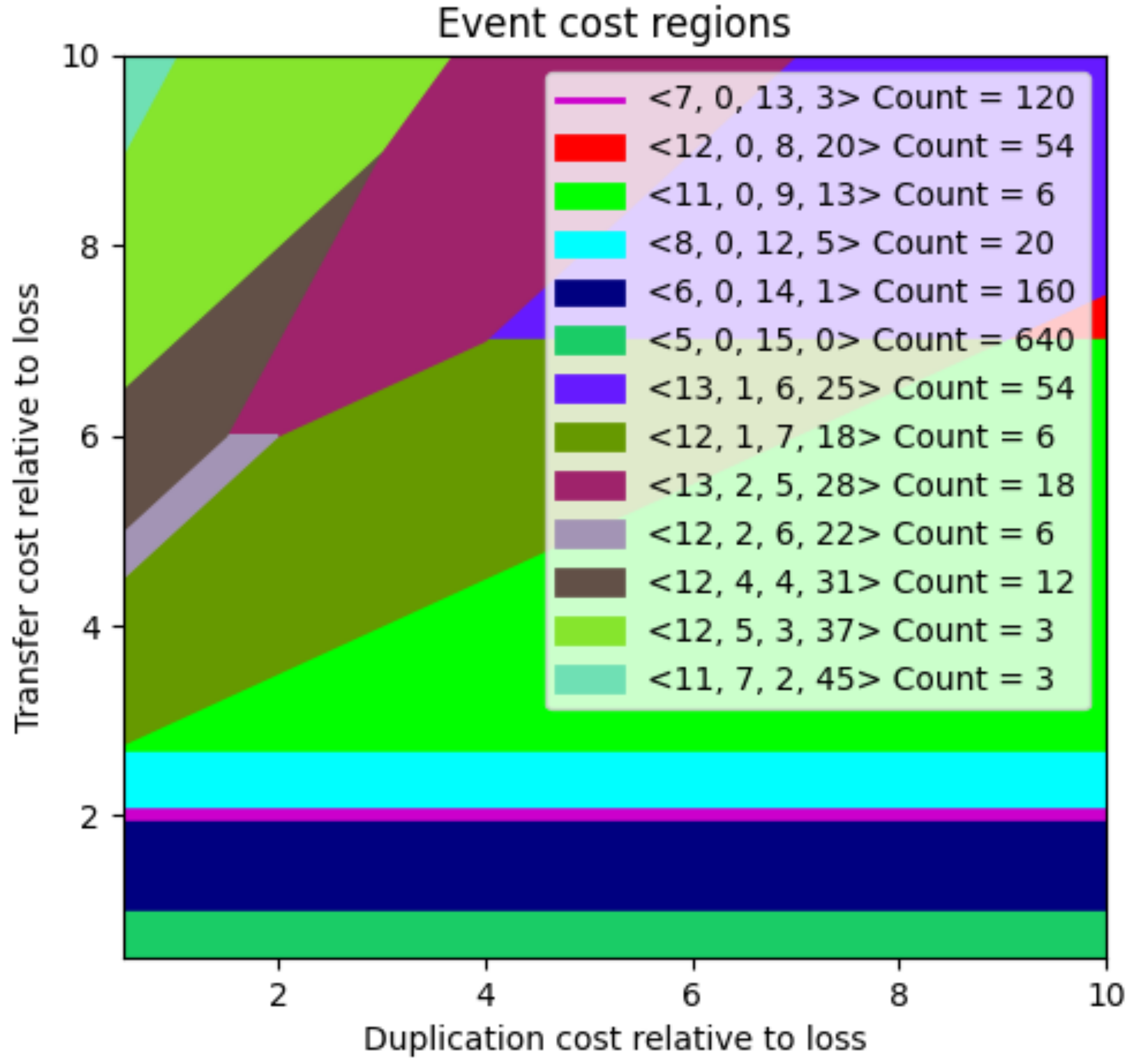
3.3. Selecting from the Large Space of MPRs
3.4. The Strength of Evidence for Tree Congruence
4. Algorithms and Tools
5. Reconciliation of Gene Trees and Species Trees
6. Conclusions and Future Work
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darwin, C. On the Origin of Species by Means of Natural Selection; Murray: London, UK, 1859. [Google Scholar]
- Legendre, P.; Desdevises, Y.; Bazin, E. A statistical test for host–parasite coevolution. Syst. Biol. 2002, 51, 217–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balbuena, J.A.; Míguez-Lozano, R.; Blasco-Costa, I. PACo: A novel procrustes application to cophylogenetic analysis. PLoS ONE 2013, 8, e61048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charleston, M.; Libeskind-Hadas, R. Event-Based Cophylogenetic Comparative Analysis. In Modern Phylogenetic Comparative Methods and Their Application in Evolutionary Biology: Concepts and Practice; Garamszegi, L.Z., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 465–480. [Google Scholar] [CrossRef]
- Filipiak, A.; Zajac, K.; Kübler, D.; Kramarz, P. Coevolution of host–parasite associations and methods for studying their cophylogeny. Invertebr. Surviv. J. 2016, 13, 56–65. [Google Scholar]
- Hayward, A.; Poulin, R.; Nakagawa, S. A broadscale analysis of host-symbiont cophylogeny reveals the drivers of phylogenetic congruence. Ecol. Lett. 2021, 24, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.L.; Poulin, R. Parasitism, commensalism, and mutualism: Exploring the many shades of symbioses. Vie Milieu/Life Environ. 2008, 58, 107–115. [Google Scholar]
- Libeskind-Hadas, R.; Wu, Y.C.; Bansal, M.S.; Kellis, M. Pareto-optimal phylogenetic tree reconciliation. Bioinformatics 2014, 30, i87–i95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorenson, M.D.; Balakrishnan, C.N.; Payne, R.B. Clade-limited colonization in brood parasitic finches (Vidua spp.). Syst. Biol. 2004, 53, 140–153. [Google Scholar] [CrossRef] [Green Version]
- Ovadia, Y.; Fielder, D.; Conow, C.; Libeskind-Hadas, R. The Cophylogeny Reconstruction Problem is NP-complete. J. Comput. Biol. 2011, 18, 59–65. [Google Scholar] [CrossRef]
- Tofigh, A.; Hallett, M.T.; Lagergren, J. Simultaneous Identification of Duplications and Lateral Gene Transfers. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 517–535. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.S.; Alm, E.J.; Kellis, M. Efficient algorithms for the reconciliation problem with gene duplication, horizontal transfer and loss. Bioinformatics 2012, 28, i283–i291. [Google Scholar] [CrossRef]
