
From the Structural and (Dys)Function of ATP Synthase to Deficiency in Age-Related Diseases



Abstract
1. Introduction
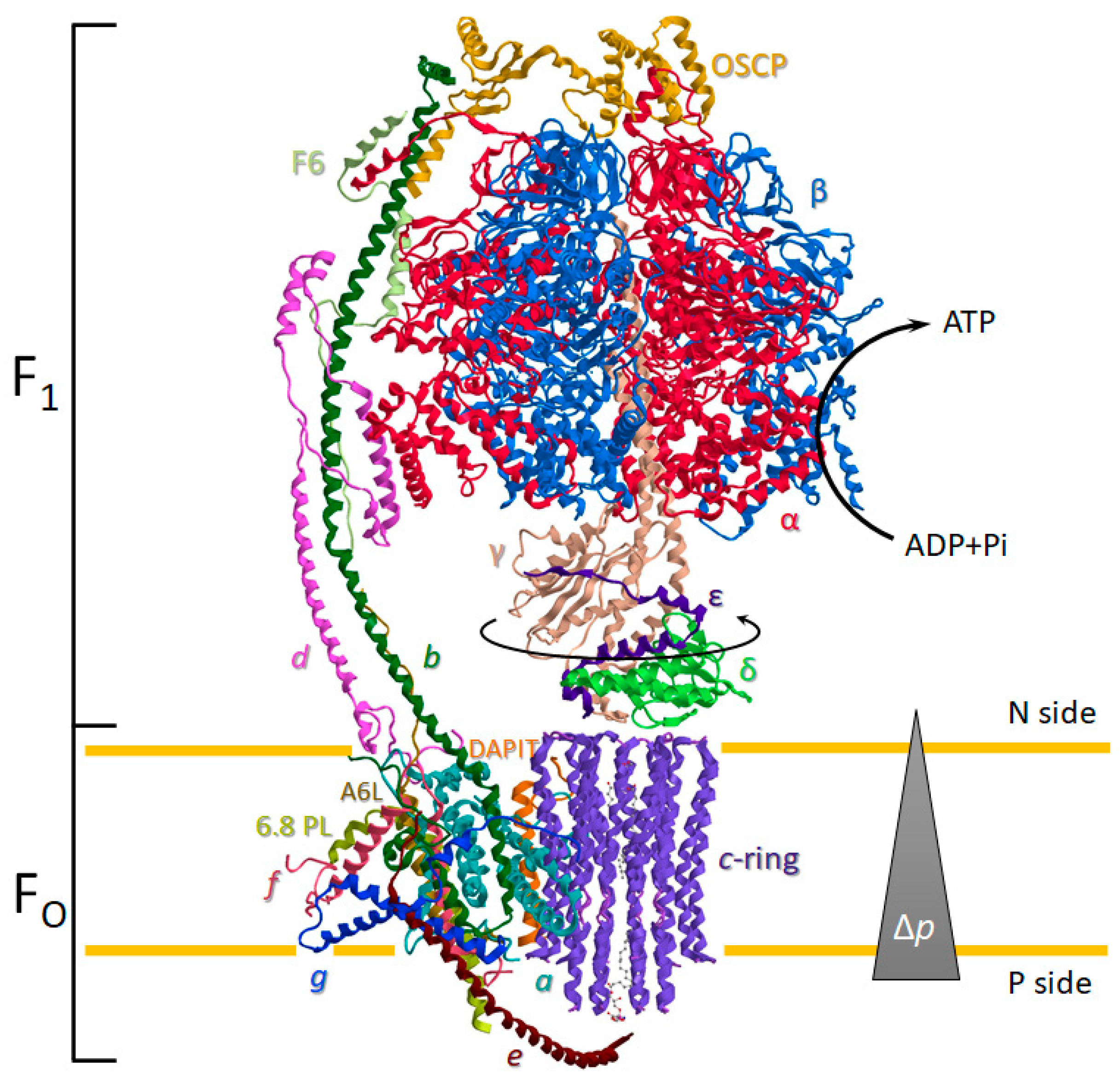
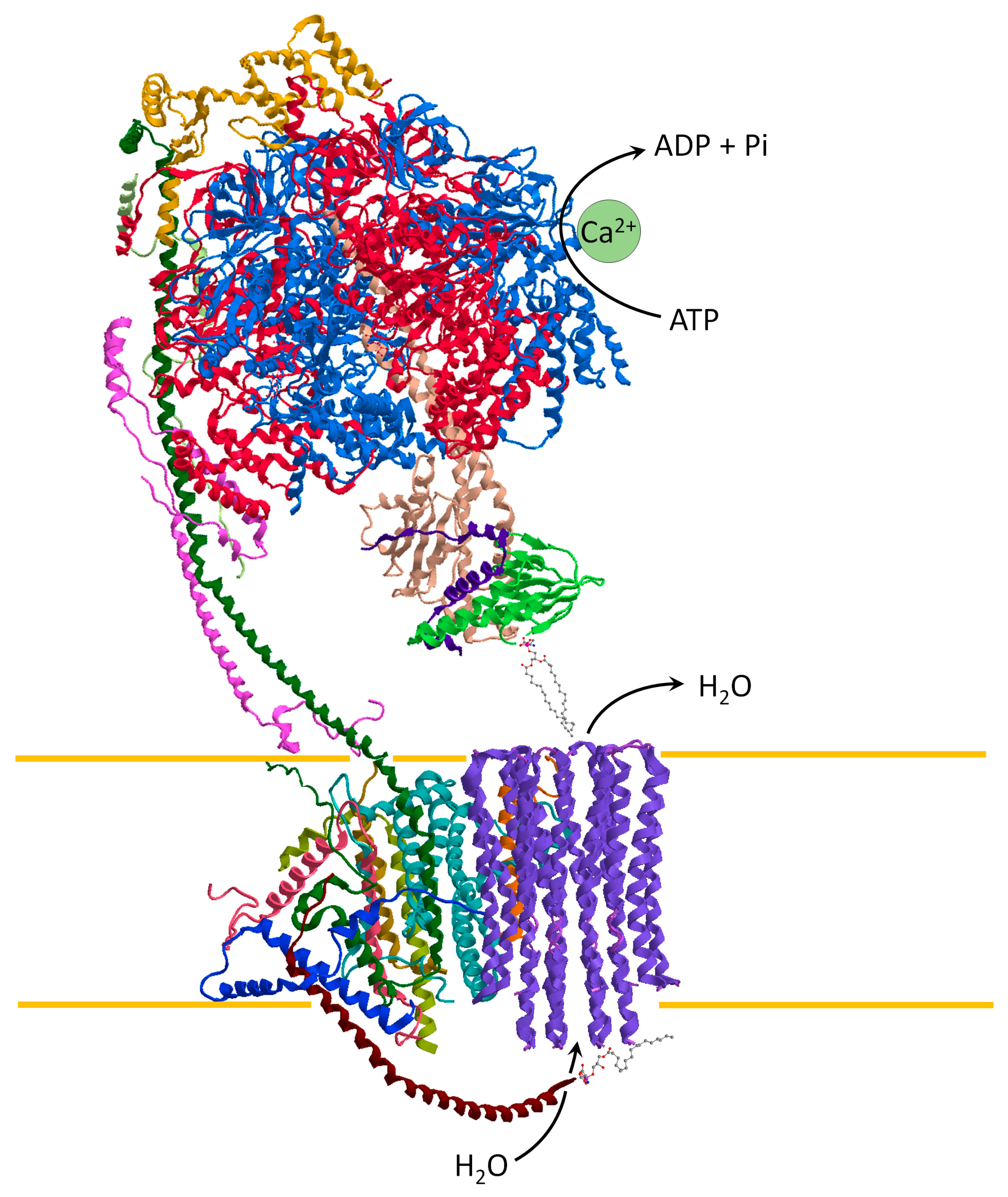
2. The Structure and Function of the ATP Synthase
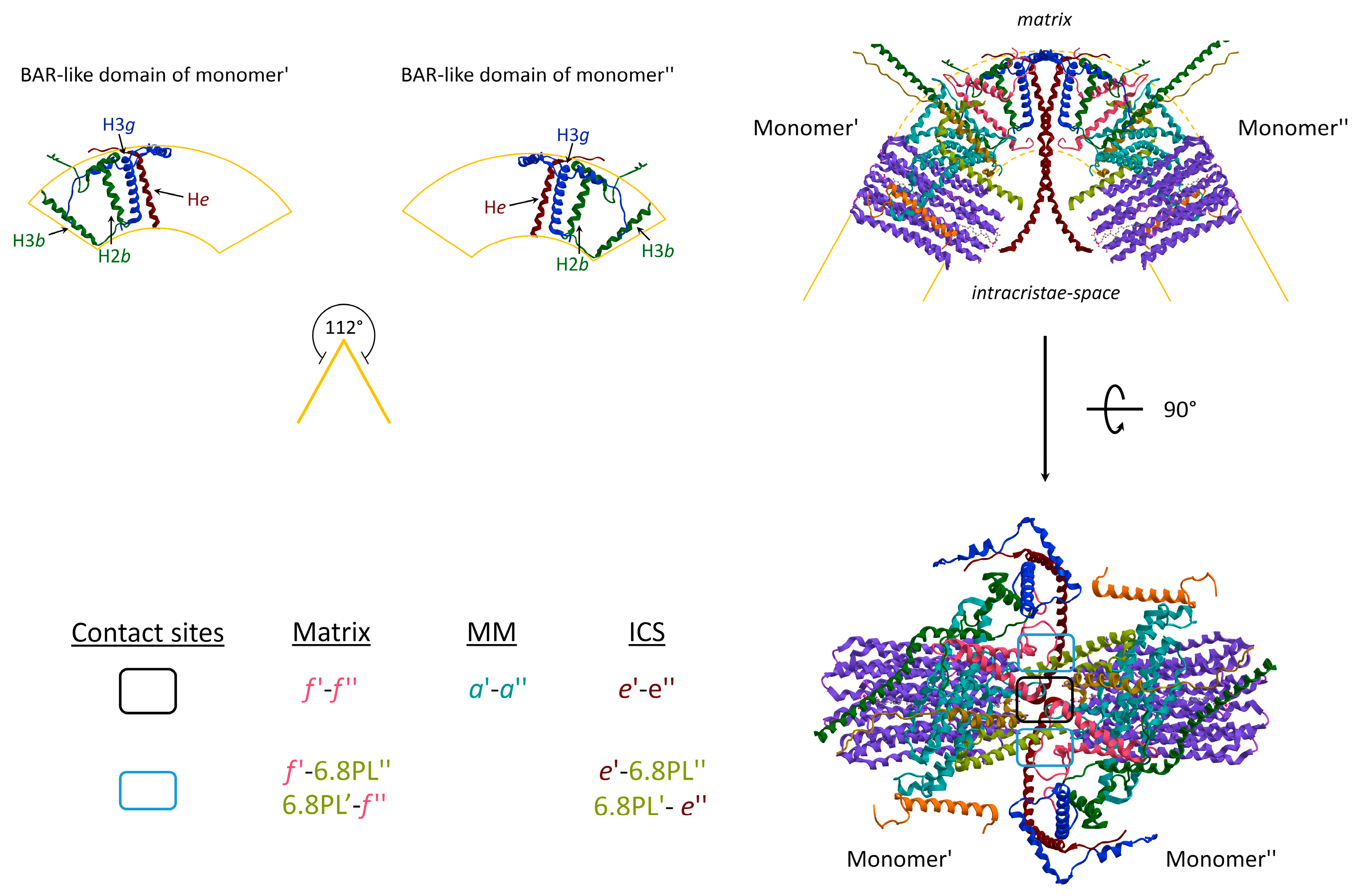
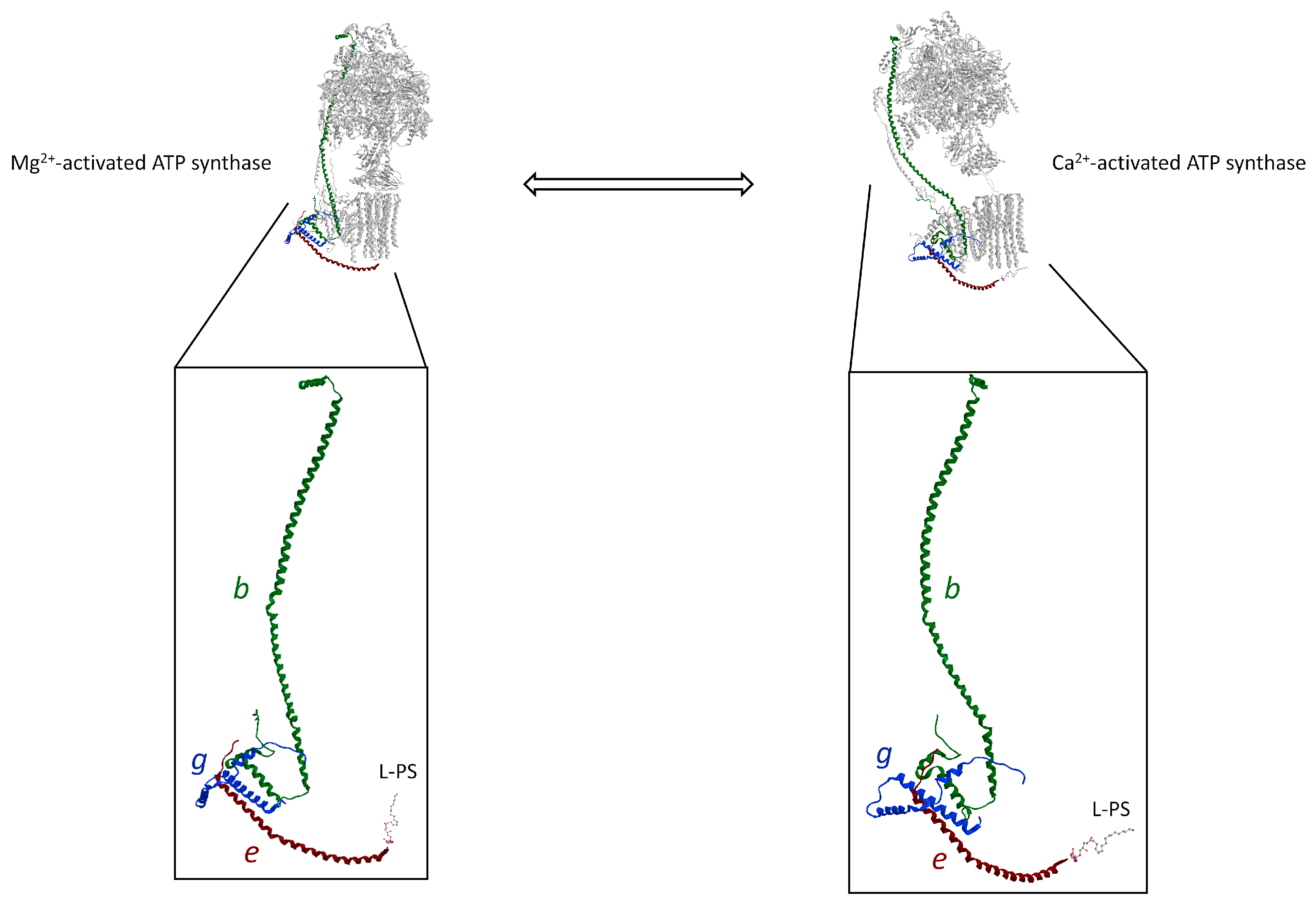
3. The Hidden Face of the ATP Synthase
4. The ATP Synthase Deficiency
5. Neurodegeneration
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saraste, M. Oxidative Phosphorylation at the Fin de Siècle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- von Ballmoos, C.; Cook, G.M.; Dimroth, P. Unique Rotary ATP Synthase and Its Biological Diversity. Annu. Rev. Biophys. 2008, 37, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Ventrella, V.; Trombetti, F.; Pirini, M.; Pagliarani, A. Mussel and Mammalian ATP Synthase Share the Same Bioenergetic Cost of ATP. J. Bioenerg. Biomembr. 2013, 45, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.-P.; Velours, J. The ATP Synthase Is Involved in Generating Mitochondrial Cristae Morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Elston, T.; Wang, H.; Oster, G. Energy Transduction in ATP Synthase. Nature 1998, 391, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.R.; Haworth, R.A. The Ca2+-Induced Membrane Transition in Mitochondria. I. The Protective Mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. The Ca2+-Induced Membrane Transition in Mitochondria. II. Nature of the Ca2+ Trigger Site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-Induced Membrane Transition in Mitochondria. III. Transitional Ca2+ Release. Arch. Biochem. Biophys. 1979, 195, 468–477. [Google Scholar] [CrossRef]
- Nesci, S. The Mitochondrial Permeability Transition Pore in Cell Death: A Promising Drug Binding Bioarchitecture. Med. Res. Rev. 2020, 40, 811–817. [Google Scholar] [CrossRef]
- Pérez, M.J.; Quintanilla, R.A. Development or Disease: Duality of the Mitochondrial Permeability Transition Pore. Dev. Biol. 2017, 426, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Mochly-Rosen, D. Mortal Engines: Mitochondrial Bioenergetics and Dysfunction in Neurodegenerative Diseases. Pharmacol. Res. 2018, 138, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Rampelt, H.; van der Laan, M. The Yin & Yang of Mitochondrial Architecture—Interplay of MICOS and F1Fo-ATP Synthase in Cristae Formation. Microb. Cell 2017, 4, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Bioenergetic Origins of Complexity and Disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, J.; Nelson, N. ATP Synthase. Annu. Rev. Biochem. 2015, 84, 631–657. [Google Scholar] [CrossRef]
- Boyer, P.D. Catalytic Site Occupancy during ATP Synthase Catalysis. FEBS Lett. 2002, 512, 29–32. [Google Scholar] [CrossRef]
- Okuno, D.; Iino, R.; Noji, H. Rotation and Structure of FoF1-ATP Synthase. J. Biochem. 2011, 149, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W.; Davies, K.M. Rotary ATPases: A New Twist to an Ancient Machine. Trends Biochem. Sci. 2016, 41, 106–116. [Google Scholar] [CrossRef]
- Nesci, S.; Trombetti, F.; Ventrella, V.; Pagliarani, A. Opposite Rotation Directions in the Synthesis and Hydrolysis of ATP by the ATP Synthase: Hints from a Subunit Asymmetry. J. Membr. Biol. 2015, 248, 163–169. [Google Scholar] [CrossRef]
- Boyer, P.D. The ATP Synthase—A Splendid Molecular Machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Parey, K.; Bublitz, M.; Mills, D.J.; Zickermann, V.; Vonck, J.; Kühlbrandt, W.; Meier, T. Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Mol. Cell 2016, 63, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Frasch, W.D. The Participation of Metals in the Mechanism of the F(1)-ATPase. Biochim. Biophys. Acta 2000, 1458, 310–325. [Google Scholar] [CrossRef]
- Weber, J.; Bowman, C.; Wilke-Mounts, S.; Senior, A.E. Alpha-Aspartate 261 Is a Key Residue in Noncatalytic Sites of Escherichia Coli F1-ATPase. J. Biol. Chem. 1995, 270, 21045–21049. [Google Scholar] [CrossRef] [PubMed]
