
Method for Accurate Detection of Amino Acids and Mycotoxins in Planetary Atmospheres


Abstract
:1. Introduction
2. Methodology
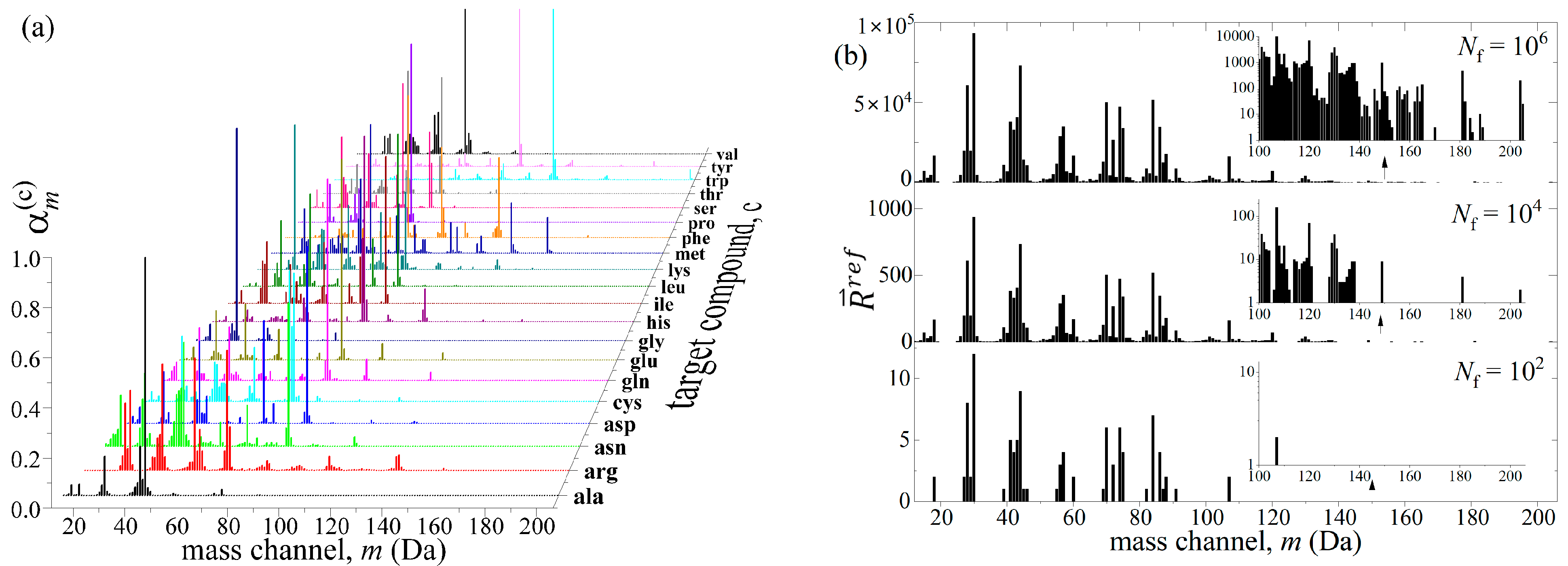
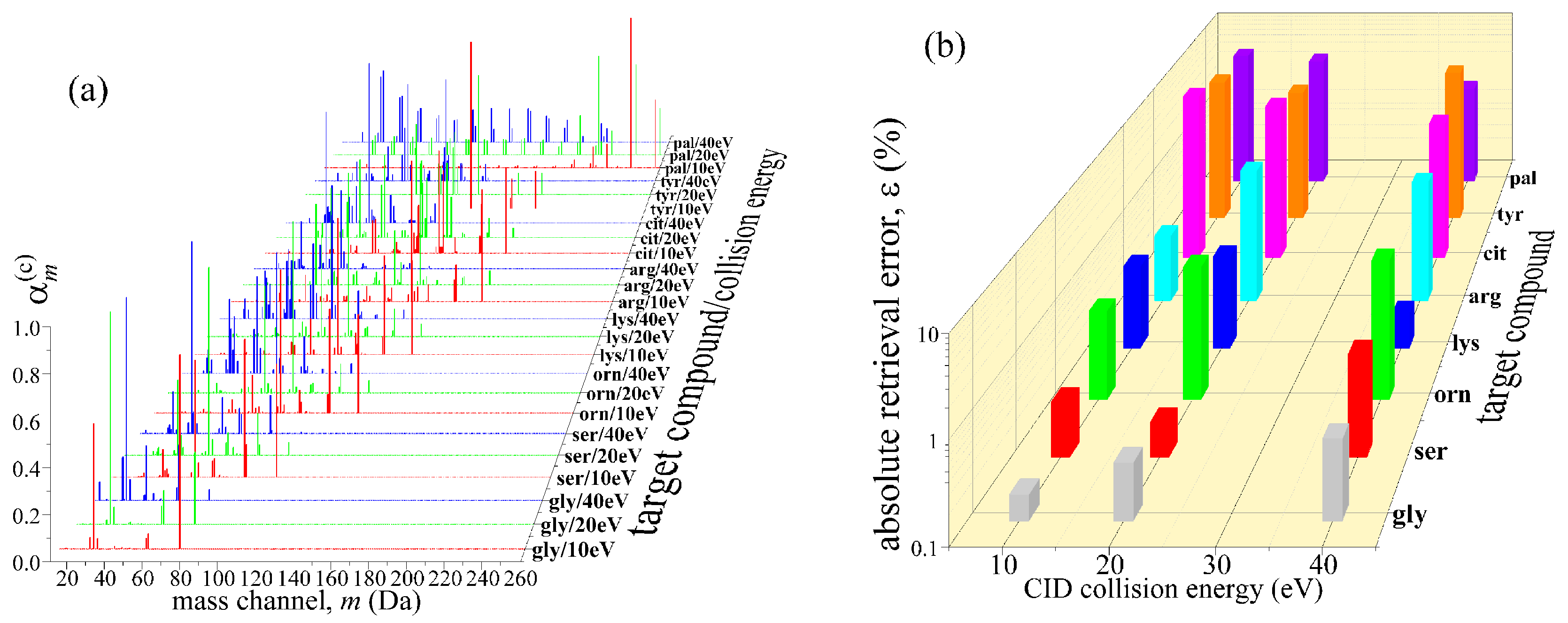
2.1. Generation of Mixture Reference Mass Spectrum
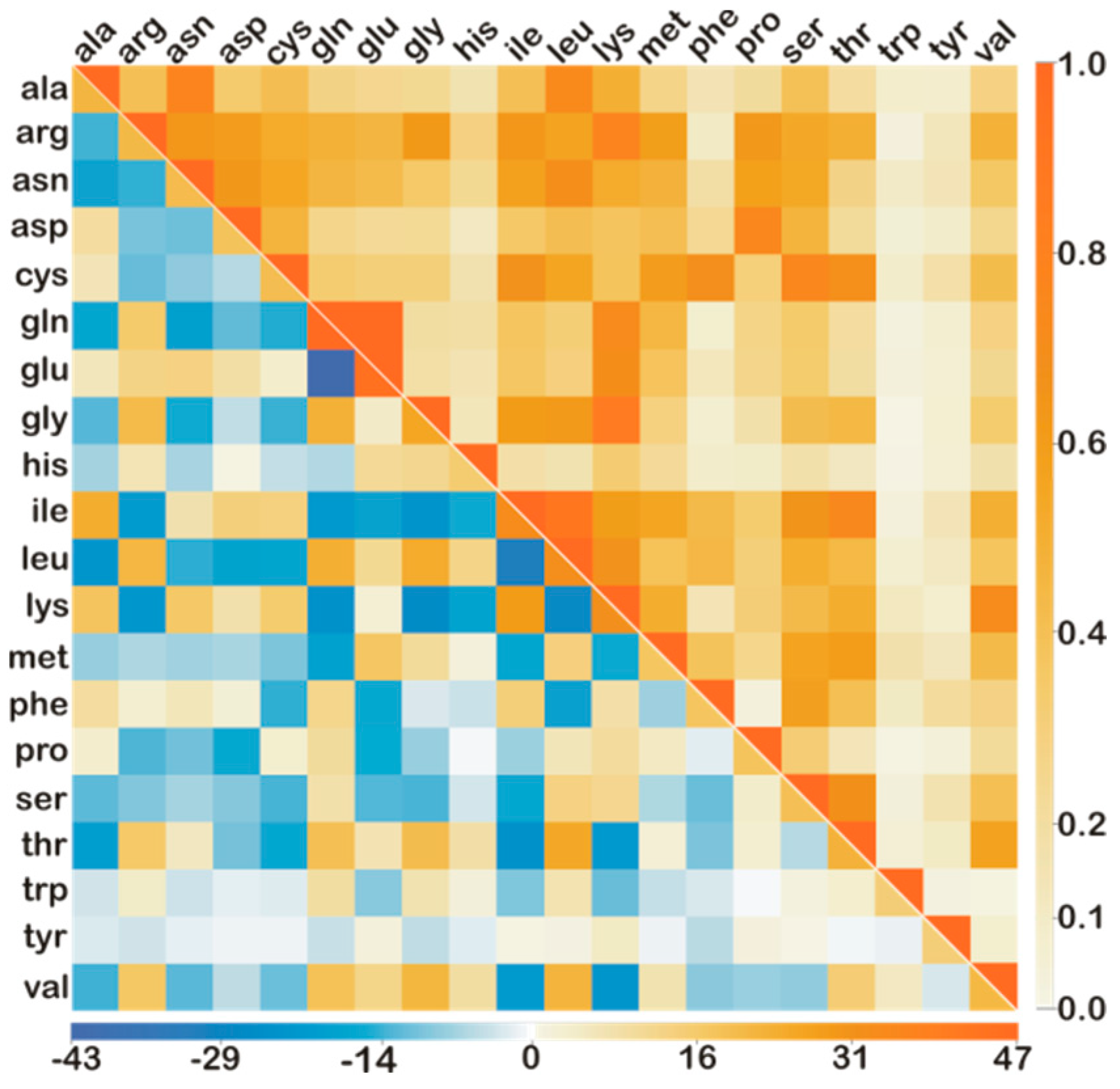
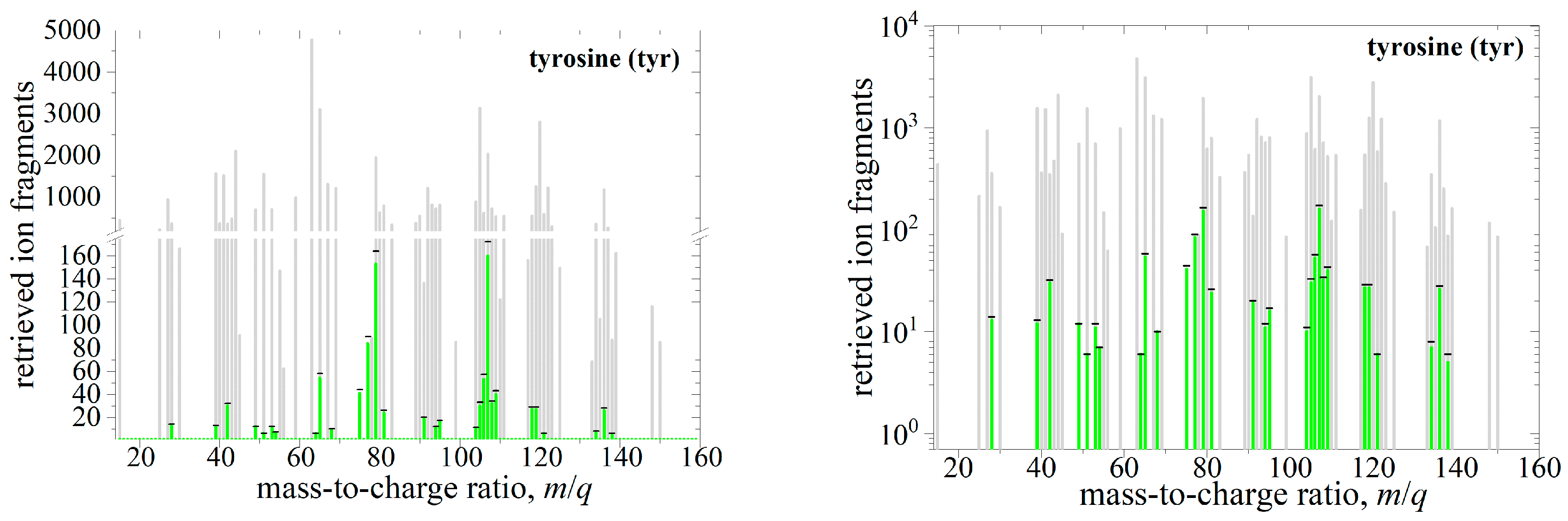
2.2. Deconvolution of Reference Mass Spectrum
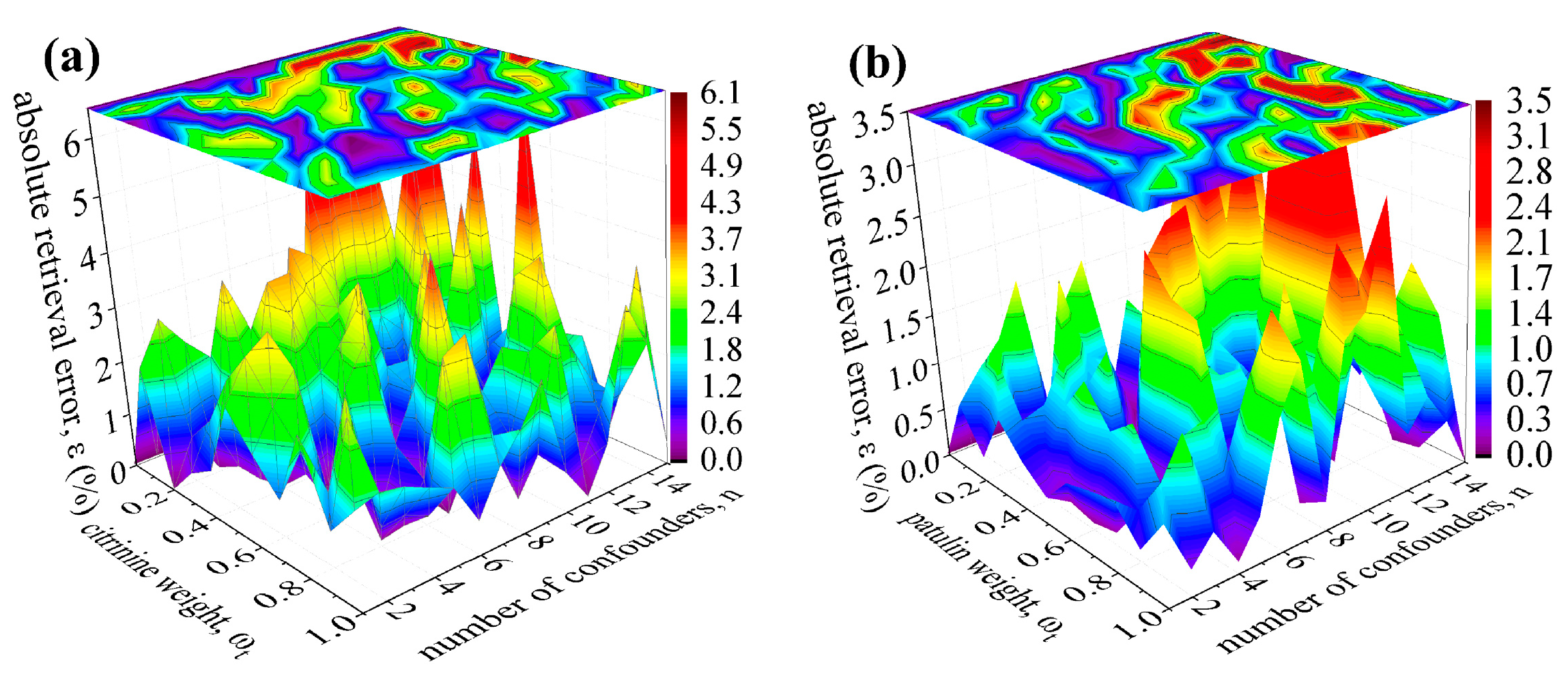
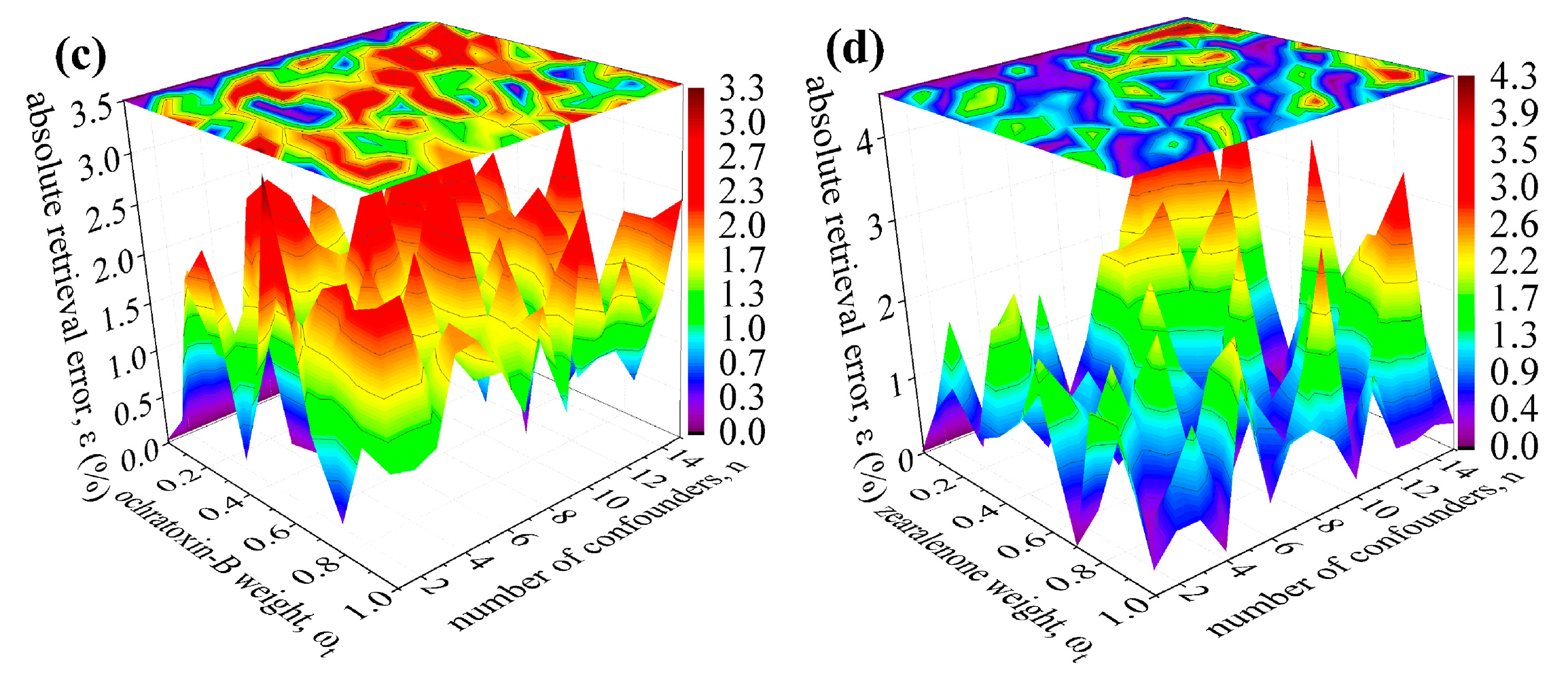
3. Robustness Tests
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Research Council Exploring Organic Environments in the Solar System; The National Academies Press: Washington, DC, USA, 2007. [CrossRef]
- Johnson, T.V.; Yeates, C.M.; Young, R. Space science reviews volume on Galileo Mission overview. Space Sci. Rev. 1992, 60, 3–21. [Google Scholar] [CrossRef]
- Smith, P.H.; Tamppari, L.; Arvidson, R.E.; Bass, D.; Blaney, D.; Boynton, W.; Carswell, A.; Catling, D.; Clark, B.; Duck, T.; et al. Introduction to special section on the Phoenix Mission: Landing Site Characterization Experiments, Mission Overviews, and Expected Science. J. Geophys. Res. Planets 2008, 113, E00A18. [Google Scholar] [CrossRef] [Green Version]