- Tofigh, A. Using Trees to Capture Reticulate Evolution: Lateral Gene Transfers and Cancer Progression. Ph.D. Thesis, KTH Royal Institute of Technology, Stockholm, Sweden, 2009. [Google Scholar]
- Szöllősi, G.J.; Tannier, E.; Lartillot, N.; Daubin, V. Lateral Gene Transfer from the Dead. Syst. Biol. 2013, 62, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Z.; Deng, F.; Wang, L. Simultaneous identification of duplications, losses, and lateral gene transfers. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1515–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdemir, A.; Sheely, M.; Bork, D.; Cheng, R.; Hulett, R.; Sung, J.; Wang, J.; Libeskind-Hadas, R. Clustering the Space of Maximum Parsimony Reconciliations in the Duplication-Transfer-Loss Model. In Algorithms for Computational Biology, Proceedings of the 4th International Conference, AlCoB 2017, Aveiro, Portugal, 5–6 June 2017; Springer International Publishing: Cham, Switzerland, 2017; pp. 127–139. [Google Scholar] [CrossRef]
- Santichaivekin, S.; Mawhorter, R.; Libeskind-Hadas, R. An efficient exact algorithm for computing all pairwise distances between reconciliations in the duplication-transfer-loss model. BMC Bioinform. 2019, 20, 636. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mary, A.; Sagot, M.F.; Sinaimeri, B. Capybara: Equivalence ClAss enumeration of coPhylogenY event-BAsed ReconciliAtions. Bioinformatics 2020, 36, 4197–4199. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Ranwez, V.; Berry, V.; Scornavacca, C. Support measures to estimate the reliability of evolutionary events predicted by reconciliation methods. PLoS ONE 2013, 8, e73667. [Google Scholar] [CrossRef] [Green Version]
- Scornavacca, C.; Paprotny, W.; Berry, V.; Ranwez, V. Representing a set of reconciliations in a compact way. J. Bioinform. Comput. Biol. 2013, 11, 1250025. [Google Scholar] [CrossRef] [Green Version]
- Grueter, M.; Duran, K.; Ramalingam, R.; Libeskind-Hadas, R. Reconciliation Reconsidered: In Search of a Most Representative Reconciliation in the Duplication-Transfer-Loss Model. In Proceedings of the 17 th Asian Pacific Bioinformatics Conference, Wuhan, China, 14–16 January 2019. [Google Scholar]
- Mawhorter, R.; Libeskind-Hadas, R. Hierarchical clustering of maximum parsimony reconciliations. BMC Bioinform. 2019, 20, 612. [Google Scholar] [CrossRef]
- Charleston, M. TreeMap 3. Available online: https://sites.google.com/site/cophylogeny/treemap (accessed on 10 January 2022).
