

Mitochondria Clumping vs. Mitochondria Fusion in CMT2A Diseases




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- (1)
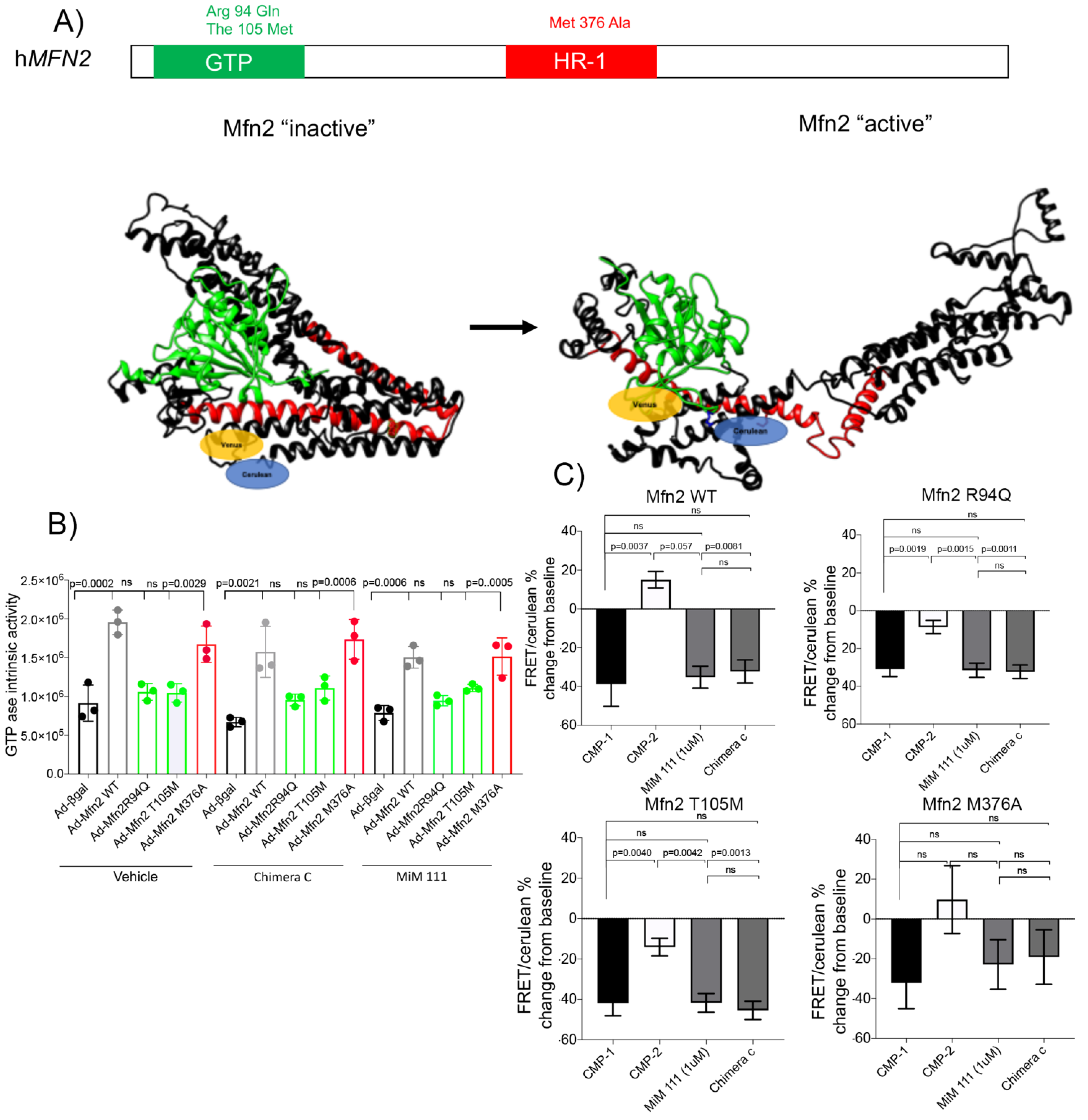
- The source of dysfunction for the MFN2 M376A mutation in the HR1 domain stems from mitofusin in the inactive conformation, notably maintaining its GTPase abilities.