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM Structure of the Entire Mammalian F-Type ATP Synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, S.; Melandri, A.B.; Solaini, G. Relevance of Divalent Cations to ATP-Driven Proton Pumping in Beef Heart Mitochondrial FOF1-ATPase. J. Bioenerg. Biomembr. 1998, 30, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Ventrella, V.; Pirini, M.; Pagliarani, A. Kinetic Properties of the Mitochondrial F1FO-ATPase Activity Elicited by Ca(2+) in Replacement of Mg(2+). Biochimie 2017, 140, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Tucker, W.C.; Schwarz, A.; Levine, T.; Du, Z.; Gromet-Elhanan, Z.; Richter, M.L.; Haran, G. Observation of Calcium-Dependent Unidirectional Rotational Motion in Recombinant Photosynthetic F1-ATPase Molecules. J. Biol. Chem. 2004, 279, 47415–47418. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Ventrella, V.; Pagliarani, A. From the Ca2+-Activated F1FO-ATPase to the Mitochondrial Permeability Transition Pore: An Overview. Biochimie 2018, 152, 85–93. [Google Scholar] [CrossRef]
- Colina-Tenorio, L.; Dautant, A.; Miranda-Astudillo, H.; Giraud, M.-F.; González-Halphen, D. The Peripheral Stalk of Rotary ATPases. Front. Physiol. 2018, 9, 1243. [Google Scholar] [CrossRef]
- Murphy, B.J.; Klusch, N.; Langer, J.; Mills, D.J.; Yildiz, Ö.; Kühlbrandt, W. Rotary Substates of Mitochondrial ATP Synthase Reveal the Basis of Flexible F1-Fo Coupling. Science 2019, 364. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Zhang, L.; Zong, S.; Guo, R.; Liu, T.; Yi, J.; Wang, P.; Zhuo, W.; Yang, M. Cryo-EM Structure of the Mammalian ATP Synthase Tetramer Bound with Inhibitory Protein IF1. Science 2019, 364, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Arnold, I.; Pfeiffer, K.; Neupert, W.; Stuart, R.A.; Schägger, H. Yeast Mitochondrial F1FO-ATP Synthase Exists as a Dimer: Identification of Three Dimer-Specific Subunits. EMBO J. 1998, 17, 7170–7178. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.J.; Kent, H.M.; Mills, I.G.; Vallis, Y.; Butler, P.J.G.; Evans, P.R.; McMahon, H.T. BAR Domains as Sensors of Membrane Curvature: The Amphiphysin BAR Structure. Science 2004, 303, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Habersetzer, J.; Larrieu, I.; Priault, M.; Salin, B.; Rossignol, R.; Brèthes, D.; Paumard, P. Human F1FO ATP Synthase, Mitochondrial Ultrastructure and OXPHOS Impairment: A (Super-)Complex Matter? PLoS ONE 2013, 8, e75429. [Google Scholar] [CrossRef] [PubMed]
- Arselin, G.; Vaillier, J.; Salin, B.; Schaeffer, J.; Giraud, M.-F.; Dautant, A.; Brèthes, D.; Velours, J. The Modulation in Subunits e and g Amounts of Yeast ATP Synthase Modifies Mitochondrial Cristae Morphology. J. Biol. Chem. 2004, 279, 40392–40399. [Google Scholar] [CrossRef]
- Salewskij, K.; Rieger, B.; Hager, F.; Arroum, T.; Duwe, P.; Villalta, J.; Colgiati, S.; Richter, C.P.; Psathaki, O.E.; Enriquez, J.A.; et al. The Spatio-Temporal Organization of Mitochondrial F1FO ATP Synthase in Cristae Depends on Its Activity Mode. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148091. [Google Scholar] [CrossRef] [PubMed]
- Almendro-Vedia, V.; Natale, P.; Valdivieso González, D.; Lillo, M.P.; Aragones, J.L.; López-Montero, I. How Rotating ATP Synthases Can Modulate Membrane Structure. Arch. Biochem. Biophys. 2021, 708, 108939. [Google Scholar] [CrossRef] [PubMed]
- Jakubke, C.; Roussou, R.; Maiser, A.; Schug, C.; Thoma, F.; Bunk, D.; Hörl, D.; Leonhardt, H.; Walter, P.; Klecker, T.; et al. Cristae-Dependent Quality Control of the Mitochondrial Genome. Sci. Adv. 2021, 7, eabi8886. [Google Scholar] [CrossRef]
- Nesci, S.; Pagliarani, A. Ca2+ as Cofactor of the Mitochondrial H+-Translocating F1FO-ATP(Hydrol)Ase. Proteins Struct. Funct. Bioinform. 2021, 89, 477–482. [Google Scholar] [CrossRef]
- Nesci, S.; Pagliarani, A. Incoming News on the F-Type ATPase Structure and Functions in Mammalian Mitochondria. BBA Adv. 2021, 1, 100001. [Google Scholar] [CrossRef]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Bernardini, C.; Fabbri, M.; Forni, M.; Nesci, S. Mitochondrial Ca2+ -Activated F1 FO -ATPase Hydrolyzes ATP and Promotes the Permeability Transition Pore. Ann. N. Y. Acad. Sci. 2019, 1457, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Nesci, S. Phenylglyoxal Inhibition of the Mitochondrial F1FO-ATPase Activated by Mg2+ or by Ca2+ Provides Clues on the Mitochondrial Permeability Transition Pore. Arch. Biochem. Biophys. 2020, 681, 108258. [Google Scholar] [CrossRef] [PubMed]
