
Heat Shock Protein 70 and 90 Family in Prostate Cancer


Abstract
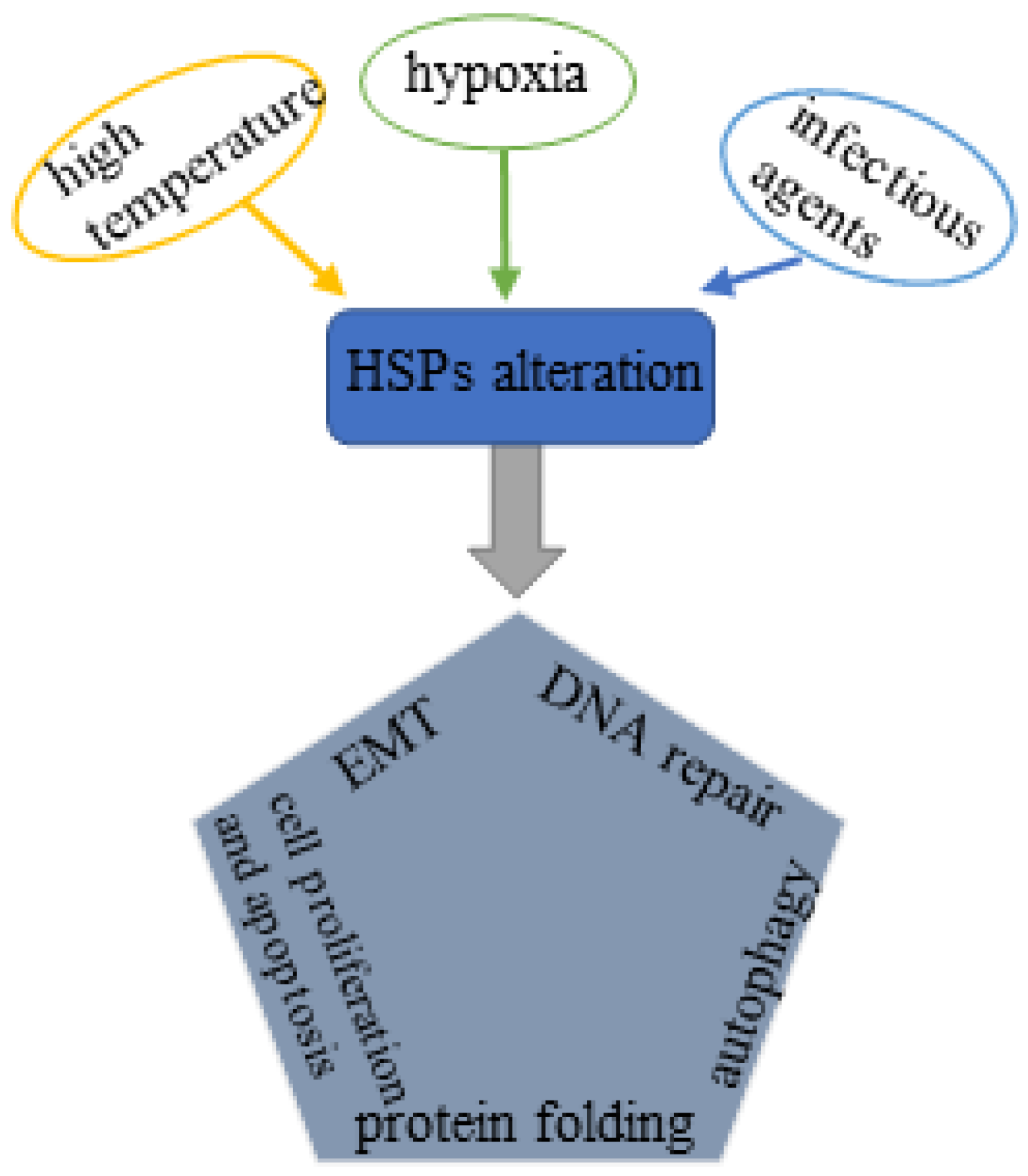
1. Introduction
2. Prostate Cancer
3. HSP Family
3.1. HSP70 Family
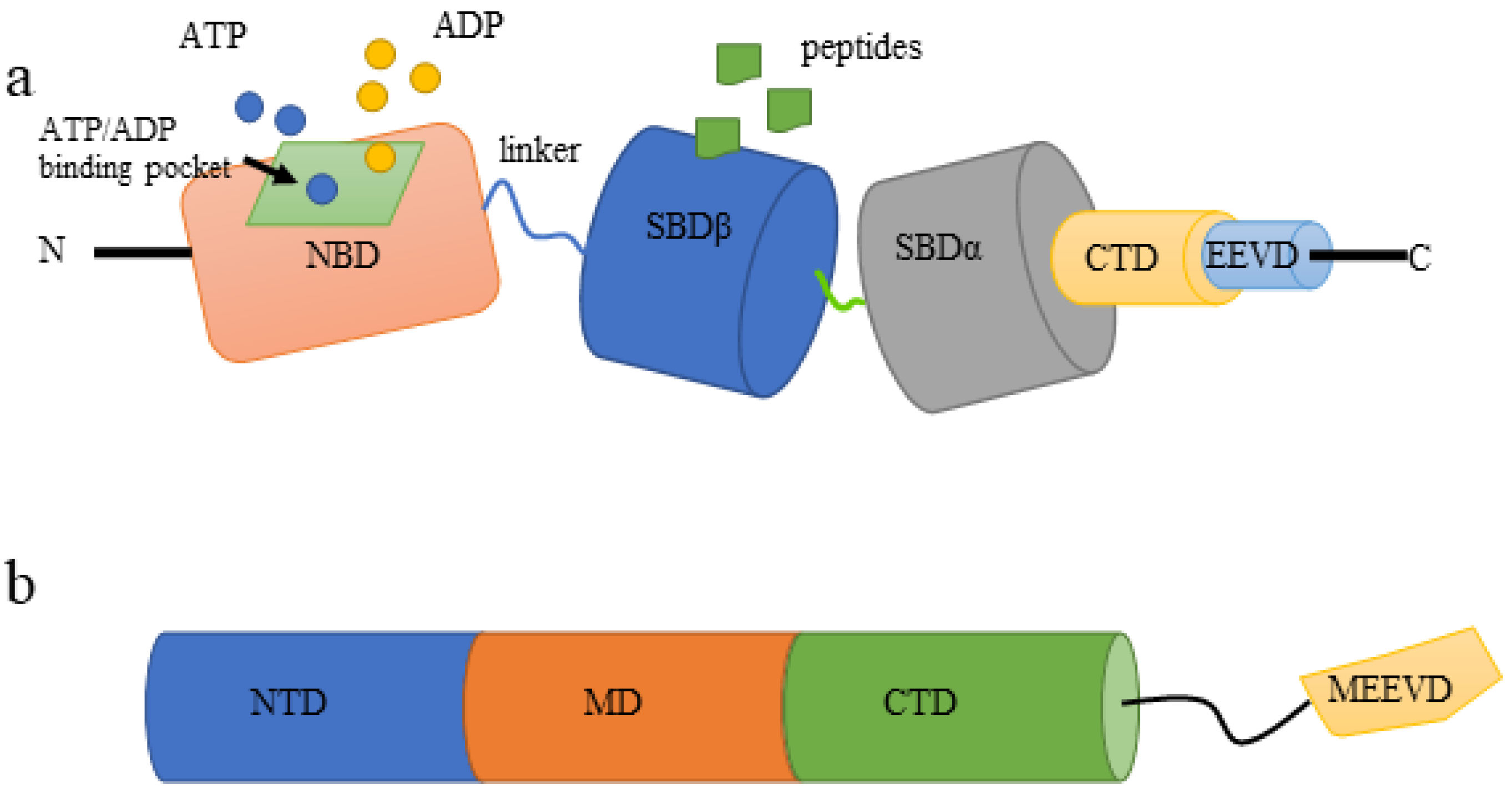
3.1.1. Structure of HSP70
3.1.2. HSP70s and AR
3.1.3. HSP70s and Cell Apoptosis and Proliferation
3.1.4. HSP70s and EMT, Migration, Invasion, and Metastasis
3.2. HSP90 Family
3.2.1. Structure of HSP90s
3.2.2. HSP90s and AR
3.2.3. HSP90s and Cell Apoptosis and Proliferation
3.2.4. HSP90s and EMT, Migration, Invasion, and Metastasis
4. HSP70 Family in Prostate Cancer Treatment
4.1. Small Molecular Inhibitors
4.2. Immunotherapeutic Approaches
4.3. A Novel Tool with High Specificity: Aptamers
5. HSP90 Family in Prostate Cancer Treatment
5.1. Natural HSP90 Inhibitors and Their Derivatives
5.2. Synthetic HSP90 Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Stein, R. Campbell Walsh Wein Urology, Chapter107: Epidemiology, Etiology, and Prevention of Prostate Cancer, PART XIV, VOLUME 3. Aktuelle Urol. 2021, 3, 25. [Google Scholar] [CrossRef]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, W.; Lubkowski, M.; Lubkowska, A. Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 897. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta 2012, 1823, 607–613. [Google Scholar] [CrossRef]
- Albakova, Z.; Mangasarova, Y.; Albakov, A.; Gorenkova, L. HSP70 and HSP90 in Cancer: Cytosolic, Endoplasmic Reticulum and Mitochondrial Chaperones of Tumorigenesis. Front. Oncol. 2022, 12, 829520. [Google Scholar] [CrossRef]
- Fu, X.; Liu, H.; Liu, J.; DiSanto, M.E.; Zhang, X. The Role of Heat Shock Protein 70 Subfamily in the Hyperplastic Prostate: From Molecular Mechanisms to Therapeutic Opportunities. Cells 2022, 11, 2052. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Milani, A.; Basirnejad, M.; Bolhassani, A. Heat-shock proteins in diagnosis and treatment: An overview of different biochemical and immunological functions. Immunotherapy 2019, 11, 215–239. [Google Scholar] [CrossRef]
- Calderwood, S.K. Heat shock proteins and cancer: Intracellular chaperones or extracellular signalling ligands? Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2018, 373, 20160524. [Google Scholar] [CrossRef] [PubMed]
- Dubrez, L.; Causse, S.; Borges Bonan, N.; Dumétier, B.; Garrido, C. Heat-shock proteins: Chaperoning DNA repair. Oncogene 2020, 39, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed]
- Thompson, I.M.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Miller, G.J.; Ford, L.G.; Lieber, M.M.; Cespedes, R.D.; Atkins, J.N.; Lippman, S.M.; et al. The influence of finasteride on the development of prostate cancer. New Engl. J. Med. 2003, 349, 215–224. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Nauseef, J.T.; Henry, M.D. Epithelial-to-mesenchymal transition in prostate cancer: Paradigm or puzzle? Nat. Rev. Urol. 2011, 8, 428–439. