
Distinguishing Evolutionary Conservation from Derivedness


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Confusion between Conservation and Derivedness Leads to Inconsistent Conclusions
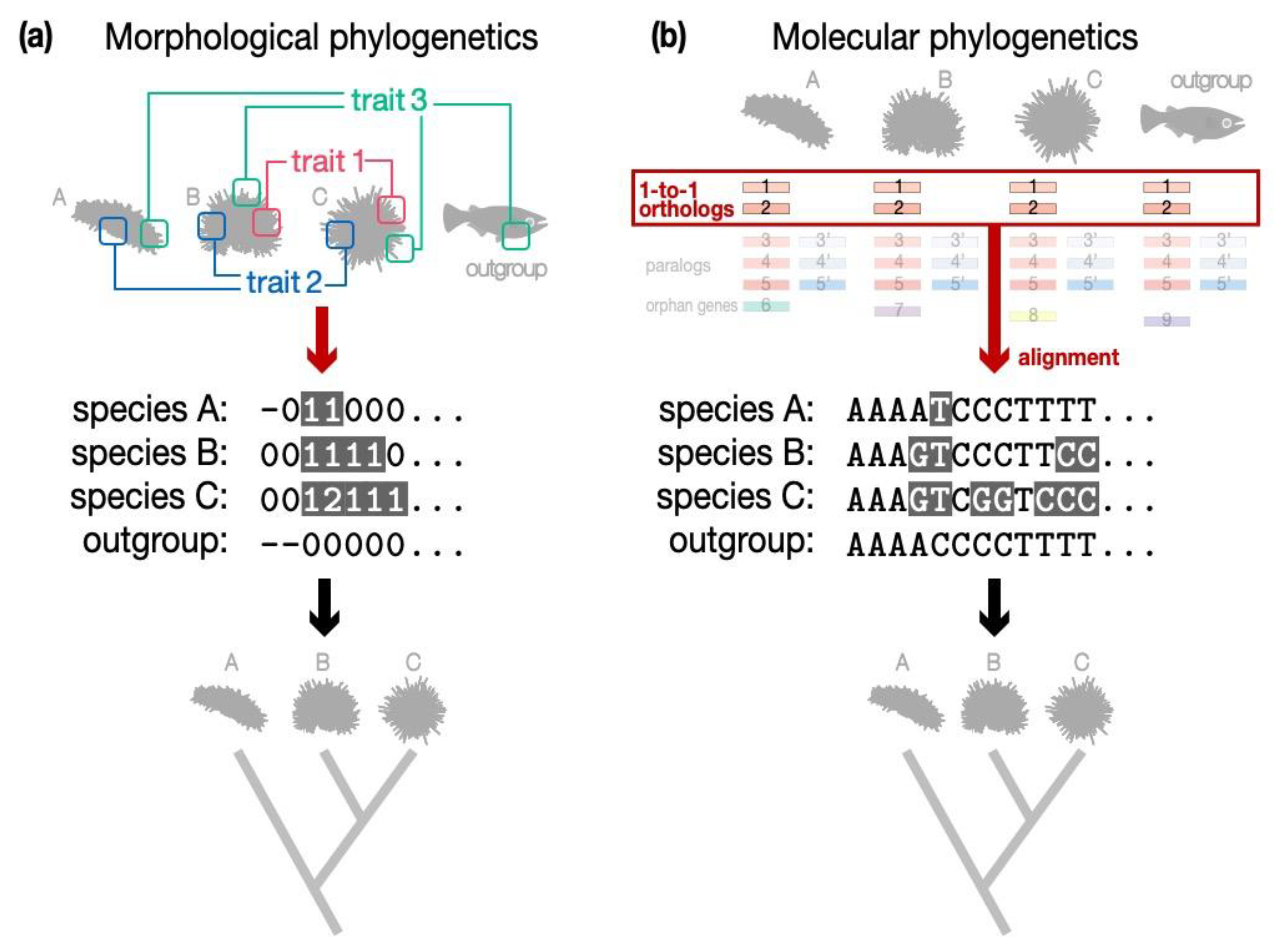
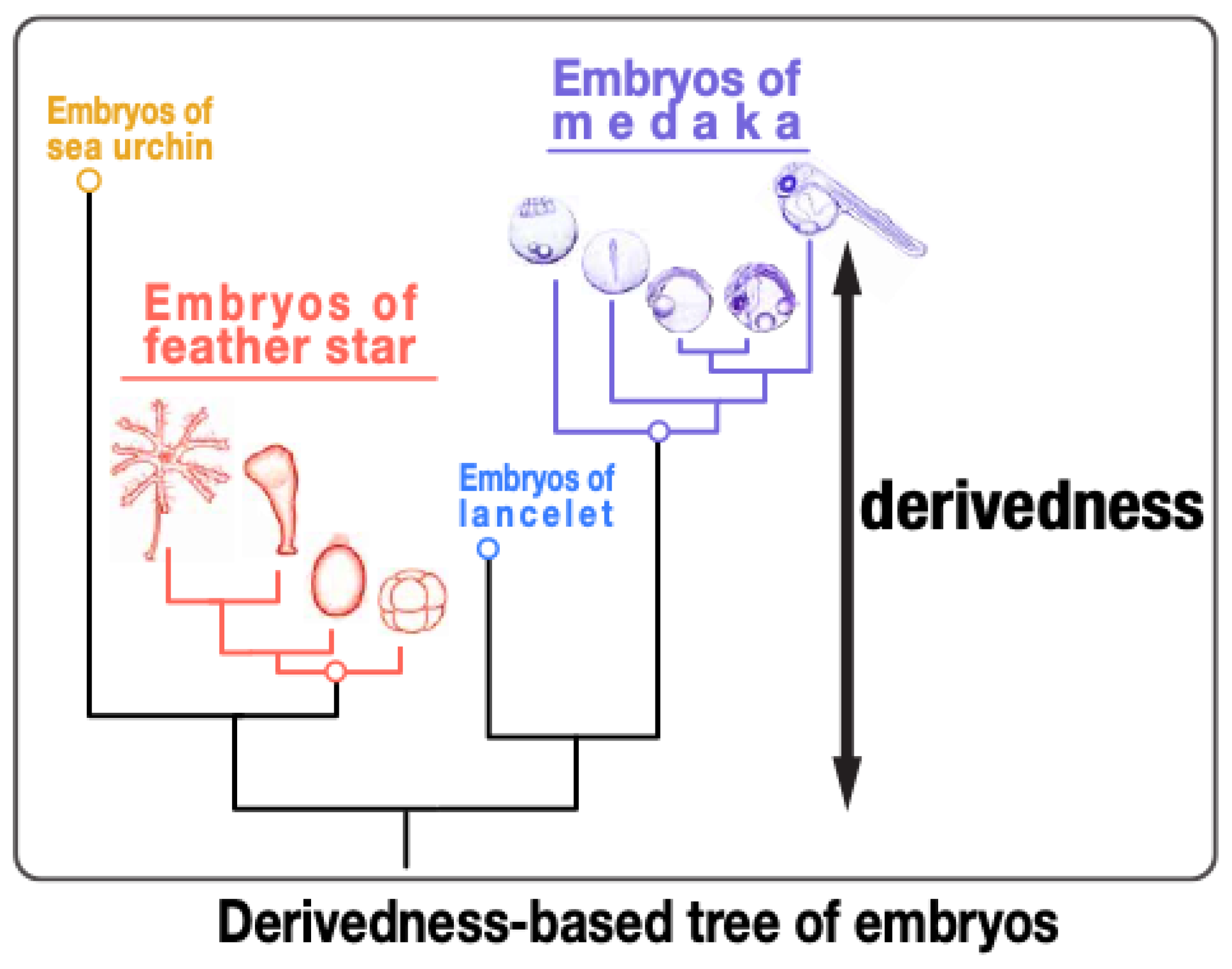
2.1. Inconsistent Results Are Often Obtained from Molecular-Based Phylogenetic Estimation and Morphology-Based Estimation
2.2. Conservation-Oriented Approach May Be Less Sufficient in Elucidating Derived Features
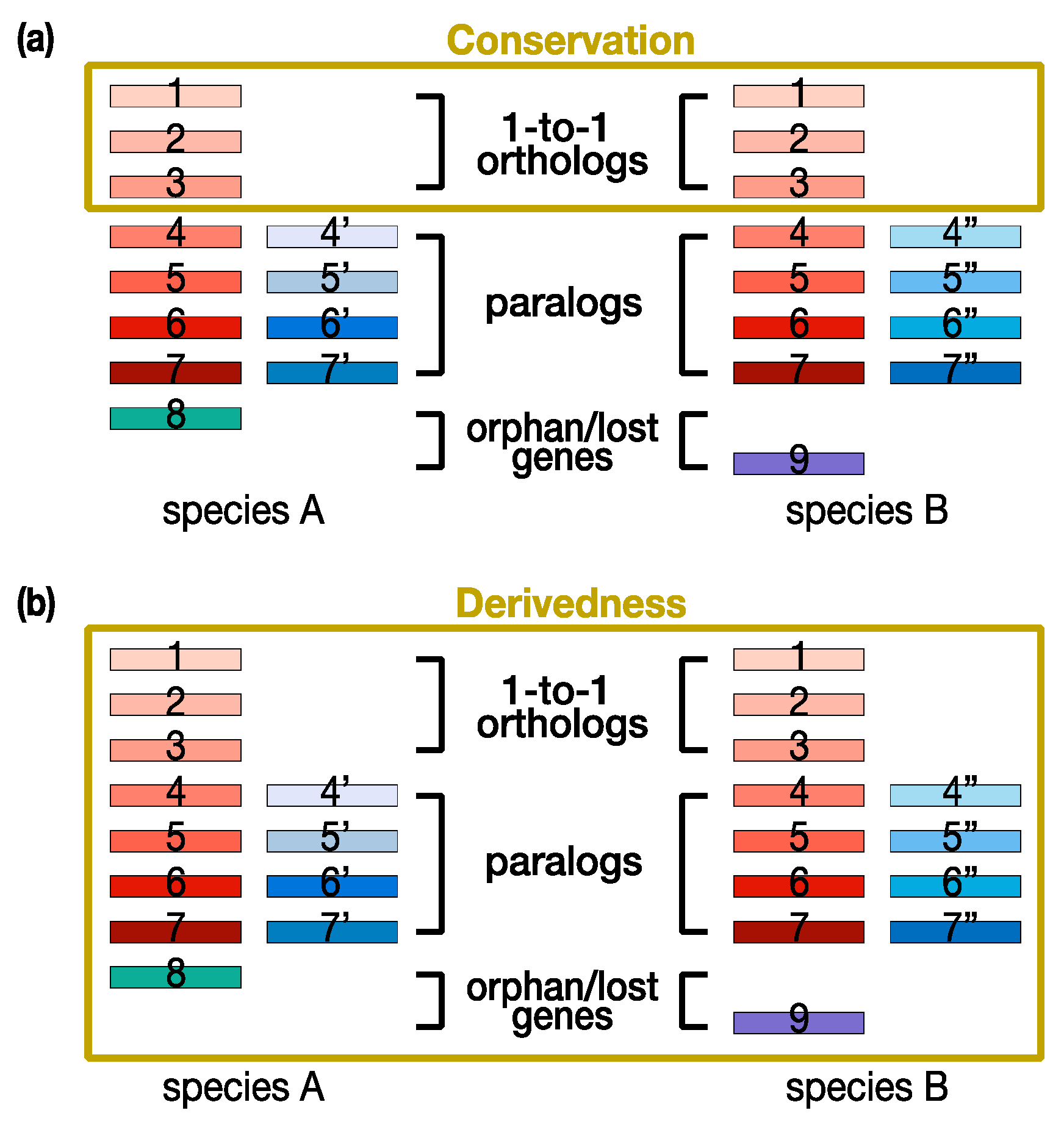
3. Technical Limitations of Current Conservation-Oriented and Derivedness-Oriented Molecular Approaches
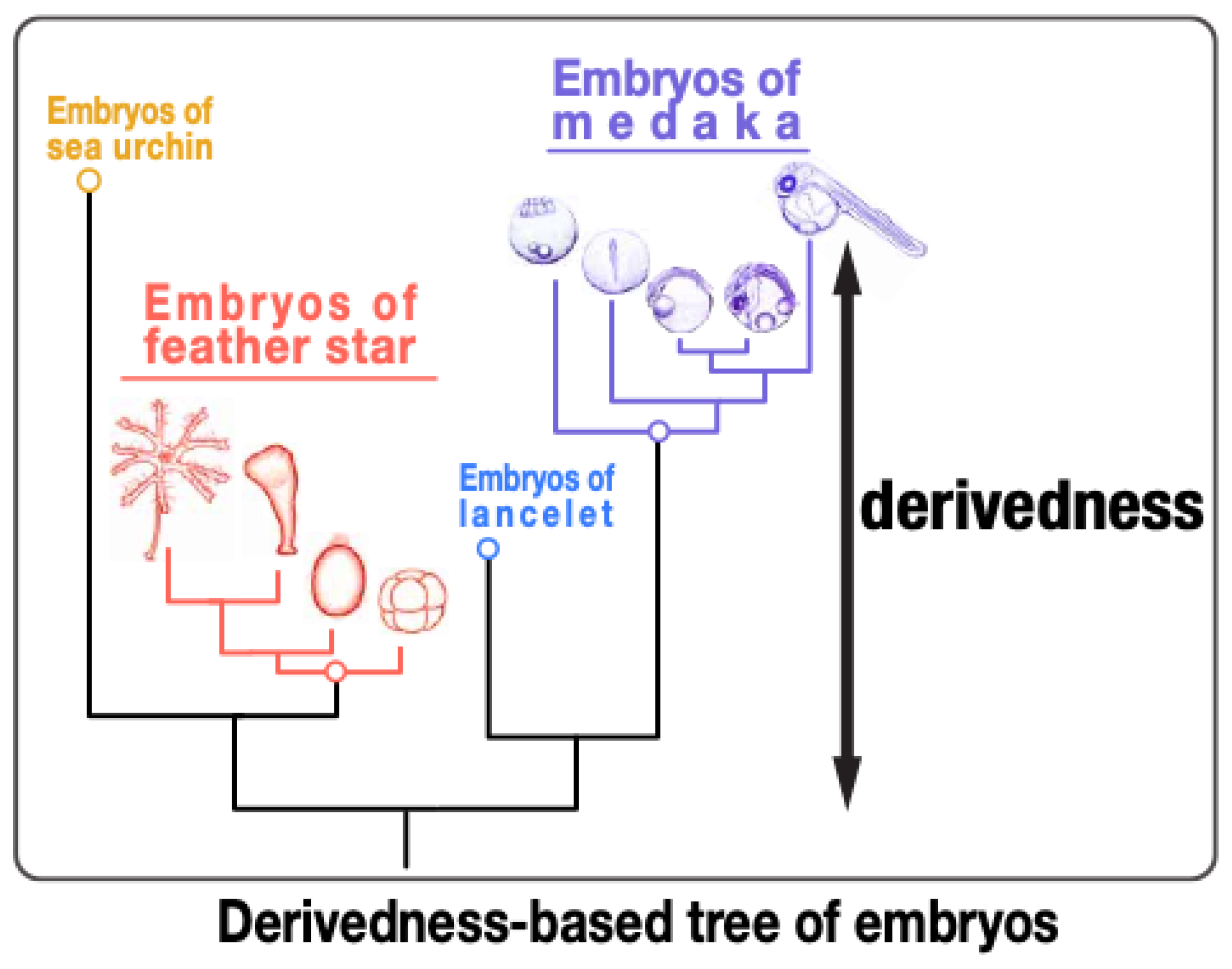
4. “Transcriptomic Derivedness Index” for Quantifying Degree of Phenotypic Evolution of Embryos
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Erwin, D.H. The Origin of Animal Body Plans: A View from Fossil Evidence and the Regulatory Genome. Development 2020, 147, dev182899. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.B.; Grenier, J.K.; Weatherbee, S.D. From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design, 2nd ed.; Blackwell Publishing: Malden, MA, USA, 2005. [Google Scholar]
- Olsen, G.J.; Woese, C.R. Ribosomal RNA: A Key to Phylogeny. FASEB J. 1993, 7, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Scotland, R.W. Deep Homology: A View from Systematics. Bioessays 2010, 32, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Kuratani, S. Comparative Transcriptome Analysis Reveals Vertebrate Phylotypic Period during Organogenesis. Nat. Commun. 2011, 2, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, I.; Peshkin, L.; Jorgensen, P.; Kirschner, M.W. Mapping Gene Expression in Two Xenopus Species: Evolutionary Constraints and Developmental Flexibility. Dev. Cell 2011, 20, 483–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Uesaka, M.; Guo, S.; Shimai, K.; Lu, T.-M.; Li, F.; Fujimoto, S.; Ishikawa, M.; Liu, S.; Sasagawa, Y.; et al. Constrained Vertebrate Evolution by Pleiotropic Genes. Nat. Ecol. Evol. 2017, 1, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Uesaka, M.; Kuratani, S.; Takeda, H.; Irie, N. Recapitulation-like Developmental Transitions of Chromatin Accessibility in Vertebrates. Zool. Lett. 2019, 5, 33. [Google Scholar] [CrossRef]
