
Screening, Docking, and Molecular Dynamics Study of Natural Compounds as an Anti-HER2 for the Management of Breast Cancer



Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Protein
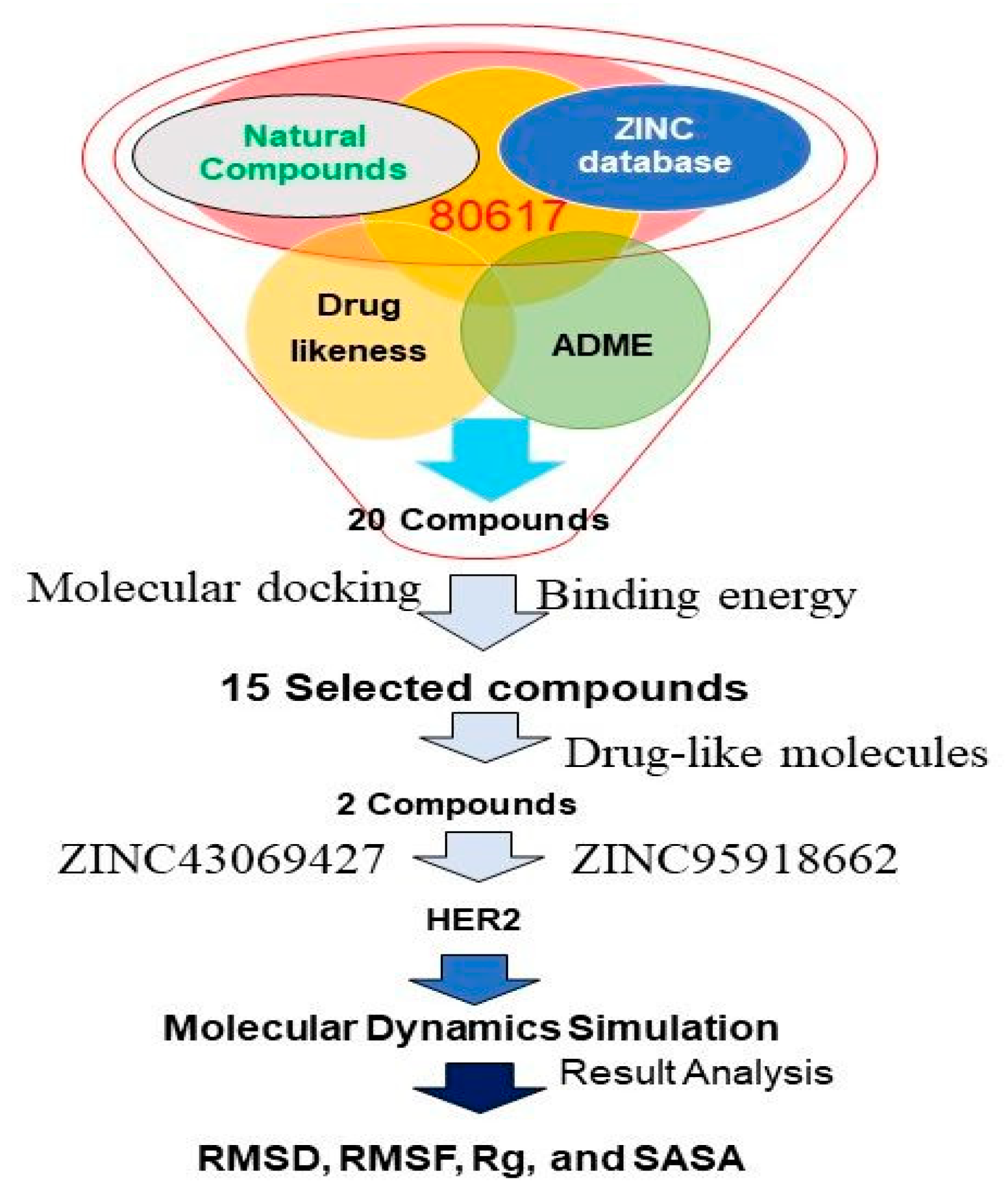
2.2. Screening for Compounds
2.3. Validation of Docking Protocol
2.4. Multiple Ligand Docking
2.5. Protein Plus Server
2.6. MD Simulation
3. Results and Discussion
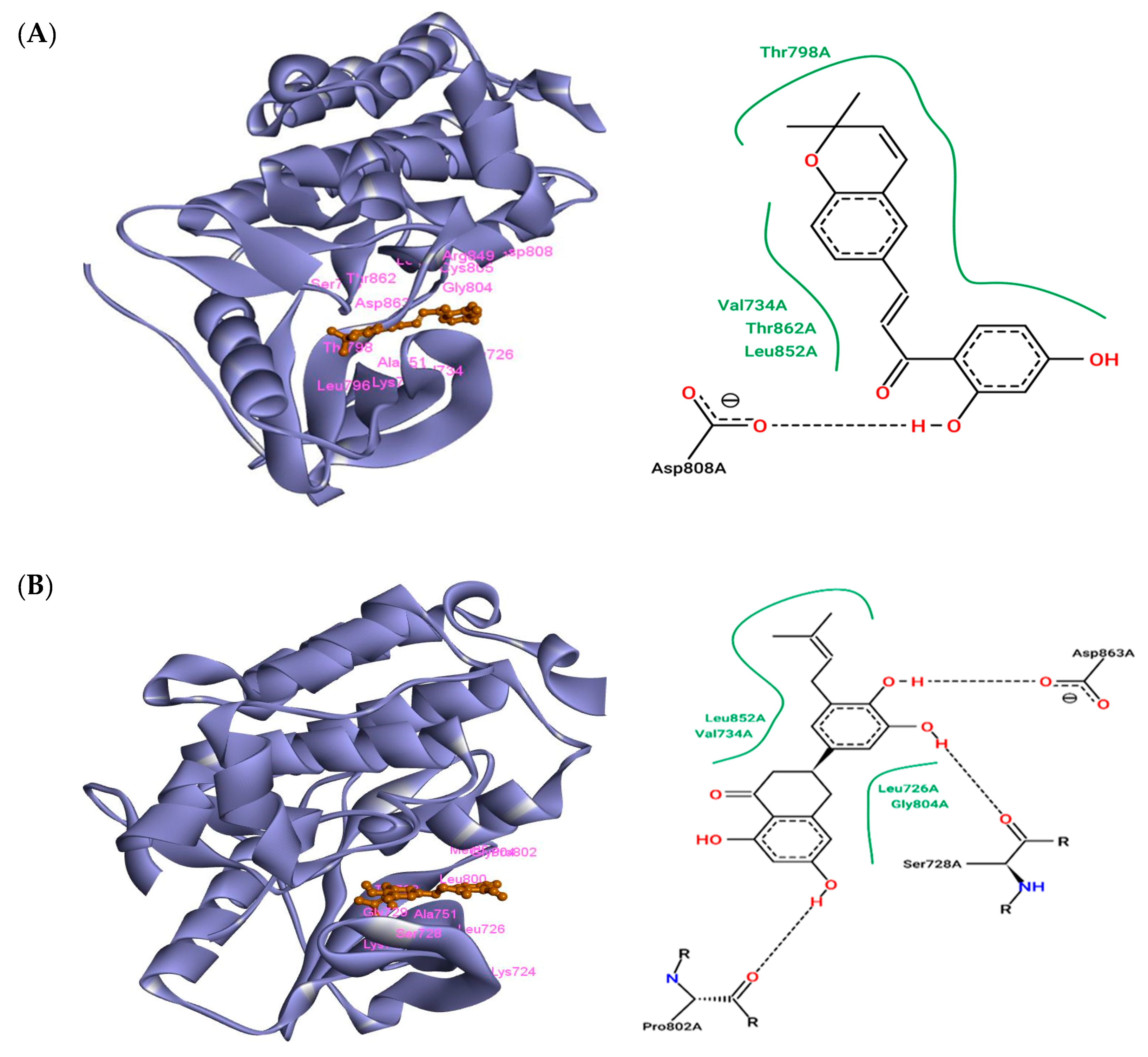
3.1. The Interaction of ZINC43069427 with HER2
3.2. The Interaction of ZINC95918662 with HER2
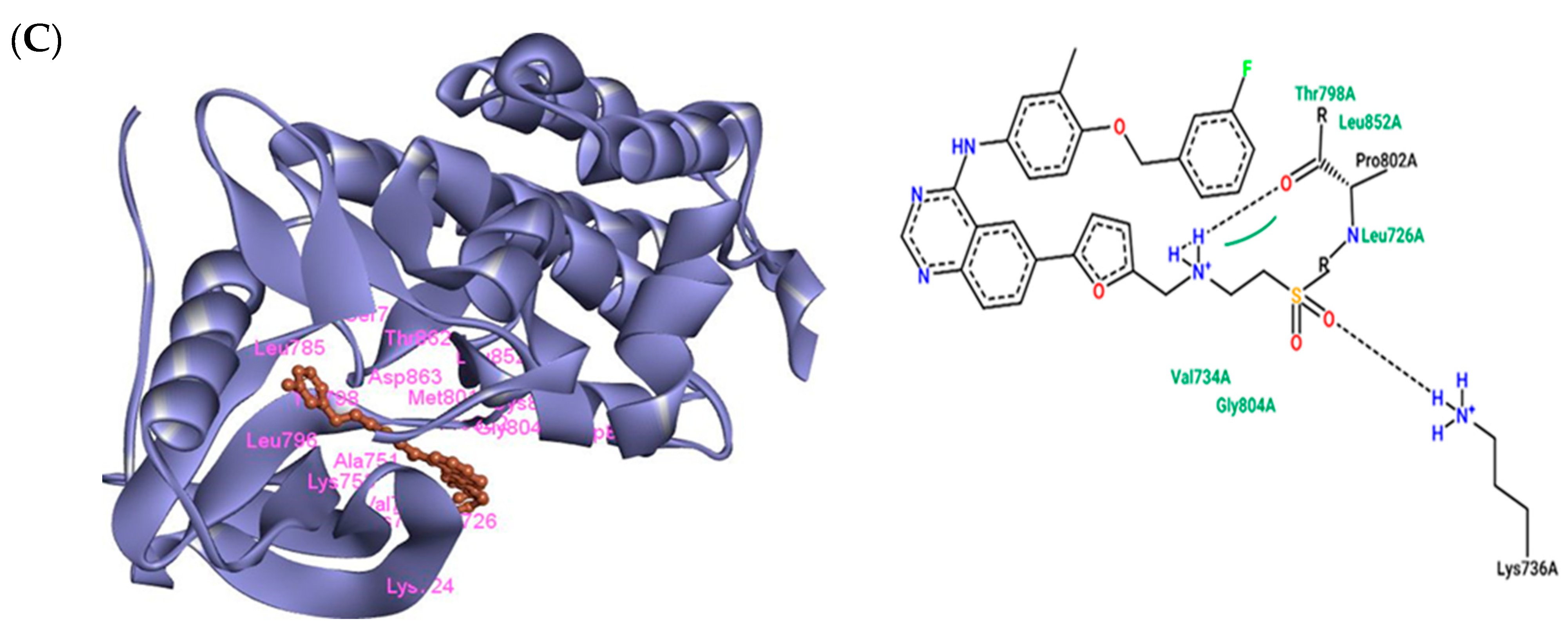
3.3. The Interaction of Lapatinib with HER2
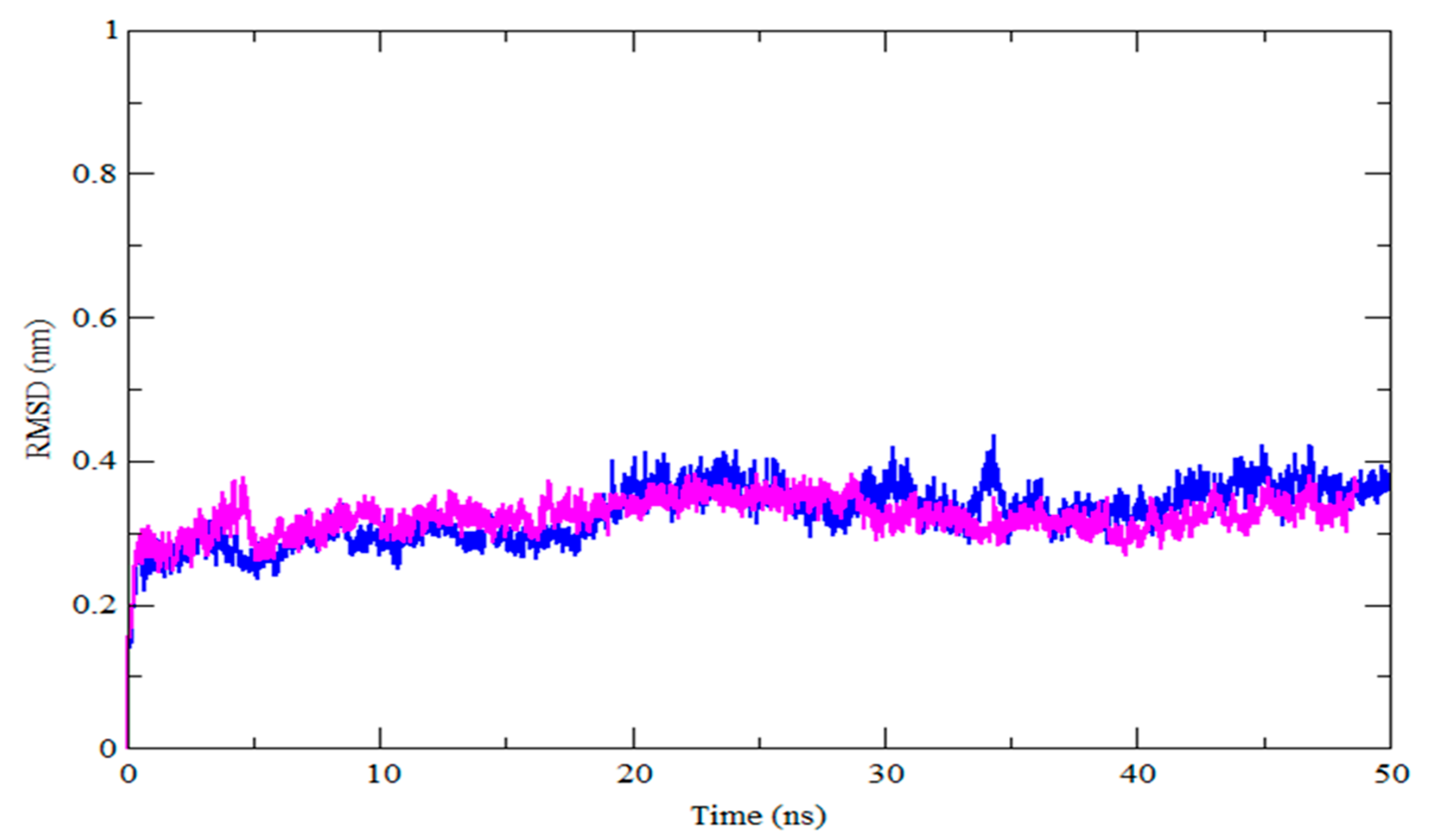
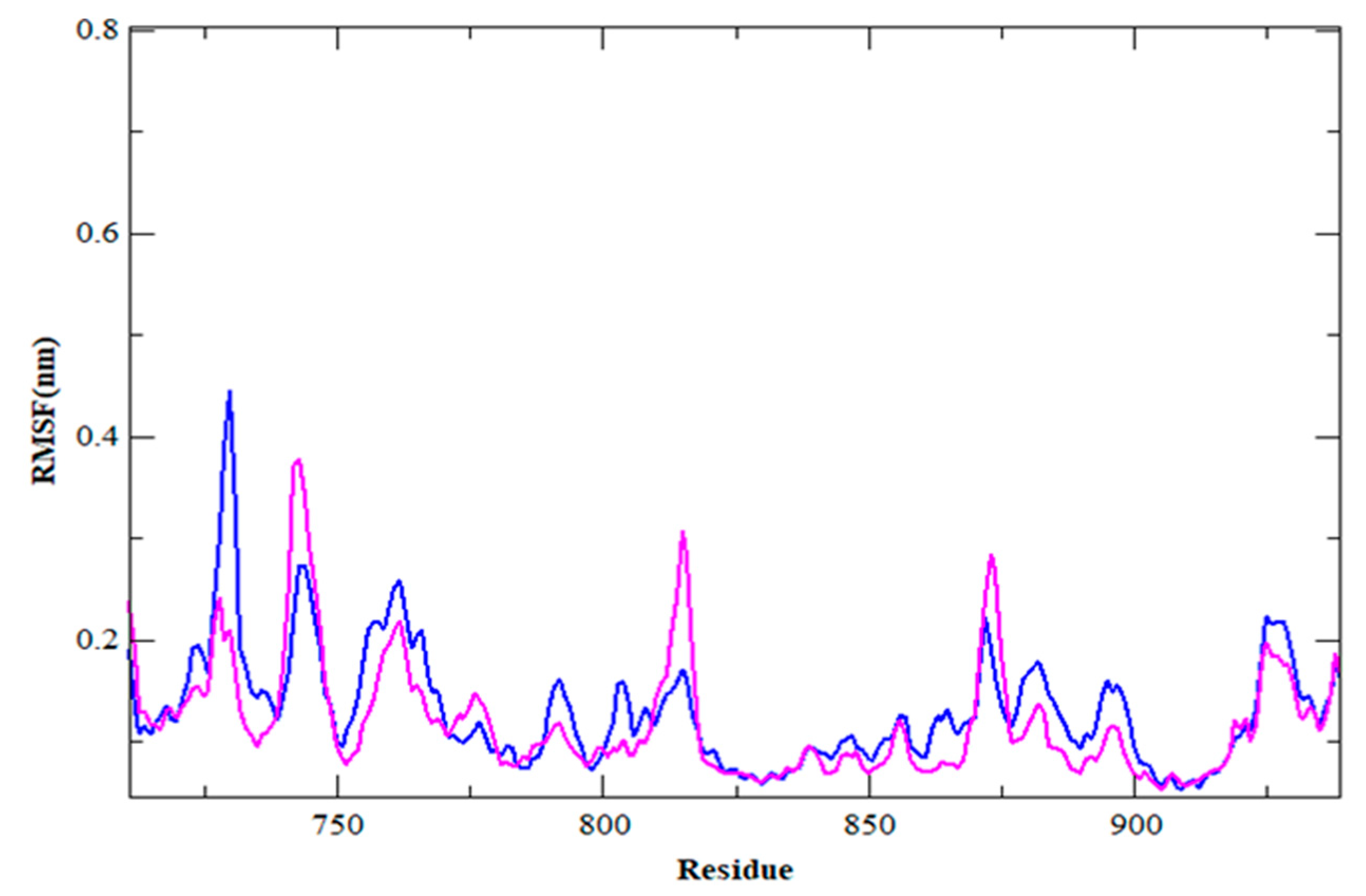
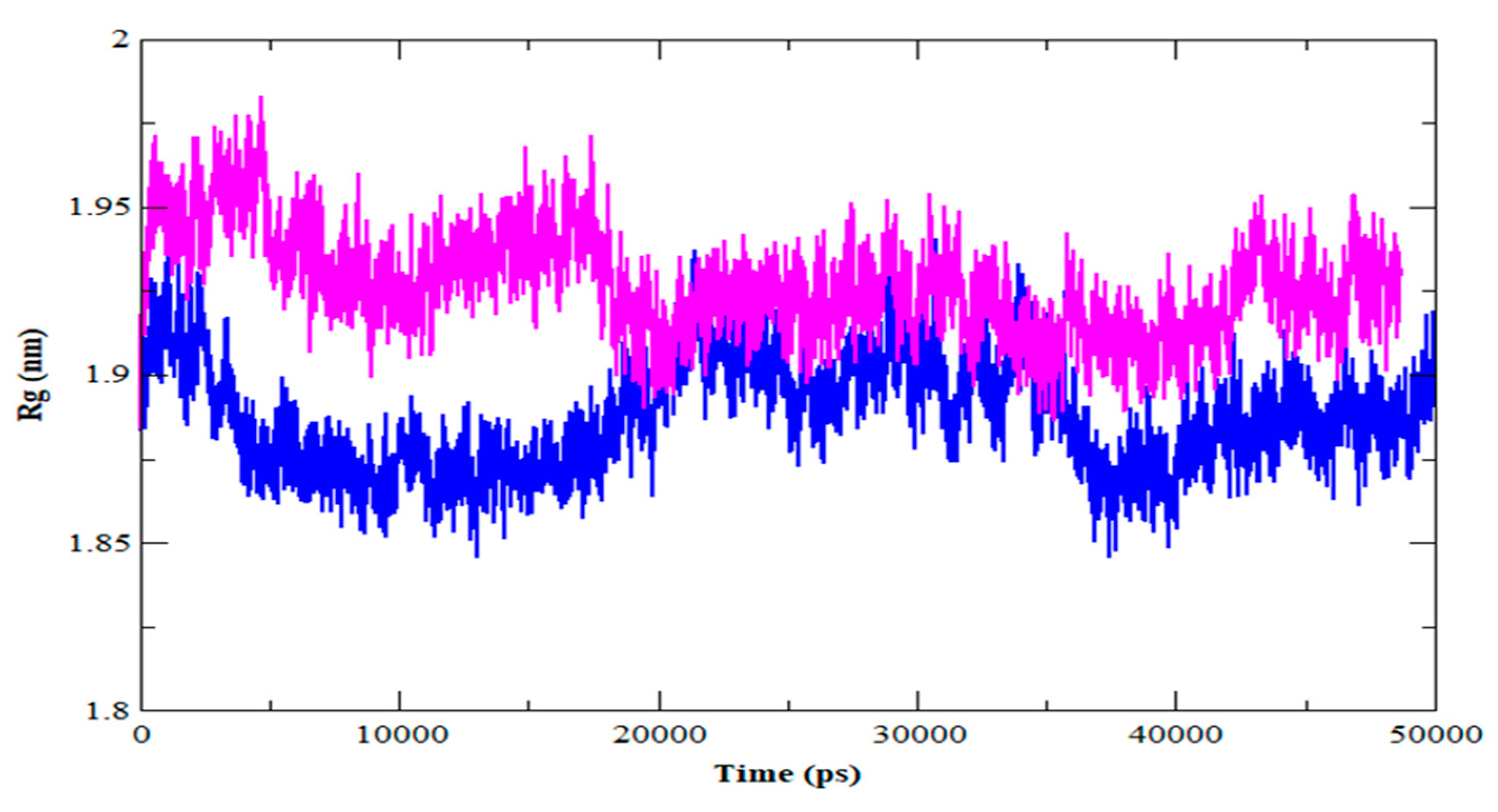
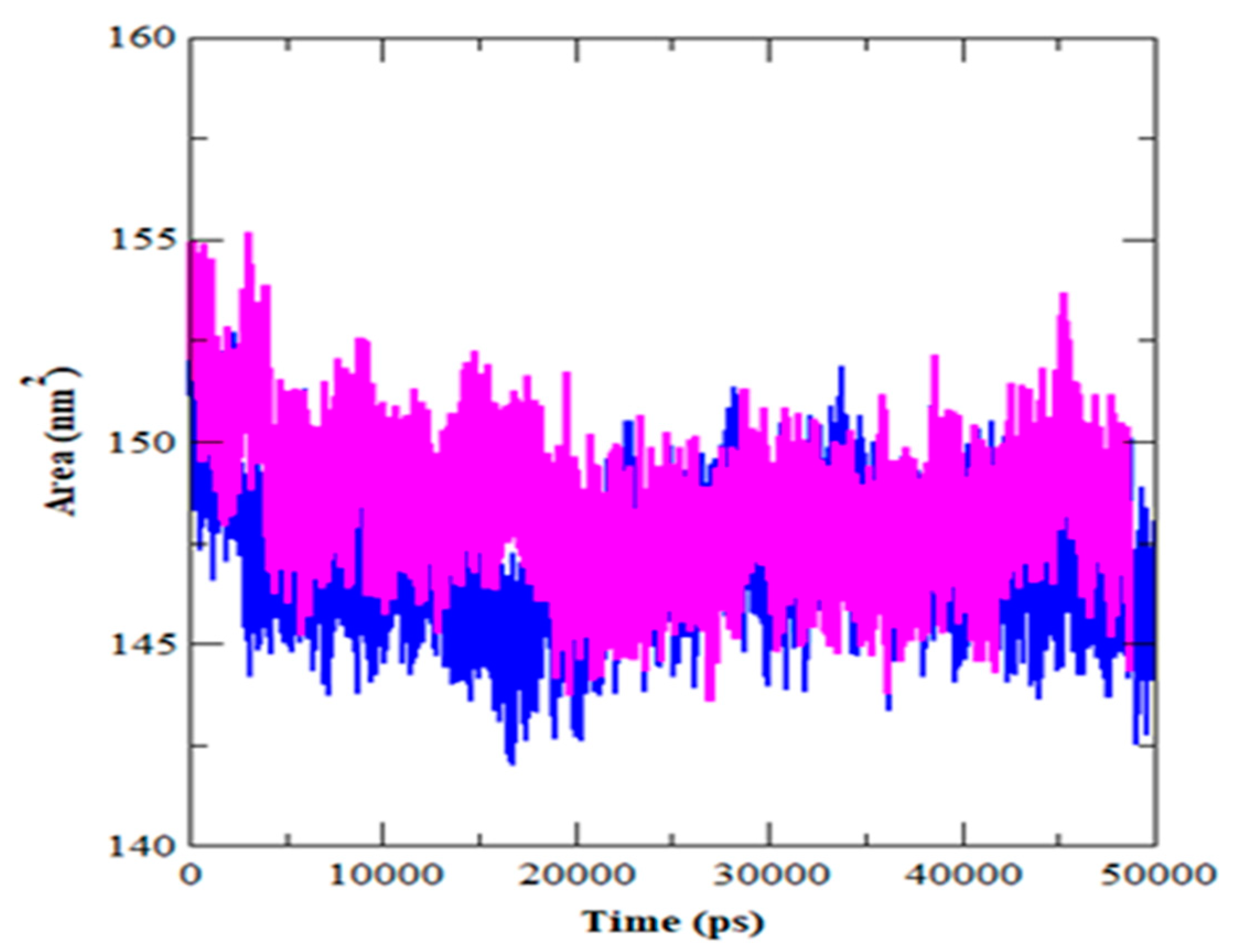
3.4. MDS Outcomes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Breast cancer |
| HER2 | Human epidermal growth factor receptor-2 |
| VS | Virtual screening |
| MDS | Dynamics simulation study |
| RMSD | Root-mean square deviation |
| RMSF | Root-mean square fluctuation |
| Rg | Radius of gyration |
| SASA | Solvent accessible surface area |
References
- Libson, S.; Lippman, M. A review of clinical aspects of breast cancer. Int. Rev. Psychiatry 2014, 26, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Kolak, A.; Kaminska, M.; Sygit, K.; Budny, A.; Surdyka, D.; Kukielka-Budny, B.; Burdan, F. Primary and secondary prevention of breast cancer. Ann. Agric. Environ. Med. 2017, 24, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- WHO. Physical Activity Fact Sheet; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Albagoush, S.A.; Limaiem, F. Her2; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Maughan, K.L.; Lutterbie, M.A.; Ham, P.S. Treatment