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Cho, K.H.; Choi, M.J.; Jeong, K.J.; Kim, J.J.; Hwang, M.H.; Shin, S.C.; Park, C.G.; Lee, H.Y. A ROS/STAT3/HIF-1α signaling cascade mediates EGF-induced TWIST1 expression and prostate cancer cell invasion. Prostate 2014, 74, 528–536. [Google Scholar] [CrossRef]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Gennigens, C.; Menetrier-Caux, C.; Droz, J.P. Insulin-Like Growth Factor (IGF) family and prostate cancer. Crit. Rev. Oncol. Hematol. 2006, 58, 124–145. [Google Scholar] [CrossRef]
- Woolf, C.M. An investigation of the familial aspects of carcinoma of the prostate. Cancer 1960, 13, 739–744. [Google Scholar] [CrossRef]
- Eeles, R.A.; Dearnaley, D.P.; Ardern-Jones, A.; Shearer, R.J.; Easton, D.F.; Ford, D.; Edwards, S.; Dowe, A. Familial prostate cancer: The evidence and the Cancer Research Campaign/British Prostate Group (CRC/BPG) UK Familial Prostate Cancer Study. Br. J. Urol. 1997, 79 (Suppl. S1), 8–14. [Google Scholar] [CrossRef] [PubMed]
- Zeegers, M.P.; Jellema, A.; Ostrer, H. Empiric risk of prostate carcinoma for relatives of patients with prostate carcinoma: A meta-analysis. Cancer 2003, 97, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wu, Z.; Wang, D.; Pascal, L.E.; Nelson, J.B.; Wipf, P.; Wang, Z. Hsp70 Binds to the Androgen Receptor N-terminal Domain and Modulates the Receptor Function in Prostate Cancer Cells. Mol. Cancer Ther. 2019, 18, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Centenera, M.M.; Carter, S.L.; Gillis, J.L.; Marrocco-Tallarigo, D.L.; Grose, R.H.; Tilley, W.D.; Butler, L.M. Co-targeting AR and HSP90 suppresses prostate cancer cell growth and prevents resistance mechanisms. Endocr. Relat. Cancer 2015, 22, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamornlert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Johng, D.; Torga, G.; Ewing, C.M.; Jin, K.; Norris, J.D.; McDonnell, D.P.; Isaacs, W.B. HOXB13 interaction with MEIS1 modifies proliferation and gene expression in prostate cancer. Prostate 2019, 79, 414–424. [Google Scholar] [CrossRef]
- Castro, E.; Eeles, R. The role of BRCA1 and BRCA2 in prostate cancer. Asian J. Androl. 2012, 14, 409–414. [Google Scholar] [CrossRef]
- Eeles, R.; Goh, C.; Castro, E.; Bancroft, E.; Guy, M.; Al Olama, A.A.; Easton, D.; Kote-Jarai, Z. The genetic epidemiology of prostate cancer and its clinical implications. Nat. Rev. Urol. 2014, 11, 18–31. [Google Scholar] [CrossRef]
- Kumar, S.; Stokes, J., 3rd; Singh, U.P.; Scissum Gunn, K.; Acharya, A.; Manne, U.; Mishra, M. Targeting Hsp70: A possible therapy for cancer. Cancer Lett. 2016, 374, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Saini, J.; Sharma, P.K. Clinical, Prognostic and Therapeutic Significance of Heat Shock Proteins in Cancer. Curr. Drug Targets 2018, 19, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Manola, J.B.; Oh, W.K.; Parslow, D.L.; George, D.J.; Austin, C.L.; Kantoff, P.W. Plasma levels of heat shock protein 70 in patients with prostate cancer: A potential biomarker for prostate cancer. Clin. Prostate Cancer 2004, 3, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Shkedi, A.; Taylor, I.R.; Echtenkamp, F.; Ramkumar, P.; Alshalalfa, M.; Rivera-Márquez, G.M.; Moses, M.A.; Shao, H.; Karnes, R.J.; Neckers, L.; et al. Selective vulnerabilities in the proteostasis network of castration-resistant prostate cancer. Cell Chem. Biol. 2022, 29, 490–501.e494. [Google Scholar] [CrossRef] [PubMed]