- Li, Y.; Kikuchi, M.; Li, X.; Gao, Q.; Xiong, Z.; Ren, Y.; Zhao, R.; Mao, B.; Kondo, M.; Irie, N.; et al. Weighted Gene Co-Expression Network Analysis Reveals Potential Genes Involved in Early Metamorphosis Process in Sea Cucumber Apostichopus japonicus. Biochem. Biophys. Res. Commun. 2018, 495, 1395–1402. [Google Scholar] [CrossRef]
- Hogan, J.D.; Keenan, J.L.; Luo, L.; Ibn-Salem, J.; Lamba, A.; Schatzberg, D.; Piacentino, M.L.; Zuch, D.T.; Core, A.B.; Blumberg, C.; et al. The Developmental Transcriptome for Lytechinus variegatus Exhibits Temporally Punctuated Gene Expression Changes. Dev. Biol. 2020, 460, 139–154. [Google Scholar] [CrossRef] [Green Version]
- Gildor, T.; Cary, G.A.; Lalzar, M.; Hinman, V.F.; de-Leon, S.B.-T. Developmental Transcriptomes of the Sea Star, Patiria miniata, Illuminate How Gene Expression Changes with Evolutionary Distance. Sci. Rep. 2019, 9, 16201. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Pascual-Anaya, J.; Zadissa, A.; Li, W.; Niimura, Y.; Huang, Z.; Li, C.; White, S.; Xiong, Z.; Fang, D.; et al. The Draft Genomes of Soft-Shell Turtle and Green Sea Turtle Yield Insights into the Development and Evolution of the Turtle-Specific Body Plan. Nat. Genet. 2013, 45, 701–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Fan, S.; Zhang, Y.; Xu, M.; Zhang, H.; Yang, Y.; Lee, A.P.; Woltering, J.M.; Ravi, V.; Gunter, H.M.; et al. The Seahorse Genome and the Evolution of Its Specialized Morphology. Nature 2016, 540, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, R.; Li, C.; Fang, Q.; Hayashi, S.; Egawa, S.; Hu, J.; Xu, L.; Pan, H.; Kondo, M.; Sato, T.; et al. Functional Roles of Aves Class-Specific Cis-Regulatory Elements on Macroevolution of Bird-Specific Features. Nat. Commun. 