- (2)
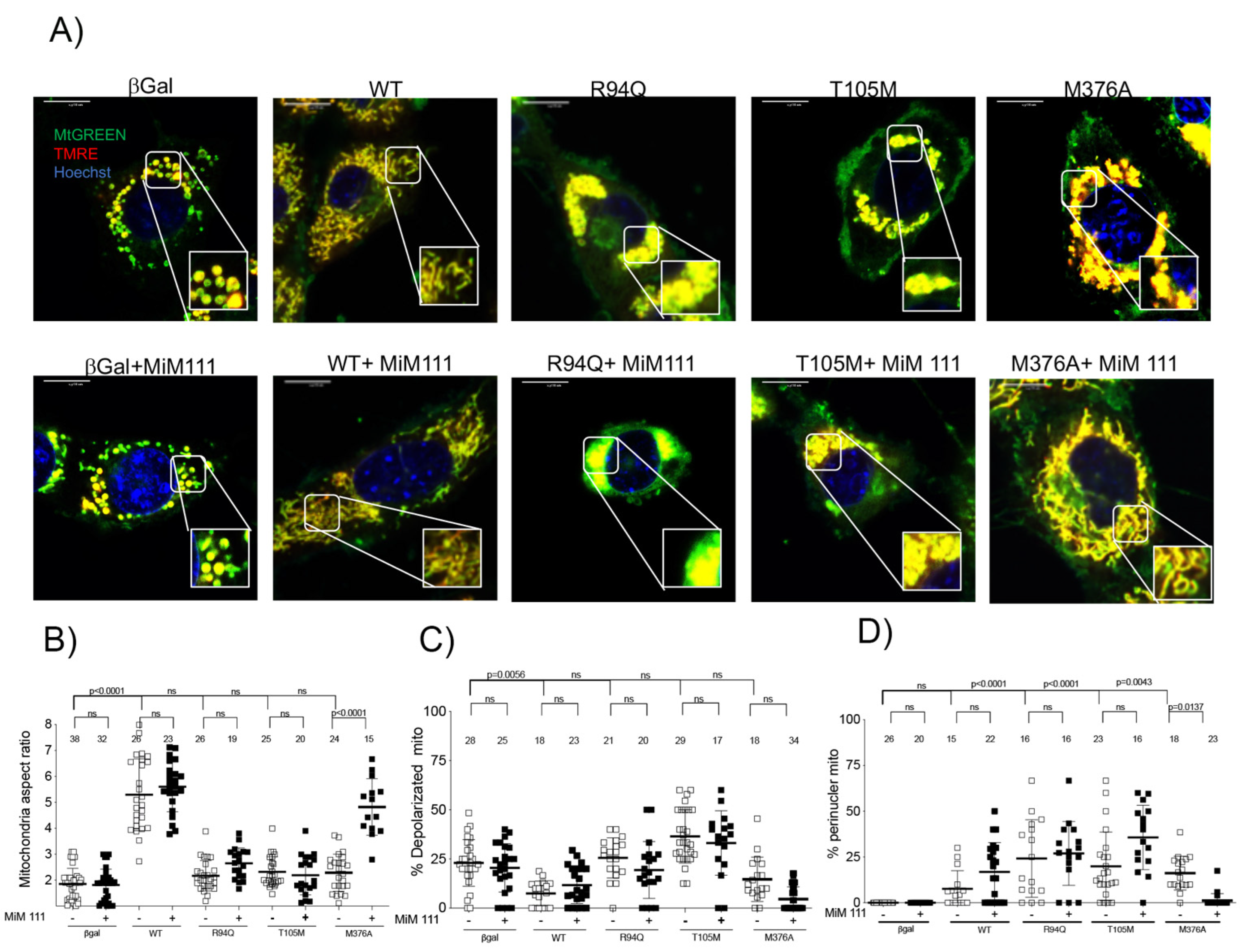
- MFN2 M376A lacks perinuclear mitochondrial clumping.