- Pootrakul, L.; Datar, R.H.; Shi, S.R.; Cai, J.; Hawes, D.; Groshen, S.G.; Lee, A.S.; Cote, R.J. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 5987–5993. [Google Scholar] [CrossRef]
- Hirth, C.G.; Vasconcelos, G.R.; da Cunha, M.; Leite, C.H.B.; Dornelas, C.A. Immunoexpression of HSPA9 and CUL2 in prostatic tissue and adenocarcinoma. Ann. Diagn. Pathol. 2022, 56, 151843. [Google Scholar] [CrossRef]
- Cornford, P.A.; Dodson, A.R.; Parsons, K.F.; Desmond, A.D.; Woolfenden, A.; Fordham, M.; Neoptolemos, J.P.; Ke, Y.; Foster, C.S. Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Res. 2000, 60, 7099–7105. [Google Scholar]
- Knapp, R.T.; Wong, M.J.; Kollmannsberger, L.K.; Gassen, N.C.; Kretzschmar, A.; Zschocke, J.; Hafner, K.; Young, J.C.; Rein, T. Hsp70 cochaperones HspBP1 and BAG-1M differentially regulate steroid hormone receptor function. PLoS ONE 2014, 9, e85415. [Google Scholar] [CrossRef]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Banduseela, V.C.; Chiesa, G.; Martínez-Cristóbal, P.; Rauch, J.N.; Nath, S.R.; Schwarz, D.M.C.; Shao, H.; Marin-Argany, M.; Di Sanza, C.; et al. Hsp70 and Hsp40 inhibit an inter-domain interaction necessary for transcriptional activity in the androgen receptor. Nat. Commun. 2019, 10, 3562. [Google Scholar] [CrossRef]
- Moses, M.A.; Kim, Y.S.; Rivera-Marquez, G.M.; Oshima, N.; Watson, M.J.; Beebe, K.E.; Wells, C.; Lee, S.; Zuehlke, A.D.; Shao, H.; et al. Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Yang, J.C.; Liu, L.; Armstrong, C.M.; Lombard, A.P.; Zhao, R.; Noel, O.D.V.; Tepper, C.G.; Chen, H.W.; et al. Proteostasis by STUB1/HSP70 complex controls sensitivity to androgen receptor targeted therapy in advanced prostate cancer. Nat. Commun. 2018, 9, 4700. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Yan, G.; You, B.; Sun, J. Down-regulation of mammalian sterile 20-like kinase 1 by heat shock protein 70 mediates cisplatin resistance in prostate cancer cells. Cancer Res. 2008, 68, 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, N.B.; Watson, R.W.; Coffey, R.N.; Brady, H.P.; Fitzpatrick, J.M. Heat-shock proteins inhibit induction of prostate cancer cell apoptosis. Prostate 2000, 45, 58–65. [Google Scholar] [CrossRef]
- Jones, E.L.; Zhao, M.J.; Stevenson, M.A.; Calderwood, S.K. The 70 kilodalton heat shock protein is an inhibitor of apoptosis in prostate cancer. Int. J. Hyperth. Off. J. Eur. Soc. Hyperthermic Oncol. N. Am. Hyperth. Group 2004, 20, 835–849. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D degradation by E3 ligases in cancer progression and treatment. Semin. Cancer Biol. 2020, 67, 159–170. [Google Scholar] [CrossRef]