- Algieri, C.; Nesci, S.; Trombetti, F.; Fabbri, M.; Ventrella, V.; Pagliarani, A. Mitochondrial F1FO-ATPase and Permeability Transition Pore Response to Sulfide in the Midgut Gland of Mytilus Galloprovincialis. Biochimie 2021, 180, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Algieri, V.; Algieri, C.; Maiuolo, L.; De Nino, A.; Pagliarani, A.; Tallarida, M.A.; Trombetti, F.; Nesci, S. 1,5-Disubstituted-1,2,3-Triazoles as Inhibitors of the Mitochondrial Ca2+ -Activated F1 FO -ATP(Hydrol)Ase and the Permeability Transition Pore. Ann. N. Y. Acad. Sci. 2021, 1485, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Fabbri, M.; Nesci, S. The Inhibition of Gadolinium Ion (Gd3+) on the Mitochondrial F1FO-ATPase Is Linked to the Modulation of the Mitochondrial Permeability Transition Pore. Int. J. Biol. Macromol. 2021, 184, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Algieri, C.; Trombetti, F.; Ventrella, V.; Fabbri, M.; Pagliarani, A. Sulfide Affects the Mitochondrial Respiration, the Ca2+-Activated F1FO-ATPase Activity and the Permeability Transition Pore but Does Not Change the Mg2+-Activated F1FO-ATPase Activity in Swine Heart Mitochondria. Pharmacol. Res. 2021, 166, 105495. [Google Scholar] [CrossRef] [PubMed]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Nesci, S. The Mitochondrial F1FO-ATPase Exploits the Dithiol Redox State to Modulate the Permeability Transition Pore. Arch. Biochem. Biophys. 2021, 712, 109027. [Google Scholar] [CrossRef] [PubMed]
- Algieri, C.; Algieri, V.; La Mantia, D.; Bernardini, C.; Maiuolo, L.; Trombetti, F.; Tallarida, M.A.; De Nino, A.; Forni, M.; Pagliarani, A.; et al. New Generation Molecules as Modulators of the Mitochondrial Permeability Transition and Potential Therapeutic Agents. FEBS Open Bio 2021, 11, 317. [Google Scholar]
- Nesci, S.; Rubattu, S. The ATP Synthase Glycine Zipper of the c Subunits: From the Structural to the Functional Role in Mitochondrial Biology of Cardiovascular Diseases. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119075. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Ventrella, V.; Trombetti, F.; Pirini, M.; Pagliarani, A. Preferential Nitrite Inhibition of the Mitochondrial F1FO-ATPase Activities When Activated by Ca(2+) in Replacement of the Natural Cofactor Mg(2+). Biochim. Biophys. Acta 2016, 1860, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The Unique Histidine in OSCP Subunit of F-ATP Synthase Mediates Inhibition of the Permeability Transition Pore by Acidic PH. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Carraro, M.; Carrer, A.; Minervini, G.; Urbani, A.; Masgras, I.; Tosatto, S.C.E.; Szabò, I.; Bernardi, P.; Lippe, G. Arg-8 of Yeast Subunit e Contributes to the Stability of F-ATP Synthase Dimers and to the Generation of the Full-Conductance Mitochondrial Megachannel. J. Biol. Chem. 2019, 294, 10987–10997. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Carraro, M.; Sartori, G.; Minervini, G.; Eriksson, O.; Petronilli, V.; Bernardi, P. Arginine 107 of Yeast ATP Synthase Subunit g Mediates Sensitivity of the Mitochondrial Permeability Transition to Phenylglyoxal. J. Biol. Chem. 2018, 293, 14632–14645. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.-A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An Uncoupling Channel within the C-Subunit Ring of the F1FO ATP Synthase Is the Mitochondrial Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c Subunit of the FO ATP Synthase in Mitochondrial Permeability Transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S. A Lethal Channel between the ATP Synthase Monomers. Trends Biochem. Sci. 2018, 43, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Jonas, E.A. ATP Synthase C-Subunit Ring as the Channel of Mitochondrial Permeability Transition: Regulator of Metabolism in Development and Degeneration. J. Mol. Cell. Cardiol. 2020, 144, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Traxler, L.; Lagerwall, J.; Eichhorner, S.; Stefanoni, D.; D’Alessandro, A.; Mertens, J. Metabolism Navigates Neural Cell Fate in Development, Aging and Neurodegeneration. Dis. Model. Mech. 2021, 14, dmm048993. [Google Scholar] [CrossRef]