of breast cancer. Am. Fam. Physician 2010, 81, 1339–1346. [Google Scholar]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Tarantino, P.; Curigliano, G.; Tolaney, S.M. Navigating the HER2-Low Paradigm in Breast Oncology: New Standards, Future Horizons. Cancer Discov. 2022, 12, 2026–2030. [Google Scholar] [CrossRef]
- Voigtlaender, M.; Schneider-Merck, T.; Trepel, M. Lapatinib. Recent Results Cancer Res. 2018, 211, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Afrose, A.; Rahman, M.R.; Chowdhury, R.; Nirzhor, S.S.R.; Khan, R.I.; Kabir, M.T. A Comprehensive Analysis into the Therapeutic Application of Natural Products as SIRT6 Modulators in Alzheimer’s Disease, Aging, Cancer, Inflammation, and Diabetes. Int. J. Mol. Sci. 2021, 22, 4180. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Sobarzo-Sanchez, E.; Santana, L.; Uriarte, E. Molecular Docking and Drug Discovery in beta-Adrenergic Receptors. Curr. Med. Chem. 2017, 24, 4340–4359. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Prabhavathi, H.; Dasegowda, K.R.; Renukananda, K.H.; Karunakar, P.; Lingaraju, K.; Raja Naika, H. Molecular docking and dynamic simulation to identify potential phytocompound inhibitors for EGFR and HER2 as anti-breast cancer agents. J. Biomol. Struct. Dyn. 2022, 40, 4713–4724. [Google Scholar] [CrossRef] [PubMed]
- Balgoname, A.A.; Alomair, S.M.; AlMubirek, A.K.; Khedr, M.A. Fragment-based Discovery of Potential Anticancer Lead: Computational and in vitro Studies. Curr. Comput. Aided Drug Des. 2021, 17, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yu, T.; Sun, R.; Wang, S.; Chen, X.Q.; Cheng, L.J.; Liu, R. Discovery of Novel Human Epidermal Growth Factor Receptor-2 Inhibitors by Structure-based Virtual Screening. Pharmacogn. Mag. 2016, 12, 139–144. [Google Scholar] [CrossRef]