- Ling, X.; Wan, J.; Peng, B.; Chen, J. Hsp70 Promotes SUMO of HIF-1α and Promotes Lung Cancer Invasion and Metastasis. J. Oncol. 2021, 2021, 7873085. [Google Scholar] [CrossRef]
- Nikotina, A.D.; Vladimirova, S.A.; Komarova, E.Y.; Alexeev, D.; Efremov, S.; Leonova, E.; Pavlov, R.; Kartsev, V.G.; Polonik, S.G.; Margulis, B.A.; et al. Prevention of High Glucose-Mediated EMT by Inhibition of Hsp70 Chaperone. Int. J. Mol. Sci. 2021, 22, 6902. [Google Scholar] [CrossRef]
- Huang, P.; Guo, Y.; Zhao, Z.; Ning, W.; Wang, H.; Gu, C.; Zhang, M.; Qu, Y.; Zhang, H.; Song, Y. UBE2T promotes glioblastoma invasion and migration via stabilizing GRP78 and regulating EMT. Aging 2020, 12, 10275–10289. [Google Scholar] [CrossRef]
- Zhang, R.; Meng, Z.; Wu, X.; Zhang, M.; Zhang, S.; Jin, T. Mortalin promotes breast cancer malignancy. Exp. Mol. Pathol. 2021, 118, 104593. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Ngoka, L.; Mei, Y.; Lesoon, L.; Cowell, J.K. HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J. Biol. Chem. 2012, 287, 10051–10059. [Google Scholar] [CrossRef] [PubMed]
- Cultrara, C.N.; Kozuch, S.D.; Ramasundaram, P.; Heller, C.J.; Shah, S.; Beck, A.E.; Sabatino, D.; Zilberberg, J. GRP78 modulates cell adhesion markers in prostate Cancer and multiple myeloma cell lines. BMC Cancer 2018, 18, 1263. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed]
- Leav, I.; Plescia, J.; Goel, H.L.; Li, J.; Jiang, Z.; Cohen, R.J.; Languino, L.R.; Altieri, D.C. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am. J. Pathol. 2010, 176, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; Rizk, S.; Naim, H.Y. The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer. Cancers 2019, 11, 1194. [Google Scholar] [CrossRef]
- Wayne, N.; Bolon, D.N. Dimerization of Hsp90 is required for in vivo function. Design and analysis of monomers and dimers. J. Biol. Chem. 2007, 282, 35386–35395. [Google Scholar] [CrossRef]
- Buchner, J. Hsp90 & Co.—A holding for folding. Trends Biochem. Sci. 1999, 24, 136–141. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Farah, E.; Sarkar, S.; Ahmad, N.; Gupta, S.; Larner, J.; Liu, X. Cotargeting HSP90 and Its Client Proteins for Treatment of Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2107–2118. [Google Scholar] [CrossRef]
- Vanaja, D.K.; Mitchell, S.H.; Toft, D.O.; Young, C.Y. Effect of geldanamycin on androgen receptor function and stability. Cell Stress Chaperones 2002, 7, 55–64. [Google Scholar] [CrossRef]