- Galber, C.; Carissimi, S.; Baracca, A.; Giorgio, V. The ATP Synthase Deficiency in Human Diseases. Life 2021, 11, 325. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Petty, R.K.; Morgan-Hughes, J.A. A New Mitochondrial Disease Associated with Mitochondrial DNA Heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar] [PubMed]
- de Vries, D.D.; van Engelen, B.G.; Gabreëls, F.J.; Ruitenbeek, W.; van Oost, B.A. A Second Missense Mutation in the Mitochondrial ATPase 6 Gene in Leigh’s Syndrome. Ann. Neurol. 1993, 34, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Sgarbi, G.; Baracca, A.; Lenaz, G.; Valentino, L.M.; Carelli, V.; Solaini, G. Inefficient Coupling between Proton Transport and ATP Synthesis May Be the Pathogenic Mechanism for NARP and Leigh Syndrome Resulting from the T8993G Mutation in MtDNA. Biochem. J. 2006, 395, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. MtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Carrozzo, R.; Tessa, A.; Vázquez-Memije, M.E.; Piemonte, F.; Patrono, C.; Malandrini, A.; Dionisi-Vici, C.; Vilarinho, L.; Villanova, M.; Schägger, H.; et al. The T9176G MtDNA Mutation Severely Affects ATP Production and Results in Leigh Syndrome. Neurology 2001, 56, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Memije, M.E.; Rizza, T.; Meschini, M.C.; Nesti, C.; Santorelli, F.M.; Carrozzo, R. Cellular and Functional Analysis of Four Mutations Located in the Mitochondrial ATPase6 Gene. J. Cell. Biochem. 2009, 106, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Tońska, K.; Kodroń, A.; Bartnik, E. Genotype-Phenotype Correlations in Leber Hereditary Optic Neuropathy. Biochim. Biophys. Acta 2010, 1797, 1119–1123. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S.; Murphy, S.M.; Cottenie, E.; Chalasani, A.; Sweeney, M.G.; Woodward, C.; Mudanohwo, E.E.; Hargreaves, I.; Heales, S.; Land, J.; et al. Genetic Dysfunction of MT-ATP6 Causes Axonal Charcot-Marie-Tooth Disease. Neurology 2012, 79, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Hejzlarová, K.; Mráček, T.; Vrbacký, M.; Kaplanová, V.; Karbanová, V.; Nůsková, H.; Pecina, P.; Houštěk, J. Nuclear Genetic Defects of Mitochondrial ATP Synthase. Physiol. Res. 2014, 63, S57–S71. [Google Scholar] [CrossRef] [PubMed]
- Mordel, P.; Schaeffer, S.; Dupas, Q.; Laville, M.-A.; Gérard, M.; Chapon, F.; Allouche, S. A 2 Bp Deletion in the Mitochondrial ATP 6 Gene Responsible for the NARP (Neuropathy, Ataxia, and Retinitis Pigmentosa) Syndrome. Biochem. Biophys. Res. Commun. 2017, 494, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Mkaouar-Rebai, E.; Kammoun, F.; Chamkha, I.; Kammoun, N.; Hsairi, I.; Triki, C.; Fakhfakh, F. A de Novo Mutation in the Adenosine Triphosphatase (ATPase) 8 Gene in a Patient with Mitochondrial Disorder. J. Child. Neurol. 2010, 25, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Felhi, R.; Mkaouar-Rebai, E.; Sfaihi-Ben Mansour, L.; Alila-Fersi, O.; Tabebi, M.; Ben Rhouma, B.; Ammar, M.; Keskes, L.; Hachicha, M.; Fakhfakh, F. Mutational Analysis in Patients with Neuromuscular Disorders: Detection of Mitochondrial Deletion and Double Mutations in the MT-ATP6 Gene. Biochem. Biophys. Res. Commun. 2016, 473, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A.M.; et al. A Complex V ATP5A1 Defect Causes Fatal Neonatal Mitochondrial Encephalopathy. Brain 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Lieber, D.S.; Calvo, S.E.; Shanahan, K.; Slate, N.G.; Liu, S.; Hershman, S.G.; Gold, N.B.; Chapman, B.A.; Thorburn, D.R.; Berry, G.T.; et al. Targeted Exome Sequencing of Suspected Mitochondrial Disorders. Neurology 2013, 80, 1762–1770. [Google Scholar] [CrossRef] [PubMed]