- Bragina, O.D.; Deyev, S.M.; Chernov, V.I.; Tolmachev, V.M. The Evolution of Targeted Radionuclide Diagnosis of HER2-Positive Breast Cancer. Acta Nat. 2022, 14, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Kwofie, S.K.; Adobor, C.; Quansah, E.; Bentil, J.; Ampadu, M.; Miller, W.A., 3rd; Wilson, M.D. Molecular docking and dynamics simulations studies of OmpATb identifies four potential novel natural product-derived anti-Mycobacterium tuberculosis compounds. Comput. Biol. Med. 2020, 122, 103811. [Google Scholar] [CrossRef]
- Zhao, W.; Xiong, M.; Yuan, X.; Li, M.; Sun, H.; Xu, Y. In Silico Screening-Based Discovery of Novel Inhibitors of Human Cyclic GMP-AMP Synthase: A Cross-Validation Study of Molecular Docking and Experimental Testing. J. Chem. Inf. Model. 2020, 60, 3265–3276. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.S.; Faver, J.C.; Micevic, G.; Muthusamy, V.; Kudalkar, S.N.; Bertoletti, N.; Anderson, K.S.; Bosenberg, M.W.; Jorgensen, W.L. Structure-Guided Identification of DNMT3B Inhibitors. ACS Med. Chem. Lett. 2020, 11, 971–976. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB Viewer (Deep View). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M., Jr.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J.; Tidor, B. Systematic placement of structural water molecules for improved scoring of protein-ligand interactions. Protein Eng. Des. Sel. 2011, 24, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Humana Press: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Fahrrolfes, R.; Bietz, S.