- Fang, Y.; Fliss, A.E.; Robins, D.M.; Caplan, A.J. Hsp90 regulates androgen receptor hormone binding affinity in vivo. J. Biol. Chem. 1996, 271, 28697–28702. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, R.; Zhao, N.; Zhang, Q.; Yan, Z.; Chang, Z.; Wei, Y.; Wu, C.; Xu, J.; Xu, Y. A comparative analysis reveals the dosage sensitivity and regulatory patterns of lncRNA in prostate cancer. Mol. Biosyst. 2016, 12, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Liu, C.; Lin, P.; Xie, H.; Wang, W.; Zhao, J.; Jiang, S.; Shi, J.; Yu, X. LncRNA AC245100.4 binds HSP90 to promote the proliferation of prostate cancer. Epigenomics 2020, 12, 1257–1271. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Zoubeidi, A.; Gleave, M.E.; Chi, K.N. Targeting heat shock proteins in metastatic castration-resistant prostate cancer. Nat. Rev. Urol. 2015, 12, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, S.; Garlick, D.S.; Bryant, K.G.; Tavecchio, M.; Mills, G.B.; Lu, Y.; Kossenkov, A.V.; Showe, L.C.; Languino, L.R.; Altieri, D.C. Transgenic Expression of the Mitochondrial Chaperone TNFR-associated Protein 1 (TRAP1) Accelerates Prostate Cancer Development. J. Biol. Chem. 2016, 291, 25247–25254. [Google Scholar] [CrossRef]
- Ono, K.; Sogawa, C.; Kawai, H.; Tran, M.T.; Taha, E.A.; Lu, Y.; Oo, M.W.; Okusha, Y.; Okamura, H.; Ibaragi, S.; et al. Triple knockdown of CDC37, HSP90-alpha and HSP90-beta diminishes extracellular vesicles-driven malignancy events and macrophage M2 polarization in oral cancer. J. Extracell. Vesicles 2020, 9, 1769373. [Google Scholar] [CrossRef]
- Zhang, A.; Qi, X.; Du, F.; Zhang, G.; Li, D.; Li, J. PNSA, a Novel C-Terminal Inhibitor of HSP90, Reverses Epithelial-Mesenchymal Transition and Suppresses Metastasis of Breast Cancer Cells In Vitro. Mar. Drugs 2021, 19, 117. [Google Scholar] [CrossRef]
- Nolan, K.D.; Franco, O.E.; Hance, M.W.; Hayward, S.W.; Isaacs, J.S. Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J. Biol. Chem. 2015, 290, 8271–8282. [Google Scholar] [CrossRef]
- Hance, M.W.; Dole, K.; Gopal, U.; Bohonowych, J.E.; Jezierska-Drutel, A.; Neumann, C.A.; Liu, H.; Garraway, I.P.; Isaacs, J.S. Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J. Biol. Chem. 2012, 287, 37732–37744. [Google Scholar] [CrossRef]
- Gyrd-Hansen, M.; Nylandsted, J.; Jaattela, M. Heat shock protein 70 promotes cancer cell viability by safeguarding lysosomal integrity. Cell Cycle 2004, 3, 1484–1485. [Google Scholar] [CrossRef]
- Leu, J.I.J.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A Small Molecule Inhibitor of Inducible Heat Shock Protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.J.; Prentice, A.G.; Hoffbrand, A.V.; Yogashangary, B.C.; Hart, S.M.; Lowdell, M.W.; Samuel, E.R.; North, J.M.; Nacheva, E.P.; Chanalaris, A.; et al. 2-phenylacetylenesulfonamide (PAS) induces p53-independent apoptotic killing of B-chronic lymphocytic leukemia (CLL) cells. Blood 2009, 114, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Kühnl, A.; Reins, J.; Fischer, S.; Ortiz-Tanchez, J.; Schlee, C.; Mochmann, L.H.; Heesch, S.; Benlasfer, O.; Hofmann, W.K.; et al. Antileukemic activity of the HSP70 inhibitor pifithrin-μ in acute leukemia. Blood Cancer J. 2011, 1, e28. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Shiota, M.; Tanaka, M.; Otsuka, A.; Matsumoto, M.; Kato, M.; Tamada, S.; Iwao, H.; Miura, K.; Nakatani, T.; et al. Heat shock protein 70 inhibitors suppress androgen receptor expression in LNCaP95 prostate cancer cells. Cancer Sci. 2017, 108, 1820–1827. [Google Scholar] [CrossRef]