- Oláhová, M.; Yoon, W.H.; Thompson, K.; Jangam, S.; Fernandez, L.; Davidson, J.M.; Kyle, J.E.; Grove, M.E.; Fisk, D.G.; Kohler, J.N.; et al. Biallelic Mutations in ATP5F1D, Which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder. Am. J. Hum. Genet. 2018, 102, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Tort, F.; García-Silva, M.T.; Ferrer-Cortès, X.; Navarro-Sastre, A.; Garcia-Villoria, J.; Coll, M.J.; Vidal, E.; Jiménez-Almazán, J.; Dopazo, J.; Briones, P.; et al. Exome Sequencing Identifies a New Mutation in SERAC1 in a Patient with 3-Methylglutaconic Aciduria. Mol. Genet. Metab. 2013, 110, 73–77. [Google Scholar] [CrossRef]
- Mayr, J.A.; Havlícková, V.; Zimmermann, F.; Magler, I.; Kaplanová, V.; Jesina, P.; Pecinová, A.; Nusková, H.; Koch, J.; Sperl, W.; et al. Mitochondrial ATP Synthase Deficiency Due to a Mutation in the ATP5E Gene for the F1 Epsilon Subunit. Hum. Mol. Genet. 2010, 19, 3430–3439. [Google Scholar] [CrossRef]
- Barca, E.; Ganetzky, R.D.; Potluri, P.; Juanola-Falgarona, M.; Gai, X.; Li, D.; Jalas, C.; Hirsch, Y.; Emmanuele, V.; Tadesse, S.; et al. USMG5 Ashkenazi Jewish Founder Mutation Impairs Mitochondrial Complex V Dimerization and ATP Synthesis. Hum. Mol. Genet. 2018, 27, 3305–3312. [Google Scholar] [CrossRef] [PubMed]
- De Meirleir, L.; Seneca, S.; Lissens, W.; de Clercq, I.; Eyskens, F.; Gerlo, E.; Smet, J.; van Coster, R. Respiratory Chain Complex V Deficiency Due to a Mutation in the Assembly Gene ATP12. J. Med. Genet. 2004, 41, 120–124. [Google Scholar] [CrossRef]
- Stojanović, V.; Doronjski, A. Mild Form of 3-Methylglutaconic Aciduria Type IV and Mutation in the TMEM70 Genes. J. Pediatr. Endocrinol. Metab. 2013, 26, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Honzík, T.; Tesarová, M.; Mayr, J.A.; Hansíková, H.; Jesina, P.; Bodamer, O.; Koch, J.; Magner, M.; Freisinger, P.; Huemer, M.; et al. Mitochondrial Encephalocardio-Myopathy with Early Neonatal Onset Due to TMEM70 Mutation. Arch. Dis. Child. 2010, 95, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Torraco, A.; Verrigni, D.; Rizza, T.; Meschini, M.C.; Vazquez-Memije, M.E.; Martinelli, D.; Bianchi, M.; Piemonte, F.; Dionisi-Vici, C.; Santorelli, F.M.; et al. TMEM70: A Mutational Hot Spot in Nuclear ATP Synthase Deficiency with a Pivotal Role in Complex V Biogenesis. Neurogenetics 2012, 13, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Huigsloot, M.; Lammens, M.; Jansen, J.; van den Heuvel, L.P.; Spiekerkoetter, U.; von Kleist-Retzow, J.-C.; Forkink, M.; Koopman, W.J.H.; Szklarczyk, R.; et al. Restoration of Complex V Deficiency Caused by a Novel Deletion in the Human TMEM70 Gene Normalizes Mitochondrial Morphology. Mitochondrion 2011, 11, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Rodenburg, R.J.T.; Jonckheere, A.; de Vries, M.C.; Huizing, M.; Heldt, K.; van den Heuvel, L.P.; Wendel, U.; Kluijtmans, L.A.; Engelke, U.F.; et al. Biochemical and Genetic Analysis of 3-Methylglutaconic Aciduria Type IV: A Diagnostic Strategy. Brain 2009, 132, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Braczynski, A.K.; Vlaho, S.; Müller, K.; Wittig, I.; Blank, A.-E.; Tews, D.S.; Drott, U.; Kleinle, S.; Abicht, A.; Horvath, R.; et al. ATP Synthase Deficiency Due to TMEM70 Mutation Leads to Ultrastructural Mitochondrial Degeneration and Is Amenable to Treatment. Biomed. Res. Int. 2015, 2015, 462592. [Google Scholar] [CrossRef] [PubMed]
- Hung, P.-C.; Wang, H.-S. A Previously Undescribed Leukodystrophy in Leigh Syndrome Associated with T9176C Mutation of the Mitochondrial ATPase 6 Gene. Dev. Med. Child Neurol. 2007, 49, 65–67. [Google Scholar] [CrossRef]
- Verny, C.; Guegen, N.; Desquiret, V.; Chevrollier, A.; Prundean, A.; Dubas, F.; Cassereau, J.; Ferre, M.; Amati-Bonneau, P.; Bonneau, D.; et al. Hereditary Spastic Paraplegia-like Disorder Due to a Mitochondrial ATP6 Gene Point Mutation. Mitochondrion 2011, 11, 70–75. [Google Scholar] [CrossRef]
- Synofzik, M.; Schicks, J.; Wilhelm, C.; Bornemann, A.; Schöls, L. Charcot-Marie-Tooth Hereditary Neuropathy Due to a Mitochondrial ATP6 Mutation. Eur. J. Neurol. 2012, 19, e114–e116. [Google Scholar] [CrossRef]
- Yu, X.-L.; Yan, C.-Z.; Ji, K.-Q.; Lin, P.-F.; Xu, X.-B.; Dai, T.-J.; Li, W.; Zhao, Y.-Y. Clinical, Neuroimaging, and Pathological Analyses of 13 Chinese Leigh Syndrome Patients with Mitochondrial DNA Mutations. Chin. Med. J. 2018, 131, 2705–2712. [Google Scholar] [CrossRef]