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Otto, T.; Volkamer, A.; Rarey, M. ProteinsPlus: A web portal for structure analysis of macromolecules. Nucleic Acids Res. 2017, 45, W337–W343. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Selick, H.E.; Beresford, A.P.; Tarbit, M.H. The emerging importance of predictive ADME simulation in drug discovery. Drug Discov. Today 2002, 7, 109–116. [Google Scholar] [CrossRef]
- Beresford, A.P.; Segall, M.; Tarbit, M.H. In silico prediction of ADME properties: Are we making progress? Curr. Opin. Drug Discov. Dev. 2004, 7, 36–42. [Google Scholar]
- Muegge, I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003, 23, 302–321. [Google Scholar] [CrossRef]
- Yang, S.C.; Chang, S.S.; Chen, C.Y. Identifying HER2 inhibitors from natural products database. PLoS ONE 2011, 6, e28793. [Google Scholar] [CrossRef]
- Spector, N.; Xia, W.; El-Hariry, I.; Yarden, Y.; Bacus, S. HER2 therapy. Small molecule HER-2 tyrosine kinase inhibitors. Breast Cancer Res. 2007, 9, 205. [Google Scholar] [CrossRef]
- Schroeder, R.L.; Stevens, C.L.; Sridhar, J. Small molecule tyrosine kinase inhibitors of ErbB2/HER2/Neu in the treatment of aggressive breast cancer. Molecules 2014, 19, 15196–15212. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Li, J.; Bao, J.; Wu, C. Discovery of a Potential HER2 Inhibitor from Natural Products for the Treatment of HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1055. [Google Scholar] [CrossRef]
- Palanisamy, C.P.; Ashafa, A.O.T. Screening of Potential Phytocompounds From Euclea crispa (Thunb.) Leaves Targeting Human Epidermal Growth Factor Receptor 2 (HER2) Signaling Pathway. J. Pharm. Bioallied. Sci. 2019, 11, 155–161. [Google Scholar] [CrossRef]