- Kumar, S.; Gurshaney, S.; Adagunodo, Y.; Gage, E.; Qadri, S.; Sharma, M.; Malik, S.; Manne, U.; Singh, U.P.; Singh, R.; et al. Hsp70 and gama-Semino protein as possible prognostic marker of prostate cancer. Front. Biosci. 2018, 23, 1987–2000. [Google Scholar] [CrossRef]
- Rodina, A.; Vilenchik, M.; Moulick, K.; Aguirre, J.; Kim, J.; Chiang, A.; Litz, J.; Clement, C.C.; Kang, Y.; She, Y.; et al. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat. Chem. Biol. 2007, 3, 498–507. [Google Scholar] [CrossRef]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef]
- Nadeau, K.; Nadler, S.G.; Saulnier, M.; Tepper, M.A.; Walsh, C.T. Quantitation of the interaction of the immunosuppressant deoxyspergualin and analogs with Hsc70 and Hsp90. Biochemistry 1994, 33, 2561–2567. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Chatterjee, M.; Andrulis, M.; Stühmer, T.; Müller, E.; Hofmann, C.; Steinbrunn, T.; Heimberger, T.; Schraud, H.; Kressmann, S.; Einsele, H.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef]
- Britten, C.D.; Rowinsky, E.K.; Baker, S.D.; Weiss, G.R.; Smith, L.; Stephenson, J.; Rothenberg, M.; Smetzer, L.; Cramer, J.; Collins, W.; et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 42–49. [Google Scholar]
- Park, S.H.; Baek, K.H.; Shin, I.; Shin, I. Subcellular Hsp70 Inhibitors Promote Cancer Cell Death via Different Mechanisms. Cell Chem. Biol. 2018, 25, 1242–1254.e1248. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E.; et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. USA 2011, 108, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Trimble, C.L.; Peng, S.; Kos, F.; Gravitt, P.; Viscidi, R.; Sugar, E.; Pardoll, D.; Wu, T.C. A phase I trial of a human papillomavirus DNA vaccine for HPV16+ cervical intraepithelial neoplasia 2/3. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 361–367. [Google Scholar] [CrossRef]
- Rérole, A.L.; Gobbo, J.; De Thonel, A.; Schmitt, E.; Pais de Barros, J.P.; Hammann, A.; Lanneau, D.; Fourmaux, E.; Demidov, O.N.; Micheau, O.; et al. Peptides and aptamers targeting HSP70: A novel approach for anticancer chemotherapy. Cancer Res. 2011, 71, 484–495. [Google Scholar] [CrossRef]
- Jego, G.; Hazoume, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef]
- Patel, H.J.; Modi, S.; Chiosis, G.; Taldone, T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin. Drug Discov. 2011, 6, 559–587. [Google Scholar] [CrossRef]
- Allan, R.K.; Mok, D.; Ward, B.K.; Ratajczak, T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: Evidence that coumarin antibiotics disrupt Hsp90 dimerization. J. Biol. Chem. 2006, 281, 7161–7171. [Google Scholar] [CrossRef]
- Heddle, J.G.; Barnard, F.M.; Wentzell, L.M.; Maxwell, A. The interaction of drugs with DNA gyrase: A model for the molecular basis of quinolone action. Nucleosides Nucleotides Nucleic Acids 2000, 19, 1249–1264. [Google Scholar] [CrossRef]