- Jakosky, B.M.; Lin, R.P.; Grebowsky, J.M.; Luhmann, J.G.; Mitchell, D.F.; Beutelschies, G.; Priser, T.; Acuna, M.; Andersson, L.; Baird, D.; et al. The Mars Atmosphere and Volatile Evolution (MAVEN) Mission. Space Sci. Rev. 2015, 195, 3–48. [Google Scholar] [CrossRef]
- Bell, J.F.; Maki, J.N.; Alwmark, S.; Ehlmann, B.L.; Fagents, S.A.; Grotzinger, J.P.; Gupta, S.; Hayes, A.; Herkenhoff, K.E.; Horgan, B.H.N.; et al. Geological, multispectral, and meteorological imaging results from the Mars 2020 Perseverance rover in Jezero crater. Sci. Adv. 2022, 8, eabo4856. [Google Scholar] [CrossRef]
- Barnes, J.W.; Turtle, E.P.; Trainer, M.G.; Lorenz, R.D.; MacKenzie, S.M.; Brinckerhoff, W.B.; Cable, M.L.; Ernst, C.M.; Freissinet, C.; Hand, K.P.; et al. Science Goals and Objectives for the Dragonfly Titan Rotorcraft Relocatable Lander. Planet. Sci. J. 2021, 2, 130. [Google Scholar] [CrossRef]
- Howell, S.M.; Pappalardo, R.T. NASA’s Europa Clipper—A mission to a potentially habitable ocean world. Nat. Commun. 2020, 11, 1311. [Google Scholar] [CrossRef] [Green Version]