- Hu, C.; Li, X.; Zhao, L.; Shi, Y.; Zhou, S.; Wu, B.; Wang, Y. Clinical and Molecular Characterization of Pediatric Mitochondrial Disorders in South of China. Eur. J. Med. Genet. 2020, 63, 103898. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, L.; Graña, M.; Mansilla, S.; Martínez, J.; Tapié, A.; Greif, G.; Montano, N.; Vaglio, A.; Gueçaimburú, R.; Robello, C.; et al. Deep Sequencing Discovery of Causal MtDNA Mutations in a Patient with Unspecific Neurological Disease. Mitochondrion 2019, 46, 337–344. [Google Scholar] [CrossRef] [PubMed]
- López-Gallardo, E.; Emperador, S.; Solano, A.; Llobet, L.; Martín-Navarro, A.; López-Pérez, M.J.; Briones, P.; Pineda, M.; Artuch, R.; Barraquer, E.; et al. Expanding the Clinical Phenotypes of MT-ATP6 Mutations. Hum. Mol. Genet. 2014, 23, 6191–6200. [Google Scholar] [CrossRef] [PubMed]
- Mkaouar-Rebai, E.; Felhi, R.; Tabebi, M.; Alila-Fersi, O.; Chamkha, I.; Maalej, M.; Ammar, M.; Kammoun, F.; Keskes, L.; Hachicha, M.; et al. Mitochondrial DNA Triplication and Punctual Mutations in Patients with Mitochondrial Neuromuscular Disorders. Biochem. Biophys. Res. Commun. 2016, 473, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Claeys, K.G.; Abicht, A.; Häusler, M.; Kleinle, S.; Wiesmann, M.; Schulz, J.B.; Horvath, R.; Weis, J. Novel Genetic and Neuropathological Insights in Neurogenic Muscle Weakness, Ataxia, and Retinitis Pigmentosa (NARP). Muscle Nerve 2016, 54, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, S.; Panaye, M.; Rabeyrin, M.; Errazuriz-Cerda, E.; Mousson de Camaret, B.; Petiot, P.; Juillard, L.; Guebre-Egziabher, F. Renal Involvement in Neuropathy, Ataxia, Retinitis Pigmentosa (NARP) Syndrome: A Case Report. Am. J. Kidney Dis. 2018, 71, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Tort, F.; Del Toro, M.; Lissens, W.; Montoya, J.; Fernàndez-Burriel, M.; Font, A.; Buján, N.; Navarro-Sastre, A.; López-Gallardo, E.; Arranz, J.A.; et al. Screening for Nuclear Genetic Defects in the ATP Synthase-Associated Genes TMEM70, ATP12 and ATP5E in Patients with 3-Methylglutaconic Aciduria. Clin. Genet. 2011, 80, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.M.; Levandovskiy, V.; Mackay, N.; Ackerley, C.; Chitayat, D.; Raiman, J.; Halliday, W.H.; Schulze, A.; Robinson, B.H. Complex V TMEM70 Deficiency Results in Mitochondrial Nucleoid Disorganization. Mitochondrion 2011, 11, 191–199. [Google Scholar] [CrossRef]
- Ortiz-González, X.R. Mitochondrial Dysfunction: A Common Denominator in Neurodevelopmental Disorders? Dev. Neurosci. 2021, 43, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.P.; Mandel, J.L. The FMR-1 Protein Is Cytoplasmic, Most Abundant in Neurons and Appears Normal in Carriers of a Fragile X Premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, K.K.; Wang, A.; Wang, L.; Tracey, M.; Kleiner, G.; Quinzii, C.M.; Sun, L.; Yang, G.; Perez-Zoghbi, J.F.; Licznerski, P.; et al. Inefficient Thermogenic Mitochondrial Respiration Due to Futile Proton Leak in a Mouse Model of Fragile X Syndrome. FASEB J. 2020, 34, 7404–7426. [Google Scholar] [CrossRef] [PubMed]
- Licznerski, P.; Park, H.-A.; Rolyan, H.; Chen, R.; Mnatsakanyan, N.; Miranda, P.; Graham, M.; Wu, J.; Cruz-Reyes, N.; Mehta, N.; et al. ATP Synthase C-Subunit Leak Causes Aberrant Cellular Metabolism in Fragile X Syndrome. Cell 2020, 182, 1170–1185.e9. [Google Scholar] [CrossRef] [PubMed]
- Carli, S.; Chaabane, L.; Butti, C.; De Palma, C.; Aimar, P.; Salio, C.; Vignoli, A.; Giustetto, M.; Landsberger, N.; Frasca, A. In Vivo Magnetic Resonance Spectroscopy in the Brain of Cdkl5 Null Mice Reveals a Metabolic Profile Indicative of Mitochondrial Dysfunctions. J. Neurochem. 2021, 157, 1253–1269. [Google Scholar] [CrossRef] [PubMed]
- de Martynoff, G.; Pohl, V.; Mercken, L.; van Ommen, G.J.; Vassart, G. Structural Organization of the Bovine Thyroglobulin Gene and of Its 5’-Flanking Region. Eur. J. Biochem. 1987, 164, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Jonas, E.A. The New Role of F1Fo ATP Synthase in Mitochondria-Mediated Neurodegeneration and Neuroprotection. Exp. Neurol. 2020, 332, 113400. [Google Scholar] [CrossRef] [PubMed]