- Wu, L.X.; Xu, J.H.; Zhang, K.Z.; Lin, Q.; Huang, X.W.; Wen, C.X.; Chen, Y.Z. Disruption of the Bcr-Abl/Hsp90 protein complex: A possible mechanism to inhibit Bcr-Abl-positive human leukemic blasts by novobiocin. Leukemia 2008, 22, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Banerji, U.; Judson, I.; Workman, P. Hsp90 inhibitors in the clinic. Handb. Exp. Pharmacol. 2006, 172, 331–358. [Google Scholar] [CrossRef]
- Kummar, S.; Gutierrez, M.E.; Gardner, E.R.; Chen, X.; Figg, W.D.; Zajac-Kaye, M.; Chen, M.; Steinberg, S.M.; Muir, C.A.; Yancey, M.A.; et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.K.; Galsky, M.D.; Stadler, W.M.; Srinivas, S.; Chu, F.; Bubley, G.; Goddard, J.; Dunbar, J.; Ross, R.W. Multicenter phase II trial of the heat shock protein 90 inhibitor, retaspimycin hydrochloride (IPI-504), in patients with castration-resistant prostate cancer. Urology 2011, 78, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.; Curry, J.; Smyth, T.; Fazal, L.; Feltell, R.; Harada, I.; Coyle, J.; Williams, B.; Reule, M.; Angove, H.; et al. The heat shock protein 90 inhibitor, AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci. 2012, 103, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Kumar, V.; Tailor, D.; Garcia-Marques, F.J.; Hsu, E.C.; Liu, S.; Bermudez, A.; Kanchustambham, V.; Shankar, V.; Inde, Z.; et al. SU086, an inhibitor of HSP90, impairs glycolysis and represents a treatment strategy for advanced prostate cancer. Cell Rep. Med. 2022, 3, 100502. [Google Scholar] [CrossRef]
- Plymate, S.R.; Sprenger, C.; Haffner, M.C. Starving lethal prostate cancer by targeting heat shock proteins and glycolytic enzymes. Cell Rep. Med. 2022, 3, 100493. [Google Scholar] [CrossRef]
- Böll, B.; Eltaib, F.; Reiners, K.S.; von Tresckow, B.; Tawadros, S.; Simhadri, V.R.; Burrows, F.J.; Lundgren, K.; Hansen, H.P.; Engert, A.; et al. Heat shock protein 90 inhibitor BIIB021 (CNF2024) depletes NF-kappaB and sensitizes Hodgkin’s lymphoma cells for natural killer cell-mediated cytotoxicity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5108–5116. [Google Scholar] [CrossRef]
- Fadden, P.; Huang, K.H.; Veal, J.M.; Steed, P.M.; Barabasz, A.F.; Foley, B.; Hu, M.; Partridge, J.M.; Rice, J.; Scott, A.; et al. Application of chemoproteomics to drug discovery: Identification of a clinical candidate targeting hsp90. Chem. Biol. 2010, 17, 686–694. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Ye, Q.; Scott, A.; Silinski, M.; Huang, K.; Fadden, P.; Partdrige, J.; Hall, S.; Steed, P.; et al. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 240–248. [Google Scholar] [CrossRef]