- Garvin, J.B.; Getty, S.A.; Arney, G.N.; Johnson, N.M.; Kohler, E.; Schwer, K.O.; Sekerak, M.; Bartels, A.; Saylor, R.S.; Elliott, V.E.; et al. Revealing the Mysteries of Venus: The DAVINCI Mission. Planet. Sci. J. 2022, 3, 117. [Google Scholar] [CrossRef]
- Krick, T.; Verstraete, N.; Alonso, L.G.; Shub, D.A.; Ferreiro, D.U.; Shub, M.; Sánchez, I.E. Amino Acid Metabolism Conflicts with Protein Diversity. Mol. Biol. Evol. 2014, 31, 2905–2912. [Google Scholar] [CrossRef] [Green Version]
- Cobb, A.K.; Pudritz, R.E. Nature’S Starships: I. Observed Abundances and Relative Frequencies of Amino Acids in Meteorites. Astrophys. J. 2014, 783, 140. [Google Scholar] [CrossRef]
- Martins, Z.; Chan, Q.H.S.; Bonal, L.; King, A.; Yabuta, H. Organic Matter in the Solar System—Implications for Future on-Site and Sample Return Missions. Space Sci. Rev. 2020, 216, 54. [Google Scholar] [CrossRef]
- Simkus, D.N.; Aponte, J.C.; Elsila, J.E.; McLain, H.L.; Parker, E.T.; Dworkin, J.P.; Glavin, D.P. Low total abundances and a predominance of n-ω-amino acids in enstatite chondrites: Implications for thermal stability of amino acids in the inner solar system. Meteorit. Planet Sci. 2021, 56, 2118–2127. [Google Scholar] [CrossRef]