- Merkle, D.; Middendorf, M.; Wieseke, N. A Parameter-adaptive Dynamic Programming Approach for Inferring Cophylogenies. BMC Bioinform. 2010, 11, S60. [Google Scholar] [CrossRef] [Green Version]
- Conow, C.; Fielder, D.; Ovadia, Y.; Libeskind-Hadas, R. Jane: A New Tool for Cophylogeny Reconstruction Problem. Algorithms Mol. Biol. 2010, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Santichaivekin, S.; Yang, Q.; Liu, J.; Mawhorter, R.; Jiang, J.; Wesley, T.; Wu, Y.C.; Libeskind-Hadas, R. eMPRess: A systematic cophylogeny reconciliation tool. Bioinformatics 2021, 37, 2481–2482. [Google Scholar] [CrossRef]
- Bansal, M.S.; Kellis, M.; Kordi, M.; Kundu, S. RANGER-DTL 2.0: Rigorous reconstruction of gene-family evolution by duplication, transfer and loss. Bioinformatics 2018, 34, 3214–3216. [Google Scholar] [CrossRef] [PubMed]
- Doyon, J.P.; Scornavacca, C.; Gorbunov, K.Y.; Szöllosi, J.G.; Ranwez, V.; Berry, V. An efficient algorithm for gene/species trees parsimonious reconciliation with losses, duplications and transfers. Comp. Genom. 2011, 6398, 93–108. [Google Scholar]
- Donati, B.; Baudet, C.; Sinaimeri, B.; Crescenzi, P.; Sagot, M.F. EUCALYPT: Efficient tree reconciliation enumerator. Algorithms Mol. Biol. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacox, E.; Chauve, C.; Szöllősi, G.J.; Ponty, Y.; Scornavacca, C. ecceTERA: Comprehensive gene tree-species tree reconciliation using parsimony. Bioinformatics 2016, 32, 2056–2058. [Google Scholar] [CrossRef] [PubMed]
- Charleston, M.A.; Perkins, S.L. Traversing the tangle: Algorithms and applications for cophylogenetic studies. J. Biomed. Inform. 2006, 39, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.S.; Alm, E.J.; Kellis, M. Reconciliation Revisited: Handling Multiple Optima when Reconciling with Duplication, Transfer, and Loss. J. Comput. Biol. 2013, 20, 738–754. [Google Scholar] [CrossRef] [Green Version]
- Doyon, J.P.; Ranwez, V.; Daubin, V.; Berry, V. Models, algorithms and programs for phylogeny reconciliation. Briefings Bioinform. 2011, 12, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.C.; Rasmussen, M.D.; Bansal, M.S.; Kellis, M. Most Parsimonious Reconciliation in the Presence of Gene Duplication, Loss, and Deep Coalescence using Labeled Coalescent Trees. Genome Res. 2014, 24, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Szöllősi, G.J.; Tannier, E.; Daubin, V.; Boussau, B. The Inference of Gene Trees with Species Trees. Syst. Biol. 2014, 64, e42–e62. [Google Scholar] [CrossRef] [Green Version]
- Dismukes, W.; Heath, T.A. treeducken: An R package for simulating cophylogenetic systems. Methods Ecol. Evol. 2021, 12, 1358–1364. [Google Scholar] [CrossRef]
- Johnson, K.P.; Clayton, D.H. Untangling Coevolutionary History. Syst. Biol. 2004, 53, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libeskind-Hadas, R. Tree Reconciliation Methods for Host-Symbiont Cophylogenetic Analyses. Life 2022, 12, 443. https://doi.org/10.3390/life12030443
Libeskind-Hadas R. Tree Reconciliation Methods for Host-Symbiont Cophylogenetic Analyses. Life. 2022; 12(3):443. https://doi.org/10.3390/life12030443
Chicago/Turabian StyleLibeskind-Hadas, Ran. 2022. "Tree Reconciliation Methods for Host-Symbiont Cophylogenetic Analyses" Life 12, no. 3: 443. https://doi.org/10.3390/life12030443
APA StyleLibeskind-Hadas, R. (2022). Tree Reconciliation Methods for Host-Symbiont Cophylogenetic Analyses. Life, 12(3), 443. https://doi.org/10.3390/life12030443