- Desai, S.B.; Moonim, M.T.; Gill, A.K.; Punia, R.S.; Naresh, K.N.; Chinoy, R.F. Hormone receptor status of breast cancer in India: A study of 798 tumours. Breast 2000, 9, 267–270. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, e2000028. [Google Scholar] [CrossRef]
- Rampogu, S.; Son, M.; Baek, A.; Park, C.; Rana, R.M.; Zeb, A.; Parameswaran, S.; Lee, K.W. Targeting natural compounds against HER2 kinase domain as potential anticancer drugs applying pharmacophore based molecular modelling approaches. Comput. Biol. Chem. 2018, 74, 327–338. [Google Scholar] [CrossRef]
- Rajagopal, K.; Sri, V.B.; Byran, G.; Gomathi, S. Pyrazole Substituted 9-Anilinoacridines as HER2 Inhibitors Targeting Breast Cancer—An in-silico approach. Curr. Drug Res. Rev. 2022, 14, 61–72. [Google Scholar] [CrossRef]
- Cheng, F.; Zhou, Y.; Li, J.; Li, W.; Liu, G.; Tang, Y. Prediction of chemical-protein interactions: Multitarget-QSAR versus computational chemogenomic methods. Mol. Biosyst. 2012, 8, 2373–2384. [Google Scholar] [CrossRef]
- Mitrasinovic, P.M. Structural elucidation of unique inhibitory activities of two thiazolo[4,5-d]pyrimidines against epidermal growth factor receptor (EGFR): Implications for successful drug design. Med. Chem. 2014, 10, 46–58. [Google Scholar] [CrossRef]
- Mitrasinovic, P.M. Progress in structure-based design of EGFR inhibitors. Curr. Drug Targets 2013, 14, 817–829. [Google Scholar] [CrossRef]
- Cortopassi, W.A.; Feital, R.J.C.; Medeiros, D.d.J.; Guizado, T.R.C.; França, T.C.C.; Pimentel, A.S. Docking and molecular dynamics studies of new potential inhibitors of the human epidermal receptor 2. Mol. Simul. 2012, 38, 1132–1142. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | List of Compounds | Binding Energy (kcal/mol) |