- Gentry, D.M.; Amador, E.S.; Cable, M.L.; Chaudry, N.; Cullen, T.; Jacobsen, M.B.; Murukesan, G.; Schwieterman, E.W.; Stevens, A.H.; Stockton, A.; et al. Correlations Between Life-Detection Techniques and Implications for Sampling Site Selection in Planetary Analog Missions. Astrobiology 2017, 17, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Klenner, F.; Postberg, F.; Hillier, J.; Khawaja, N.; Cable, M.L.; Abel, B.; Kempf, S.; Glein, C.R.; Lunine, J.I.; Hodyss, R.; et al. Discriminating Abiotic and Biotic Fingerprints of Amino Acids and Fatty Acids in Ice Grains Relevant to Ocean Worlds. Astrobiology 2020, 20, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Kopplin, P.; Gabelica, Z.; Gougeon, R.D.; Fekete, A.; Kanawati, B.; Harir, M.; Gebefuegi, I.; Eckel, G.; Hertkorn, N. High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall. Proc. Natl. Acad. Sci. USA 2010, 107, 2763–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, A.; D’Hendecourt, L.L.S.; Schmitt-Kopplin, P. Data-Driven Astrochemistry: One Step Further within the Origin of Life Puzzle. Life 2018, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Arevalo, J.R.; Ni, Z.; Danell, R.M. Mass spectrometry and planetary exploration: A brief review and future projection. J. Mass Spectrom. 2020, 55, e4454. [Google Scholar] [CrossRef] [Green Version]
- Zubarev, R.A.; Makarov, A. Orbitrap Mass Spectrometry. Anal. Chem. 2013, 85, 5288–5296. [Google Scholar] [CrossRef]
- Selliez, L.; Briois, C.; Carrasco, N.; Thirkell, L.; Thissen, R.; Ito, M.; Orthous-Daunay, F.-R.; Chalumeau, G.; Colin, F.; Cottin, H.; et al. Identification of organic molecules with a laboratory prototype based on the Laser Ablation-CosmOrbitrap. Planet. Space Sci. 2019, 170, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Alelyunas, Y.; Wrona, M.; Cook, K.; McDonald, S.; Rainville, P.D. Effect of MS Scan Speed on UPLC Peak Separation and Metabolite Identification: Time-of-Flight HRMS vs. Orbitrap. Waters. 2013. Application Note 720004762EN. Available online: https://www.waters.com/content/dam/waters/en/app-notes/2013/720004762/720004762-en.pdf (accessed on 16 June 2022).