2017, 8, 14229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, C.R.; Wegrzyn, J.L.; Jockusch, E.L. Co-Option of Wing-Patterning Genes Underlies the Evolution of the Treehopper Helmet. Nat. Ecol. Evol. 2020, 4, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Martinez, D.; de Magalhães, J.P.; Opazo, J.C. Positive Selection and Gene Duplications in Tumour Suppressor Genes Reveal Clues about How Cetaceans Resist Cancer. Proc. R. Soc. B 2021, 288, 20202592. [Google Scholar] [CrossRef]
- Lü, Z.; Gong, L.; Ren, Y.; Chen, Y.; Wang, Z.; Liu, L.; Li, H.; Chen, X.; Li, Z.; Luo, H.; et al. Large-Scale Sequencing of Flatfish Genomes Provides Insights into the Polyphyletic Origin of Their Specialized Body Plan. Nat. Genet. 2021, 53, 742–751. [Google Scholar] [CrossRef]
- Chan, Y.F.; Marks, M.E.; Jones, F.C.; Villarreal, G.; Shapiro, M.D.; Brady, S.D.; Southwick, A.M.; Absher, D.M.; Grimwood, J.; Schmutz, J.; et al. Adaptive Evolution of Pelvic Reduction in Sticklebacks by Recurrent Deletion of a Pitx1 Enhancer. Science 2010, 327, 302–305. [Google Scholar] [CrossRef] [Green Version]
- McLean, C.Y.; Reno, P.L.; Pollen, A.A.; Bassan, A.I.; Capellini, T.D.; Guenther, C.; Indjeian, V.B.; Lim, X.; Menke, D.B.; Schaar, B.T.; et al. Human-Specific Loss of Regulatory DNA and the Evolution of Human-Specific Traits. Nature 2011, 471, 216–219. [Google Scholar] [CrossRef]
- Sadier, A.; Sears, K.E.; Womack, M. Unraveling the Heritage of Lost Traits. J. Exp. Zool. Part B Mol. Dev. Evol. 2021, 338, 107–118. [Google Scholar] [CrossRef]
- Parker, J.; Tsagkogeorga, G.; Cotton, J.A.; Liu, Y.; Provero, P.; Stupka, E.; Rossiter, S.J. Genome-Wide Signatures of Convergent Evolution in Echolocating Mammals. Nature 2013, 502, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Castoe, T.A.; de Koning, A.P.J.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese Python Genome Reveals the Molecular Basis for Extreme Adaptation in Snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallant, J.R.; Traeger, L.L.; Volkening, J.D.; Moffett, H.; Chen, P.