|---|---|---|
| 1. | ZINC43069427 | −11.0 |
| 2. | ZINC95918662 | −8.5 |
| 3. | ZINC1550477 (Lapatinib) | −7.65 |
| 4. | ZINC3976838 (Afatinib) | −7.80 |
| 5. | ZINC34587071 (Sapitinib) | −7.15 |
| 6. | Salvianolic acid C | −7.10 |
| 7. | ZINC000000006256 | −8.3 |
| 8. | ZINC000000000052 | −8.1 |
| 9. | ZINC000000000056 | −8.1 |
| 10. | ZINC000000000446 | −8.0 |
| 11. | ZINC000000001288 | −8.0 |
| 12. | ZINC000000001161 | −7.9 |
| 13. | ZINC000095918662 | −7.8 |
| 14. | ZINC000095918662 | −7.5 |
| 15. | ZINC000095918662 | −7.4 |
| 16. | ZINC000095918662 | −7.2 |
| 17. | ZINC000095918662 | −7.0 |
| 18. | ZINC000095918662 | −6.8 |
| 19. | ZINC000000000290 | −6.9 |
| 20. | ZINC000000006606 | −6.7 |
| 21. | ZINC000000001288 | −6.0 |
| 22. | ZINC000043069427 | −5.8 |
| 23. | ZINC000043069427 | −5.5 |
| Property | Parameters | ZINC43069427 | ZINC95918662 | ||||
|---|---|---|---|---|---|---|---|
| Absorption | Water solubility (log mol/L)) | −4.135 | −3.811 | ||||
| Caco2 permeability | 1.024 | 1.015 | |||||
| Intestinal absorption (human) (% absorbed) | 92.481 | 82.451 | |||||
| Skin permeability (log Kp) | −3.121 | −2.739 | |||||
| P-glycoprotein | substrate | (Yes/No) | Yes | Yes | |||
| I inhibitor | Yes | No | |||||
| II inhibitor | No | No | |||||
| Distribution | VDss (human) (log L/kg) | 0.064 | 0.036 | ||||
| Fraction unbound (human) (Fu) | 0.092 | 0.104 | |||||
| BBB permeability (log BB) | −0.123 | −0.991 | |||||
| CNS permeability (log PS) | −1.954 | −2.243 | |||||