- (3)
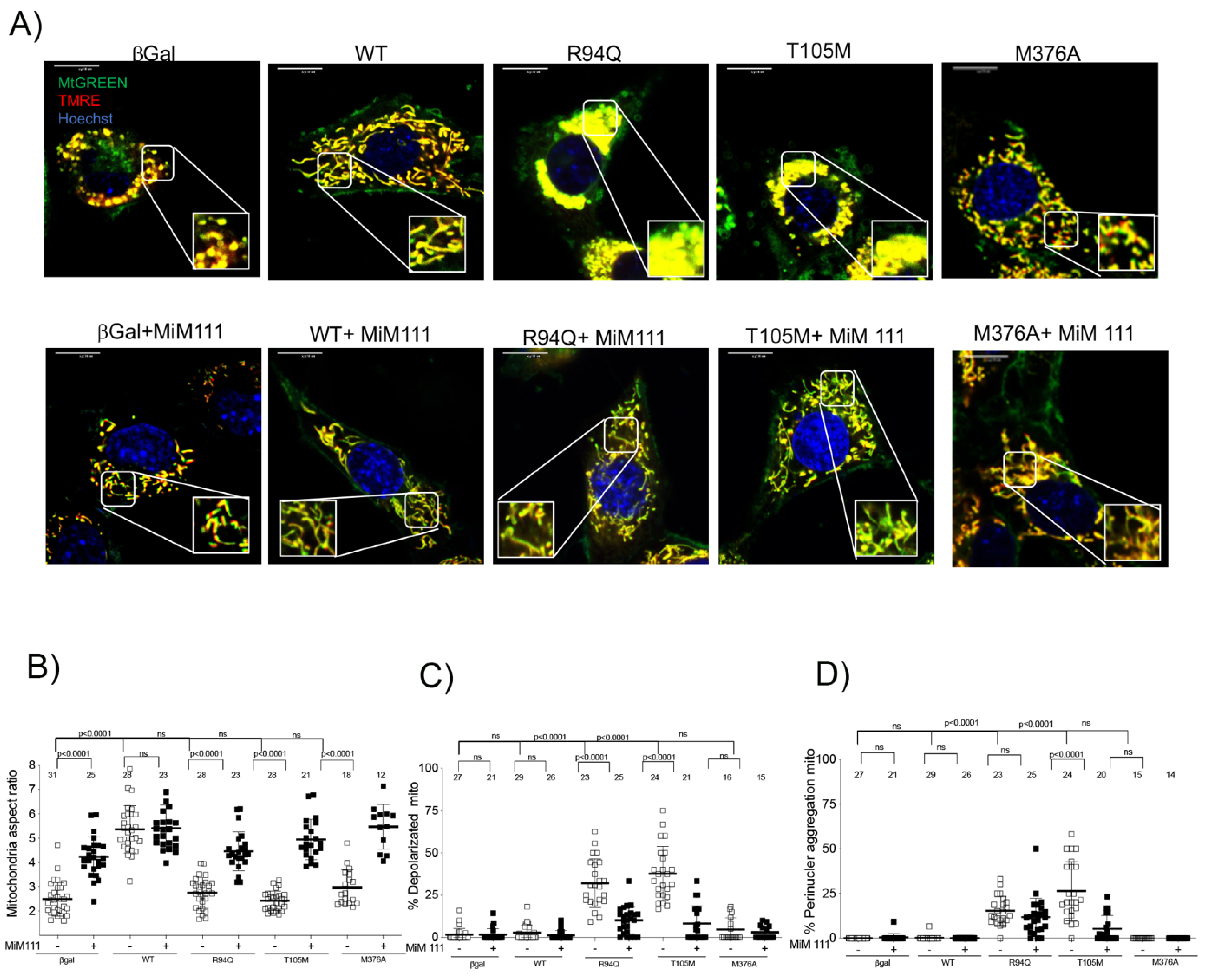
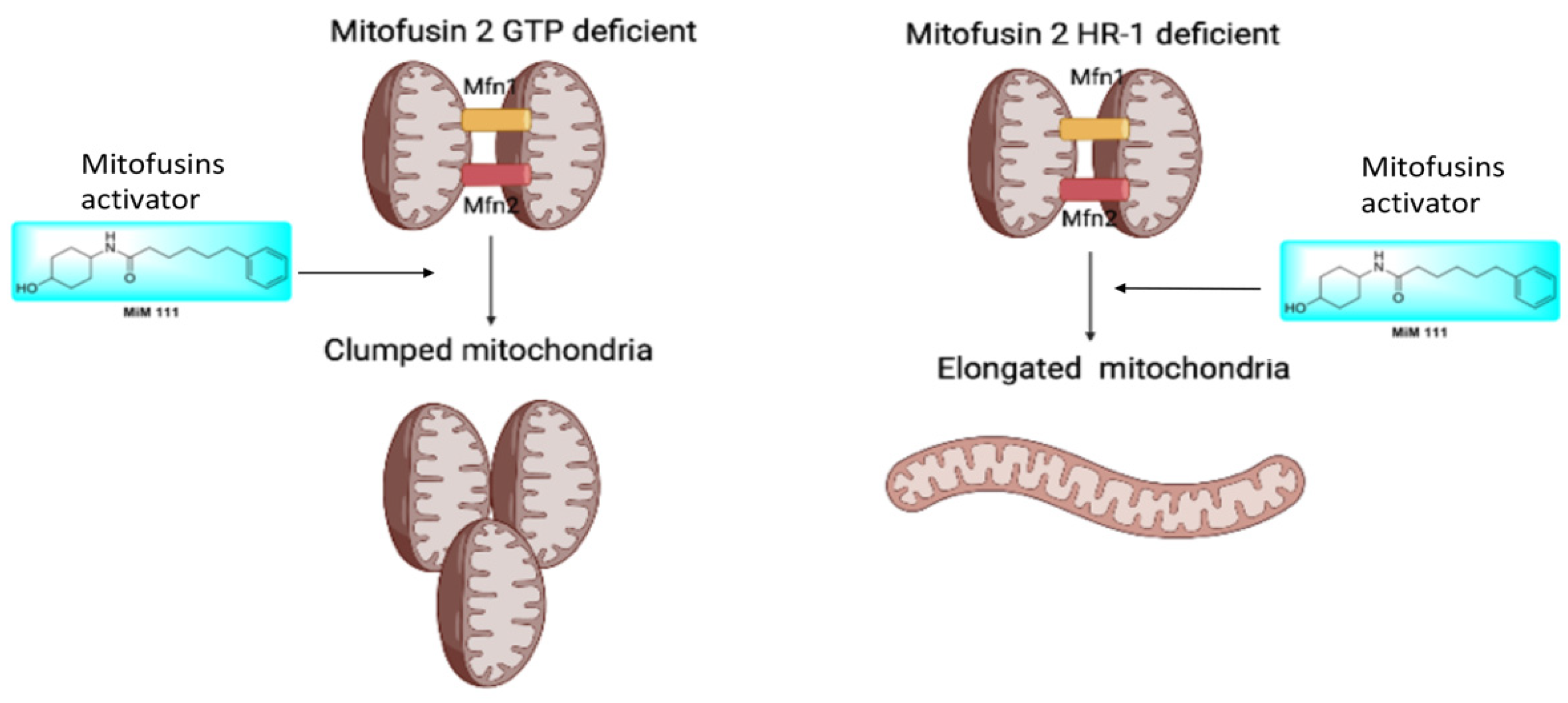
- Mitofusin agonists MiM 111 and Chimera C open the MFN2 M376A mutant protein and reverse its dysfunctional mitochondrial fusion abilities.