-H.; Novina, C.D.; Phillips, G.N.; Anand, R.; Wells, G.B.; Pinch, M.; et al. Genomic Basis for the Convergent Evolution of Electric Organs. Science 2014, 344, 1522–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foote, A.D.; Liu, Y.; Thomas, G.W.C.; Vinař, T.; Alföldi, J.; Deng, J.; Dugan, S.; van Elk, C.E.; Hunter, M.E.; Joshi, V.; et al. Convergent Evolution of the Genomes of Marine Mammals. Nat. Genet. 2015, 47, 272–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Omori, A.; Flores, R.L.; Satterfield, S.; Nguyen, C.; Ota, T.; Tsurugaya, T.; Ikuta, T.; Ikeo, K.; Kikuchi, M.; et al. Genomic Insights of Body Plan Transitions from Bilateral to Pentameral Symmetry in Echinoderms. Commun. Biol. 2020, 3, 371. [Google Scholar] [CrossRef]
- Leong, J.C.K.; Li, Y.; Uesaka, M.; Uchida, Y.; Omori, A.; Hao, M.; Wan, W.; Dong, Y.; Ren, Y.; Zhang, S.; et al. Derivedness Index for Estimating Degree of Phenotypic Evolution of Embryos: A Study of Comparative Transcriptomic Analyses of Chordates and Echinoderms. Front. Cell Dev. Biol. 2021, 9, 749963. [Google Scholar] [CrossRef]
- Koonin, E.V. Orthologs, Paralogs, and Evolutionary Genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef] [Green Version]
- Hedges, S.B. Amniote Phylogeny and the Position of Turtles. BMC Biol. 2012, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Green, R.E.; Braun, E.L.; Armstrong, J.; Earl, D.; Nguyen, N.; Hickey, G.; Vandewege, M.W.; John, J.A.S.; Capella-Gutierrez, S.; Castoe, T.A.; et al. Three Crocodilian Genomes Reveal Ancestral Patterns of Evolution among Archosaurs. Science 2014, 346, 1254449. [Google Scholar] [CrossRef] [Green Version]
- Lyson, T.R.; Bever, G.S. Origin and Evolution of the Turtle Body Plan. Annu. Rev. Ecol. Evol. Syst. 2020, 51, 1–24. [Google Scholar] [CrossRef]
- Hillis, D.M. Molecular versus Morphological Approaches to Systematics. Annu. Rev. Ecol. Syst. 1987, 18, 23–42. [Google Scholar] [CrossRef]