| Metabolism | CYP | 2D6 | substrate | (Yes/No) | No | No | |
| 3A4 | No | No | |||||
| 1A2 | inhibitor | Yes | Yes | ||||
| 2C19 | Yes | Yes | |||||
| 2C9 | Yes | Yes | |||||
| 2D6 | No | No | |||||
| 3A4 | Yes | No | |||||
| Excretion | Total clearance (log ml/min/kg) | 0.143 | 0.202 | ||||
| Renal OCT2 substrate (Yes/No) | No | No | |||||
| Toxicity | AMES toxicity (Yes/No) | No | Yes | ||||
| Max. tolerated dose (human) (log mg/kg/day) | 0.004 | 0.318 | |||||
| hERG I inhibitor (Yes/No) | No | No | |||||
| hERG II inhibitor (Yes/No) | Yes | No | |||||
| Oral Rat Acute Toxicity (LD50) (mol/kg) | 2.301 | 2.215 | |||||
| Oral Rat Chronic Toxicity (LOAEL) (log mg/kg_bw/day) | 1.793 | 2.16 | |||||
| Hepatotoxicity | (Yes/No) | No | No | ||||
| Skin sensitisation | No | No | |||||
| T. Pyriformis toxicity (log ug/L) | 0.83 | 0.38 | |||||
| Minnow toxicity (log mM) | 0.513 | 0.575 | |||||
| Physicochemical | Formula | C20H18O4 | C21H22O5 | ||||
| Molecular weight (g/mol) | 322.35 | 354.40 | |||||
| No. heavy atoms | 24 | 26 | |||||
| No. arom. heavy atoms | 12 | 12 | |||||
| No. rotatable bonds | 3 | 3 | |||||
| No. H-bond | acceptors | 4 | 5 | ||||
| donors | 2 | 4 | |||||
| Molar Refractivity | 94.42 | 100.59 | |||||
| TPSA (Ų) | 66.76 | 97.99 | |||||
| Druglikeness | Lipinski | Yes | Yes | ||||
| Ghose | Yes | Yes | |||||
| Veber | Yes | Yes | |||||
| Egan | Yes | Yes | |||||
| Muegge | Yes | Yes | |||||
| Drug Like Properties | Cutoff Range | Nature of Selected Compounds | |
|---|---|---|---|
| ZINC43069427 | ZINC95918662 | ||
| Lipinski Rule of Five | 1. Molecular mass: <500 Dalton 2. High lipophilicity: LogP <5 3. Hydrogen bond donors: <5 4. Hydrogen bond acceptors: <10 5. Molar refractivity: 40 to130 | Yes | Yes |