- Nikolić, D.; Madzunkov, S.M.; Darrach, M.R. Computer Modeling of an Ion Trap Mass Analyzer, Part I: Low Pressure Regime. J. Am. Soc. Mass Spectrom. 2015, 26, 2115–2124. [Google Scholar] [CrossRef]
- Paul, W.; Steinwedel, H. Notizen: Ein neues Massenspektrometer ohne Magnetfeld. Z. Für Nat. A 1953, 8, 448–450. [Google Scholar] [CrossRef]
- Paul, W. Electromagnetic traps for charged and neutral particles. Rev. Mod. Phys. 1990, 62, 531–540. [Google Scholar] [CrossRef]
- Avice, G.; Belousov, A.; Farley, K.A.; Madzunkov, S.M.; Simcic, J.; Nikolić, D.; Darrach, M.R.; Sotin, C. High-precision measurements of krypton and xenon isotopes with a new static-mode quadrupole ion trap mass spectrometer. J. Anal. At. Spectrom. 2019, 34, 104–117. [Google Scholar] [CrossRef]
- Madzunkov, S.M.; Nikolić, D.; Simcic, J.; Belousov, A.; Gonzalez, M.P.; Darrach, M.R. Data analysis and isotopic ratios measured onboard the Spacecraft Atmosphere Monitor. Int. J. Mass Spectrom. 2022, 477, 116847. [Google Scholar] [CrossRef]
- Darrach, M.; Madzunkov, S.; Kidd, R.; Bae, B.; Zhong, F.; Simcic, J.; Malone, C.; Belousov, A.; Maiwald, F.; Gonzales, M.; et al. Update on the Spacecraft Atmosphere Monitor Technology Demonstration Project. In Proceedings of the 50th International Conference on Environmental Systems, Lisbon, Portugal, 12–16 July 2020; Available online: https://hdl.handle.net/2346/86313 (accessed on 11 July 2022).
- Belousov, A.; Miller, M.; Continetti, R.; Madzunkov, S.; Simcic, J.; Nikolic, D.; Maiwald, F.; Waller, S.; Malaska, M.; Cable, M. Sampling Accelerated Micron Scale Ice Particles with a Quadrupole Ion Trap Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2021, 32, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Waller, S.E.; Belousov, A.; Kidd, R.D.; Nikolić, D.; Madzunkov, S.M.; Wiley, J.S.; Darrach, M.R. Chemical Ionization Mass Spectrometry: Applications for the In Situ Measurement of Nonvolatile Organics at Ocean Worlds. Astrobiology 2019, 19, 1196–1210. [Google Scholar] [CrossRef]
- Naraoka, H.; Hashiguchi, M.; Sato, Y.; Hamase, K. New Applications of High-Resolution Analytical Methods to Study Trace Organic Compounds in Extraterrestrial Materials. Life 2019, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- De Middeleer, G.; Leys, N.; Sas, B.; De Saeger, S. Fungi and Mycotoxins in Space—A Review. Astrobiology 2019, 19, 915–926. [Google Scholar] [CrossRef]
- Mora, M.; Wink, L.; Kögler, I.; Mahnert, A.; Rettberg, P.; Schwendner, P.; Demets, R.; Cockell, C.; Alekhova, T.; Klingl, A.; et al. Space Station conditions are selective but do not alter microbial characteristics relevant to human health. Nat. Commun. 2019, 10, 3990. [Google Scholar] [CrossRef] [Green Version]
- Onofri, S.; Selbmann, L.; Pacelli, C.; De Vera, J.P.; Horneck, G.; Hallsworth, J.E.; Zucconi, L. Integrity of the DNA and Cellular Ultrastructure of Cryptoendolithic Fungi in Space or Mars Conditions: A 1.5-Year Study at the International Space Station. Life 2018, 8, 23. [Google Scholar] [CrossRef]
- Satoh, K.; Alshahni, M.M.; Umeda, Y.; Komori, A.; Tamura, T.; Nishiyama, Y.; Yamazaki, T.; Makimura, K. Seven years of progress in determining fungal diversity and characterization of fungi isolated from the Japanese Experiment Module KIBO, International Space Station. Microbiol. Immunol. 2021, 65, 463–471. [Google Scholar] [CrossRef]
- Cortesão, M.; Schütze, T.; Marx, R.; Moeller, R.; Meyer, V.; Nevalainen, H. Fungal Biotechnology in Space: Why and How? Grand Chall. Fungal. Biotechnol. 2020, 18, 501–535. [Google Scholar] [CrossRef] [Green Version]
- Cortesão, M.; de Haas, A.; Unterbusch, R.; Fujimori, A.; Schütze, T.; Meyer, V.; Moeller, R. Aspergillus niger Spores Are Highly Resistant to Space Radiation. Front. Microbiol. 2020, 11, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, R.L. Microbial diversity and its relationship to planetary protection. Appl. Environ. Microbiol. 2005, 71, 4163–4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIST Standard Reference Database 1A, “NIST/EPA/NIH Mass Spectral Library with Search Program”, Data Version: NIST 20, Software Version: 2.4. Available online: https://www.nist.gov/srd/nist-standard-reference-database-1a (accessed on 14 August 2022).