- Lee, M.S.Y.; Palci, A. Morphological Phylogenetics in the Genomic Age. Curr. Biol. 2015, 25, R922–R929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, M.G.; Norman, D.B.; Barrett, P.M. A New Hypothesis of Dinosaur Relationships and Early Dinosaur Evolution. Nature 2017, 543, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Langer, M.C.; Ezcurra, M.D.; Rauhut, O.W.M.; Benton, M.J.; Knoll, F.; McPhee, B.W.; Novas, F.E.; Pol, D.; Brusatte, S.L. Untangling the Dinosaur Family Tree. Nature 2017, 551, E1–E3. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.G.; Norman, D.B.; Barrett, P.M. Baron et al. Reply. Nature 2017, 551, E4–E5. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rannala, B. Molecular Phylogenetics: Principles and Practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Inferring Phylogenies; Sinauer Associates: Sunderland, MA, USA, 2004; ISBN 0878931775. [Google Scholar]
- Kapli, P.; Yang, Z.; Telford, M.J. Phylogenetic Tree Building in the Genomic Age. Nat. Rev. Genet. 2020, 21, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the Reconstruction of the Tree of Life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Muse, S.V.; Gaut, B.S. A Likelihood Approach for Comparing Synonymous and Nonsynonymous Nucleotide Substitution Rates, with Application to the Chloroplast Genome. Mol. Biol. Evol. 1994, 11, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, N.; Yang, Z. A Codon-Based Model of Nucleotide Substitution for Protein-Coding DNA Sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. Maximum Likelihood Phylogenetic Estimation from DNA Sequences with Variable Rates over Sites: Approximate Methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas, M. Trends in Substitution Models of Molecular Evolution. Front. Genet. 2015, 6, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichio, B.T.L.; Marchaukoski, J.N.; Raittz, R.T. New Tools in Orthology Analysis: A Brief Review of Promising Perspectives. Front. Genet. 