- Amodeo, G.F.; Solesio, M.E.; Pavlov, E.V. From ATP Synthase Dimers to C-Ring Conformational Changes: Unified Model of the Mitochondrial Permeability Transition Pore. Cell Death Dis. 2017, 8, e2662. [Google Scholar] [CrossRef] [PubMed]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17.e2. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Amodeo, G.F.; Lee, B.Y.; Krilyuk, N.; Filice, C.T.; Valyuk, D.; Otzen, D.E.; Noskov, S.; Leonenko, Z.; Pavlov, E.V. C Subunit of the ATP Synthase Is an Amyloidogenic Calcium Dependent Channel-Forming Peptide with Possible Implications in Mitochondrial Permeability Transition. Sci. Rep. 2021, 11, 8744. [Google Scholar] [CrossRef]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s Disease: Genetics and Pathogenesis. Annu. Rev. Pathol. 2011, 6, 193–222. [Google Scholar] [CrossRef]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial Dysfunction Associated with Increased Oxidative Stress and α-Synuclein Accumulation in PARK2 IPSC-Derived Neurons and Postmortem Brain Tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Yokota, M.; Kakuta, S.; Shiga, T.; Ishikawa, K.-I.; Okano, H.; Hattori, N.; Akamatsu, W.; Koike, M. Establishment of an in Vitro Model for Analyzing Mitochondrial Ultrastructure in PRKN-Mutated Patient IPSC-Derived Dopaminergic Neurons. Mol. Brain 2021, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial Dynamics in Parkinson’s Disease: A Role for α-Synuclein? Dis Model. Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Rango, M.; Bresolin, N. Brain Mitochondria, Aging, and Parkinson’s Disease. Genes 2018, 9, E250. [Google Scholar] [CrossRef] [PubMed]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, Prefibrillar α-Synuclein Oligomers Promote Complex I-Dependent, Ca2+-Induced Mitochondrial Dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Chen, R.; Park, H.-A.; Mnatsakanyan, N.; Niu, Y.; Licznerski, P.; Wu, J.; Miranda, P.; Graham, M.; Tang, J.; Boon, A.J.W.; et al. Parkinson’s Disease Protein DJ-1 Regulates ATP Synthase Protein Components to Increase Neuronal Process Outgrowth. Cell Death Dis. 2019, 10, 469. [Google Scholar] [CrossRef]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of Mitochondrial F1FO-ATP Synthase via OSCP in Alzheimer’s Disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [PubMed]
- Daum, B.; Walter, A.; Horst, A.; Osiewacz, H.D.; Kühlbrandt, W. Age-Dependent Dissociation of ATP Synthase Dimers and Loss of Inner-Membrane Cristae in Mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 15301–15306. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.-Y.; Cho, H.J.; Kim, C.; Jung, Y.O.; Kang, M.J.; Murray, M.E.; Hong, H.S.; Choi, Y.-J.; Choi, H.; Kim, D.K.; et al. Mitochondrial ATP Synthase Activity Is Impaired by Suppressed O-GlcNAcylation in Alzheimer’s Disease. Hum. Mol. Genet. 2015, 24, 6492–6504. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of Nitrated Proteins in Alzheimer’s Disease Brain Using a Redox Proteomics Approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.; Perluigi, M.; Sultana, R.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Coccia, R.; Markesbery, W.R.; Butterfield, D.A. Redox Proteomic Identification of 4-Hydroxy-2-Nonenal-Modified Brain Proteins in Amnestic Mild Cognitive Impairment: Insight into the Role of Lipid Peroxidation in the Progression and Pathogenesis of Alzheimer’s Disease. Neurobiol. Dis. 2008, 30, 107–120. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variants | CNS | PNS | Muscle | Heart | EYE | Brain MRI | Metabolic Alterations | Other | References |
|---|---|---|---|---|---|---|---|---|---|---|
| ATP5A1 | c.985C>T (p.Arg329Cys) c.962A>G (p.Tyr321Cys) | Encephalopathy Seizures Hypotonia | - | - | Pulmonary hypertension heart failure | - | Hyperintensity of thalamus and subcortical density Progressive cortical, subcortical, cerebellum and pons damage | Hyperalaninemia | Hypoplastic lungs renal cysts Microcephaly | [74,75] |