- Madzunkov, S.M.; MacAskill, J.A.; Chutjian, A.; Ehrenfreund, P.; Darrach, M.R.; Vidali, G.; Shortt, B.J. Formation of Formaldehyde and Carbon Dioxide on an Icy Grain Analog Using Fast Hydrogen Atoms. Astrophys. J. 2009, 697, 801–806. [Google Scholar] [CrossRef]
- Nikolić, D.; Madzunkov, S.M. Mapping of spacecraft atmosphere monitor signal to major constituent abundances. In Proceedings of the 46th International Conference on Environmental Systems, Vienna, Austria, 10 July 2016; Volume 285, pp. 1–7. Available online: https://hdl.handle.net/2014/46134 (accessed on 11 July 2022).
- Allen, F.; Pon, A.; Greiner, R.; Wishart, D. Computational Prediction of Electron Ionization Mass Spectra to Assist in GC/MS Compound Identification. Anal. Chem. 2016, 88, 7689–7697. [Google Scholar] [CrossRef] [PubMed]
- André, A.; Müller, N.; Chetschik, I. Occurrence of Zearalenone and Enniatin B in Swiss Wheat Grains and Wheat Flours. Appl. Sci. 2022, 12, 10566. [Google Scholar] [CrossRef]
- Limaye, S.S.; Mogul, R.; Smith, D.J.; Ansari, A.H.; Słowik, G.P.; Vaishampayan, P. Venus’ Spectral Signatures and the Potential for Life in the Clouds. Astrobiology 2018, 18, 1181–1198. [Google Scholar] [CrossRef]
- Sasaki, S.; Yamagishi, A.; Yoshimura, Y.; Enya, K.; Miyakawa, A.; Ohno, S.; Fujita, K.; Usui, T.; Limaye, S.S. In situ biochemical characterization of Venus cloud particles using a life-signature detection microscope. Can. J. Microbiol. 2022, 68, 413–425. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (c) | Name | NIST EII # | Formula | ||
|---|---|---|---|---|---|
| 1 | alanine (ala) | 228084 | C3H7NO2 | 0.076 | 2.89 |
| 2 | arginine (arg) | 154822 | C6H14N4O2 | 0.058 | 4.90 |
| 3 | asparagine (asn) | 229288 | C4H8N2O3 | 0.052 | 4.52 |
| 4 | aspartic acid (asp) | 230141 | C4H7NO4 | 0.057 | 3.06 |
| 5 | cysteine (cys) | 228086 | C3H7NO2S | 0.010 | 4.42 |
| 6 | glutamine (gln) | 228123 | C5H10N2O3 | 0.055 | 3.31 |
| 7 | glutamic acid (glu) | 228122 | C5H9NO4 | 0.063 | 3.08 |
| 8 | glycine (gly) | 229287 | C2H5NO2 | 0.076 | 3.09 |
| 9 | histidine (his) | 228152 | C6H9N3O2 | 0.018 | 0.96 |
| 10 | isoleucine (ile) | 228158 | C6H13NO2 | 0.066 | 5.18 |
| 11 | leucine (leu) | 228159 | C6H13NO2 | 0.067 | 4.78 |
| 12 | lysine (lys) | 26152 | C6H14N2O2 | 0.040 | 5.36 |
| 13 | methionine (met) | 191948 | C5H11NO2S | 0.023 | 3.66 |
| 14 | phenylalanine (phe) | 107173 | C9H11NO2 | 0.044 | 2.09 |
| 15 | proline (pro) | 228120 | C5H9NO2 | 0.066 | 2.43 |
| 16 | serine (ser) | 228085 | C3H7NO3 | 0.065 | 4.29 |
| 17 | threonine (thr) | 26149 | C4H9NO3 | 0.058 | 3.64 |
| 18 | tryptophan (trp) | 127959 | C11H12N2O2 | 0.007 | 0.32 |
| 19 | tyrosine (tyr) | 228180 | C9H11NO3 | 0.030 | 0.54 |
| 20 | valine (val) | 26146 | C5H11NO2 | 0.069 | 2.90 |