- Okawa, Y.; Hideshima, T.; Steed, P.; Vallet, S.; Hall, S.; Huang, K.; Rice, J.; Barabasz, A.; Foley, B.; Ikeda, H.; et al. SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell growth, angiogenesis, and osteoclastogenesis in multiple myeloma and other hematologic tumors by abrogating signaling via Akt and ERK. Blood 2009, 113, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.; Kelly, R.J.; Trepel, J.B.; Kim, Y.S.; Alarcon, S.V.; Kummar, S.; Gutierrez, M.; Crandon, S.; Zein, W.M.; Jain, L.; et al. A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 6831–6839. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.K.; Heilbrun, L.K.; Sheng, S.; Stein, M.; Liu, G.; Antonarakis, E.S.; Vaishampayan, U.; Dzinic, S.H.; Li, X.; Freeman, S.; et al. A phase II trial of ganetespib, a heat shock protein 90 Hsp90) inhibitor, in patients with docetaxel-pretreated metastatic castrate-resistant prostate cancer (CRPC)-a prostate cancer clinical trials consortium (PCCTC) study. Investig. New Drugs 2016, 34, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Eskew, J.D.; Sadikot, T.; Morales, P.; Duren, A.; Dunwiddie, I.; Swink, M.; Zhang, X.; Hembruff, S.; Donnelly, A.; Rajewski, R.A.; et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 468. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Agents | Target Site | Clinical Evaluation | Reference |
|---|---|---|---|
| 1. Small molecular inhibitors | |||
| (1) PES | SBD | YES | [70,71,72,73,74,75] |
| (2) 15-DSG | NBD | NO | [78] |
| (3) MAL3-101 | NBD | NO | [76,79] |
| (4) VER-155008 | NBD | NO | [77,80] |
| (5) MKT-077 | NBD | Halted | [81] |
| (6) AZ | NBD | NO | [82,83] |
| (7) Az-TPP-O3 | NBD | NO | [82] |
| 2. Immunotherapeutic agents | |||
| (1) cmHsp70.1 | TDK | Phase II (on going) | [84] |
| (2) pNGVL4a-Sig/E7(detox) /HSP70 DNA | - | Phase I | [85] |
| 3. Aptamers | |||
| A17 | NBD | NO | [86,87] |
| Agents | Targeting Site | Clinical Evaluation | Reference |
|---|---|---|---|
| 1. Natural inhibitors and derivatives | |||
| (1) GM | NTD | Halted | [88] |
| (2) Novobiocin | CTD | NO | [89,90,91] |
| (3) 17-AAG | NTD | Phase II | [92] |
| (4) 17-DMAG | NTD | NO | [93] |
| (5) IPI-504 | NTD | Halted | [94] |
| (6) AT13387 | NTD | Phase I and II | [64,79,95] |
| (7) SU086 | NO | [96,97] | |
| 2. Synthetic inhibitors | |||
| (1) CNF-2024 | NTD | NO | [98] |
| (2) SNX-5422 | NTD | Phase I | [99,100,101,102] |
| (3) STA-9090 | NTD | Phase II | [103] |
| (4) KU174 | CTD | NO | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, X.; Liu, J.; Yan, X.; DiSanto, M.E.; Zhang, X. Heat Shock Protein 70 and 90 Family in Prostate Cancer. Life 2022, 12, 1489. https://doi.org/10.3390/life12101489
Fu X, Liu J, Yan X, DiSanto ME, Zhang X. Heat Shock Protein 70 and 90 Family in Prostate Cancer. Life. 2022; 12(10):1489. https://doi.org/10.3390/life12101489
Chicago/Turabian StyleFu, Xun, Jiang Liu, Xin Yan, Michael E. DiSanto, and Xinhua Zhang. 2022. "Heat Shock Protein 70 and 90 Family in Prostate Cancer" Life 12, no. 10: 1489. https://doi.org/10.3390/life12101489
APA StyleFu, X., Liu, J., Yan, X., DiSanto, M. E., & Zhang, X. (2022). Heat Shock Protein 70 and 90 Family in Prostate Cancer. Life, 12(10), 1489. https://doi.org/10.3390/life12101489