2. Material and Methods
2.1. Cell Lines
2.2. Viral Vectors
2.3. Protein Structure Modeling
2.4. Imaging
2.5. GTPase Activity Assay
2.6. MFN2 FRET Assays
2.7. Data Presentation and Statistical Analyses
3. Results
3.1. Novel Mechanism of Dysfunction in CMT2A HR1 vs. GTPase Domain Mutation
3.2. MFN2 M376A Induces Short Mitochondria without Depolarization
3.3. Dominant MFN2 M376A Mutation Suppresses Mitochondrial Fusion
3.4. M376A Is a New Shape Shifting Mitofusin 2 Conformational Mutant
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dang, X.; Franco, A.; Dorn, G.W., 2nd. Reciprocal Regulation of Mitofusin 2-Mediated Mitophagy and Mitochondrial Fusion by Different PINK1 Phosphorylation Events. Front. Cell Dev. Biol. 2022, 10, 868465. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W., 2nd; Scorrano, L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 2013, 342, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Ballard, A.; Zeng, R.; Zarei, A.; Shao, C.; Cox, L.; Yan, H.; Franco, A.; Dorn, G.W., 2nd; Faccio, R.; Veis, D.J. The tethering function of mitofusin2 controls osteoclast differentiation by modulating the Ca2+-NFATc1 axis. J. Biol. Chem. 2020, 295, 6629–6640. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Dang, X.; Walton, E.K.; Ho, J.N.; Zablocka, B.; Ly, C.; Miller, T.M.; Baloh, R.H.; Shy, M.E.; Yoo, A.S.; et al. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. Elife 2020, 9, e61119. [Google Scholar] [CrossRef] [PubMed]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana-Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 mutations in Charcot-Marie-Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Carmona, S.; Muhammad, A.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W., 2nd; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2019, 129, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Low, H.H.; Löwe, J. A bacterial dynamin-like protein. Nature 2006, 444, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Zhang, L.; Franco, A.; Li, J.; Rocha, A.G.; Devanathan, S.; Dolle, R.E.; Bernstein, P.R.; Dorn, G.W., 2nd. Discovery of 6-Phenylhexanamide Derivatives as Potent Stereoselective Mitofusin Activators for the Treatment of Mitochondrial Diseases. J. Med. Chem. 2020, 63, 7033–7051. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Williams, S.B.; Devanathan, S.; Franco, A.; Fu, L.; Bernstein, P.R.; Walters, D.; Dorn, G.W., 2nd. Pharmacophore-Based Design of Phenyl-[hydroxycyclohexyl] Cycloalkyl-Carboxamide Mitofusin Activators with Improved Neuronal Activity. J. Med. Chem. 2021, 64, 12506–12524. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Walton, E.K.; Zablocka, B.; Baloh, R.H.; Shy, M.E.; Dorn, G.W., 2nd. Mitochondrial Phenotypes in Genetically Diverse Neurodegenerative Diseases and Their Response to Mitofusin Activation. Cells 2022, 11, 1053. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Dang, X.; Zhang, L.; Molinoff, P.B.; Dorn, G.W. Mitochondrial Dysfunction and Pharmacodynamics of Mitofusin Activation in Murine Charcot-Marie-Tooth Disease Type 2A. J. Pharmacol. Exp. Ther. 2022, 383, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Li, X.; Liu, L.; Hu, Z.; Huang, S.; Zhan, Y.; Zi, X.; Xia, K.; Tang, B.; Zhang, R. MFN2-related genetic and clinical features in a cohort of Chinese CMT2 patients. J. Peripher. Nerv. Syst. 2016, 21, 38–44. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, A.; Walton, C.E.; Dang, X. Mitochondria Clumping vs. Mitochondria Fusion in CMT2A Diseases. Life 2022, 12, 2110. https://doi.org/10.3390/life12122110
Franco A, Walton CE, Dang X. Mitochondria Clumping vs. Mitochondria Fusion in CMT2A Diseases. Life. 2022; 12(12):2110. https://doi.org/10.3390/life12122110
Chicago/Turabian StyleFranco, Antonietta, Caroline E. Walton, and Xiawei Dang. 2022. "Mitochondria Clumping vs. Mitochondria Fusion in CMT2A Diseases" Life 12, no. 12: 2110. https://doi.org/10.3390/life12122110
APA StyleFranco, A., Walton, C. E., & Dang, X. (2022). Mitochondria Clumping vs. Mitochondria Fusion in CMT2A Diseases. Life, 12(12), 2110. https://doi.org/10.3390/life12122110