2017, 8, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmuth, M.; Wieseke, N.; Lechner, M.; Lenhof, H.-P.; Middendorf, M.; Stadler, P.F. Phylogenomics with Paralogs. Proc. Natl. Acad. Sci. USA 2015, 112, 2058–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevosti, F.J.; Chemisquy, M.A. The Impact of Missing Data on Real Morphological Phylogenies: Influence of the Number and Distribution of Missing Entries. Cladistics 2010, 26, 326–339. [Google Scholar] [CrossRef]
- Zou, Z.; Zhang, J. Morphological and Molecular Convergences in Mammalian Phylogenetics. Nat. Commun. 2016, 7, 12758. [Google Scholar] [CrossRef] [Green Version]
- Amemiya, C.T.; Alföldi, J.; Lee, A.P.; Fan, S.; Philippe, H.; MacCallum, I.; Braasch, I.; Manousaki, T.; Schneider, I.; Rohner, N.; et al. The African Coelacanth Genome Provides Insights into Tetrapod Evolution. Nature 2013, 496, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Gemmell, N.J.; Rutherford, K.; Prost, S.; Tollis, M.; Winter, D.; Macey, J.R.; Adelson, D.L.; Suh, A.; Bertozzi, T.; Grau, J.H.; et al. The Tuatara Genome Reveals Ancient Features of Amniote Evolution. Nature 2020, 584, 403–409. [Google Scholar] [CrossRef]
- Hay, J.M.; Subramanian, S.; Millar, C.D.; Mohandesan, E.; Lambert, D.M. Rapid Molecular Evolution in a Living Fossil. Trends Genet. 2008, 24, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Casane, D.; Laurenti, P. Why Coelacanths Are Not ‘Living Fossils’. Bioessays 2013, 35, 332–338. [Google Scholar] [CrossRef]
- Cavin, L.; Guinot, G. Coelacanths as “Almost Living Fossils”. Front. Ecol. Evol. 2014, 2, 49. [Google Scholar] [CrossRef] [Green Version]
- Creevey, C.J.; Muller, J.; Doerks, T.; Thompson, J.D.; Arendt, D.; Bork, P. Identifying Single Copy Orthologs in Metazoa. PLoS Comput. Biol. 2011, 7, e1002269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, V.E.; Pickett, F.B. Splitting Pairs: The Diverging Fates of Duplicated Genes. Nat. Rev. Genet. 2002, 3, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Krinsky, B.H.; Long, M. New Genes as Drivers of Phenotypic Evolution. Nat. Rev. Genet. 