| (c) | (c) | (c) | (c) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ala | 0.12(15) | 661(62) | 6 | gln | 3.8(7) | 878(131) | 11 | leu | 1.83(84) | 931(175) | 16 | ser | 0.15(14) | 655(51) |
| 2 | arg | 0.1(3) | 1321(132) | 7 | glu | 2.1(3) | 798(77) | 12 | lys | 2.6(9) | 1302(265) | 17 | thr | 0.2(3) | 905(91) |
| 3 | asn | 0.44(19) | 1077(83) | 8 | gly | 0.57(17) | 596(46) | 13 | met | 0.22(19) | 622(77) | 18 | trp | 0.10(8) | 275(28) |
| 4 | asp | 0.28(15) | 497(54) | 9 | his | 0.26(18) | 686(54) | 14 | phe | 0.12(7) | 514(44) | 19 | tyr | 0.10(7) | 249(20) |
| 5 | cys | 1.36(82) | 3063(237) | 10 | ile | 1.88(86) | 1286(188) | 15 | pro | 0.15(11) | 519(63) | 20 | val | 0.52(35) | 628(85) |
| (t) | Target Name | Target Formula | Confounders (Canonical SMILES) | |||||
|---|---|---|---|---|---|---|---|---|
| c-1 | c-2 | c-3 | c-4 | c-5 | c-6 | |||
| (pal) | palmitic acid | C16H32O2 | CCCCC(CC)C(=O)C(O)C(CC)CCCC | CCCCCCCCCCCCC(C)(O)C(C)=O | CCCCCCCCCCC(C)(C(=O)O)C(C)C | CCCC(C)CC(C)CC(CC)(CCC)C(=O)O | CCCCCCCC(CC)(CCCC)C(=O)O | CCCCCCCCCC(C)CC(C)CC(=O)O |
| (tyr) | tyrosine | C9H11NO3 | CC(=O)c1c(C)[nH]c(C(=O)O)c1C | Cc1cc(C)n(C)c(=O)c1C(=O)O | Nc1cc(CCC(=O)O)ccc1O | NCCOc1ccc(C(=O)O)cc1 | COc1ccc(NCC(=O)O)cc1 | -- |
| (cit) | citrulline | C6H13N3O3 | [H][C@](O)(CCCNC(=N)N)C(=O)O | [H][C@]1(O)CN(C(=N)N)C[C@@]([H])(O)C1O | COCC(N)C(=O)NCC(N)=O | COC(CN)CC(=O)NC(N)=O | C(CNC(=O)CNC(=O)N)OC | -- |
| (arg) | arginine | C6H14N4O2 | NC(=O)NCCCCNC(N)=O | NC(=O)CC(N)C(N)CC(N)=O | CC(NCC(N)C(N)=O)C(N)=O | NC(=O)NCCCC(N)C(N)=O | N=C(N)NCCC(O)C(N)C=O | -- |
| (lys) | lysine | C6H14N2O2 | CN(C)CCCNC(=O)O | COCCNC(=O)[C@@H](C)N | CN(CC(C)(C)O)C(N)=O | CCC[C@H](N)C(O)C(N)=O | -- | -- |
| (orn) | ornithine | C5H12N2O2 | CN(C)CCNC(=O)O | [H][C@](C)(CN)NC(=O)OC | CN(C)C(=O)[C@@H](N)CO | N[C@@H]1COCOC[C@@H]1N | COC[C@@H](C)NC(N)=O | -- |
| (gly) | glycine | C2H5NO2 | NC(=O)CO | COC(N)=O | CNC(=O)O | -- | -- | -- |
| (ser) | serine | C3H7NO3 | n1ccccc1C=C | c1(cccnc1)C=C | c1(ccncc1)C=C | C1=CC=C(C=C1)C=N | N#Cc1nccnc1 | C(C#N)C(C#N)C#N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madzunkova, S.; Nikolić, D. Method for Accurate Detection of Amino Acids and Mycotoxins in Planetary Atmospheres. Life 2022, 12, 2122. https://doi.org/10.3390/life12122122
Madzunkova S, Nikolić D. Method for Accurate Detection of Amino Acids and Mycotoxins in Planetary Atmospheres. Life. 2022; 12(12):2122. https://doi.org/10.3390/life12122122
Chicago/Turabian StyleMadzunkova, Sigrid, and Dragan Nikolić. 2022. "Method for Accurate Detection of Amino Acids and Mycotoxins in Planetary Atmospheres" Life 12, no. 12: 2122. https://doi.org/10.3390/life12122122
APA StyleMadzunkova, S., & Nikolić, D. (2022). Method for Accurate Detection of Amino Acids and Mycotoxins in Planetary Atmospheres. Life, 12(12), 2122. https://doi.org/10.3390/life12122122