| ATP5D | c.245C>T (p.Pro82Leu) c.317T>G (p.Val106Gly) | Encephalopathy Lethargy Motor delay Ataxia Speech delay Myoclonic seizure Hypotonia | - | Proximal muscle weakness exercise intolerance | Dilatative cardiomyopathy | - | Transitory widespread cortical and subcortical oedema | Severe acidosis, Hypoglicaemia, Hyperammonaemia, 3-MGA1 Ketoacidosis | Short stature | [76] |
| ATP5E | c.35A>G (p.Tyr12Cys) | Ataxia | Peripheral neuropathy | Weakness exercise intolerance | Mild hypertrophy of left ventricle | - | - | Hyperlacticaemia, 3MGA1, hyperammonaemia | Respiratory distress | [78] |
| DAPIT-UMSG5 | c.87+1G>C (p.?) | Motor delay Ataxia, Ophthalmoplegia | - | - | NA | - | Brainstem and basal ganglia lesions | - | NA | [79] |
| MT-ATP6 | m.9176T>C (p.Leu217Pro) m.9185T>C (p.Leu220Pro) m.9127_9128delAT (p.Ile201ProfsX2) m.8993T>C (p.Leu156Pro) m.9185T>C (p.Leu220Pro) m.8993T>G (p.Leu156Arg) m.8993T>C (p.Leu156Pro) m.9017T>C (p.Ile164Thr) and m.9010G>A (p.Ala162Thr) m.9025G>A (p.Gly167Ser) m.9020A>G (p.His168Arg) m.9032T>C (p.Leu169Pro) m.9157G>A (p.Ala211Thr) m.8618insT (p.Ile31IlefsX64) m.8993T>G (p.Leu156Arg) m.8993T>G (p.Leu156Arg) | Cerebellar ataxia Myoclonic seizure Developmental delay Cognitive deficit Motor delay Seizures Hypotonia Spastic paraplegy Dystonia Dysarthria | Paraesthesia Motor and sensorineural neuropathy Perypheral polyneuropathy | Muscle weakness Muscle atrophy | Hypertrophic cardiomyopathy Supraventricular arrhythmia Atrioventricular block Bradycardia | Optic atrophy Retinal degeneration Retinitis pigmentosa Nystagmus, Strabismus, Ptosis Cataract Atypical LHON Blindness | Basal ganglia lesions Pituitary atrophy Medulla, pons and brain stem lesions White matter abnormalities Cerebellar vermis atrophy Cortical atrophy Cerebellar atrophy | Increase CSF lactic acid Lactic acidosis Increase of plasma pyruvate and alanine Metabolic acidosis Hyperammonaemia Increase of creatinine kinase | Sensorineural hearing loss Diabetes mellitus Hypogonadism HypothyroidismSurrenalic failure Short stature Respiratory distress Dysmorphisms Cafè-au-lait spot Deafness Headache Renal failure | [69,71,87,88,89,90,91,92,93,94,95,96] |
| MT-ATP8 | m.8411A>G (p.Met16Val) m.8393C>T (Pro136Ser) | Developmental delay Ataxia Seizure Lethargy Encephalopathy Tetraplegia | Neuropathy | Weakness | Hypertrophic cardiomyopathy arrhythmia | Blindness | Cerebellar atrophy, White matter alterations | Lactic acidosis Hyperammonaemia Hyperalaninemia Hypoglicaemia | Deafness Anorexia Respiratory distress | [70,72] |
| MT-ATP6 /MT-ATP8 | m.8528T>C m.8529G>A m.8527A>G | Developmental delay Seizures | Global and axial hypotony | - | - | - | Lenticular nucleus and white matter anomalies | - | - | [73] |
| ATPAF2 | c.280T>A (p.Trp94Arg) | NA | - | - | - | - | Cerebral atrophy Corpus callosum dysgenesis Hypomyelination Basal ganglia and thalamus atrophy | Urinary and plasma lactic acidosis | Dysmorphisms Hepatomegaly Renal hypoplasia | [80] |
| TMEM70 | c.317-2A>G (p.?); c.628A>C (p.Thr210Pro) c.118_119insGT(p.Ser40CysfsTer11) c.317-2A>G (p.?) c.317-2A>G (p.?) Exon2 deletion | Encephalopathy Seizures Developmental delay Motor delay Ataxia Hypotonia | - | - | Dilated cardiomyopathy Arrhythmia Non-compact cardiomyopathy Hypertrophic cardiomyopathy | Cataract Ptosis | Cortical atrophy | 3-MGA 1 Lactic acidosis, Hyperalaninemia hyperammonaemia Increase of ornithine | Dysmorphisms Microcephaly Hepatomegaly Umbilical hernia | [81,82,83,84,85,86,97,98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garone, C.; Pietra, A.; Nesci, S. From the Structural and (Dys)Function of ATP Synthase to Deficiency in Age-Related Diseases. Life 2022, 12, 401. https://doi.org/10.3390/life12030401
Garone C, Pietra A, Nesci S. From the Structural and (Dys)Function of ATP Synthase to Deficiency in Age-Related Diseases. Life. 2022; 12(3):401. https://doi.org/10.3390/life12030401
Chicago/Turabian StyleGarone, Caterina, Andrea Pietra, and Salvatore Nesci. 2022. "From the Structural and (Dys)Function of ATP Synthase to Deficiency in Age-Related Diseases" Life 12, no. 3: 401. https://doi.org/10.3390/life12030401
APA StyleGarone, C., Pietra, A., & Nesci, S. (2022). From the Structural and (Dys)Function of ATP Synthase to Deficiency in Age-Related Diseases. Life, 12(3), 401. https://doi.org/10.3390/life12030401