2013, 14, 645–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albalat, R.; Cañestro, C. Evolution by Gene Loss. Nat. Rev. Genet. 2016, 17, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Anavy, L.; Cole, A.G.; Winter, E.; Mostov, N.; Khair, S.; Senderovich, N.; Kovalev, E.; Silver, D.H.; Feder, M.; et al. The Mid-Developmental Transition and the Evolution of Animal Body Plans. Nature 2016, 531, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Guschanski, K.; Warnefors, M.; Kaessmann, H. The Evolution of Duplicate Gene Expression in Mammalian Organs. Genome Res. 2017, 27, 1461–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, K.; Pollock, D.D. Amalgamated Cross-Species Transcriptomes Reveal Organ-Specific Propensity in Gene Expression Evolution. Nat. Commun. 2020, 11, 4459. [Google Scholar] [CrossRef] [PubMed]
- Hyman, L.H. The Invertebrates: Echinodermata: The Coelomate Bilateria; McGraw-Hill: New York, NY, USA, 1955; Volume 4. [Google Scholar]
- Brusca, R.C.; Moore, W.; Shuster, S.M. Invertebrates, 3rd ed.; Sinauer Associates: Sunderland, MA, USA, 2016; ISBN 1605353752. [Google Scholar]
- Hazkani-Covo, E.; Wool, D.; Graur, D. In Search of the Vertebrate Phylotypic Stage: A Molecular Examination of the Developmental Hourglass Model and von Baer’s Third Law. J. Exp. Zool. Part B Mol. Dev. Evol. 2005, 304B, 150–158. [Google Scholar] [CrossRef]
- Irie, N.; Sehara-Fujisawa, A. The Vertebrate Phylotypic Stage and an Early Bilaterian-Related Stage in Mouse Embryogenesis Defined by Genomic Information. BMC Biol. 2007, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Domazet-Lošo, T.; Tautz, D. A Phylogenetically Based Transcriptome Age Index Mirrors Ontogenetic Divergence Patterns. Nature 2010, 468, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Kalinka, A.T.; Varga, K.M.; Gerrard, D.T.; Preibisch, S.; Corcoran, D.L.; Jarrells, J.; Ohler, U.; Bergman, C.M.; Tomancak, P. Gene Expression Divergence Recapitulates the Developmental Hourglass Model. Nature 2010, 468, 811–814. [Google Scholar] [CrossRef]