| Ghose Rule | 1. clogP: −0.4 to 5.6 2. MW: 160 to 480 3. Molar refractivity: 40 to 130 4. Total number of atoms: 20 to 70 | Yes | Yes |
| Veber Rule | 1. Rotatable bonds: ≤10 2. Polar surface area: ≤140 Å2 | Yes | Yes |
| Egan Rule | 1. WlogP: ≤5.88 2. TPSA: ≤131.6 | Yes | Yes |
| Muegge Rule | 1. MW: 200 to 600 2. XlogP: −2 to 5 3. TPSA: ≤150 4. Number of rings: ≤7 5. Number of carbon: >4 6. Number of heteroatoms: >1 7. Number of rotatable bonds: ≤15 8. Hydrogen bond acceptors: ≤10 9. Hydrogen bond donors: ≤5 | Yes | Yes |
| Parameters | HER2-ZINC43069427 | HER2-ZINC95918662 |
|---|---|---|
| RMSD (nm) | 0.32 | 0.32 |
| RMSF (nm) | 0.12 | 0.12 |
| Rg (nm) | 1.88 | 1.92 |
| SASA | 146.99 | 148.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sohrab, S.S.; Kamal, M.A. Screening, Docking, and Molecular Dynamics Study of Natural Compounds as an Anti-HER2 for the Management of Breast Cancer. Life 2022, 12, 1729. https://doi.org/10.3390/life12111729
Sohrab SS, Kamal MA. Screening, Docking, and Molecular Dynamics Study of Natural Compounds as an Anti-HER2 for the Management of Breast Cancer. Life. 2022; 12(11):1729. https://doi.org/10.3390/life12111729
Chicago/Turabian StyleSohrab, Sayed Sartaj, and Mohammad Amjad Kamal. 2022. "Screening, Docking, and Molecular Dynamics Study of Natural Compounds as an Anti-HER2 for the Management of Breast Cancer" Life 12, no. 11: 1729. https://doi.org/10.3390/life12111729
APA StyleSohrab, S. S., & Kamal, M. A. (2022). Screening, Docking, and Molecular Dynamics Study of Natural Compounds as an Anti-HER2 for the Management of Breast Cancer. Life, 12(11), 1729. https://doi.org/10.3390/life12111729