- Liu, J.; Viales, R.R.; Khoueiry, P.; Reddington, J.P.; Girardot, C.; Furlong, E.; Robinson-Rechavi, M. The Hourglass Model of Evolutionary Conservation during Embryogenesis Extends to Developmental Enhancers with Signatures of Positive Selection. Genome Res. 2021, 31, gr.275212.121. [Google Scholar] [CrossRef]
- Levin, M.; Hashimshony, T.; Wagner, F.; Yanai, I. Developmental Milestones Punctuate Gene Expression in the Caenorhabditis Embryo. Dev. Cell 2012, 22, 1101–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Domazet-Lošo, T.; Fan, D.; Dunwell, T.L.; Li, L.; Fang, X.; Zhang, G. High Expression of New Genes in Trochophore Enlightening the Ontogeny and Evolution of Trochozoans. Sci. Rep. 2016, 6, 34664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Tabari, E.; Su, Z. PorthoMCL: Parallel Orthology Prediction Using MCL for the Realm of Massive Genome Availability. Big Data Anal. 2017, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Berná, L.; Alvarez-Valin, F. Evolutionary Genomics of Fast Evolving Tunicates. Genome Biol. Evol. 2014, 6, 1724–1738. [Google Scholar] [CrossRef] [Green Version]
- Holland, L.Z. Tunicates. Curr. Biol. 2016, 26, R146–R152. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leong, J.C.K.; Uesaka, M.; Irie, N. Distinguishing Evolutionary Conservation from Derivedness. Life 2022, 12, 440. https://doi.org/10.3390/life12030440
Leong JCK, Uesaka M, Irie N. Distinguishing Evolutionary Conservation from Derivedness. Life. 2022; 12(3):440. https://doi.org/10.3390/life12030440
Chicago/Turabian StyleLeong, Jason Cheok Kuan, Masahiro Uesaka, and Naoki Irie. 2022. "Distinguishing Evolutionary Conservation from Derivedness" Life 12, no. 3: 440. https://doi.org/10.3390/life12030440
APA StyleLeong, J. C. K., Uesaka, M., & Irie, N. (2022). Distinguishing Evolutionary Conservation from Derivedness. Life, 12(3), 440. https://doi.org/10.3390/life12030440