
Direct RNA Nanopore Sequencing of SARS-CoV-2 Extracted from Critical Material from Swabs

 , , , ,
, , , ,  ,
,  , ,
, ,  ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Input RNA Collection and Quality Control Steps
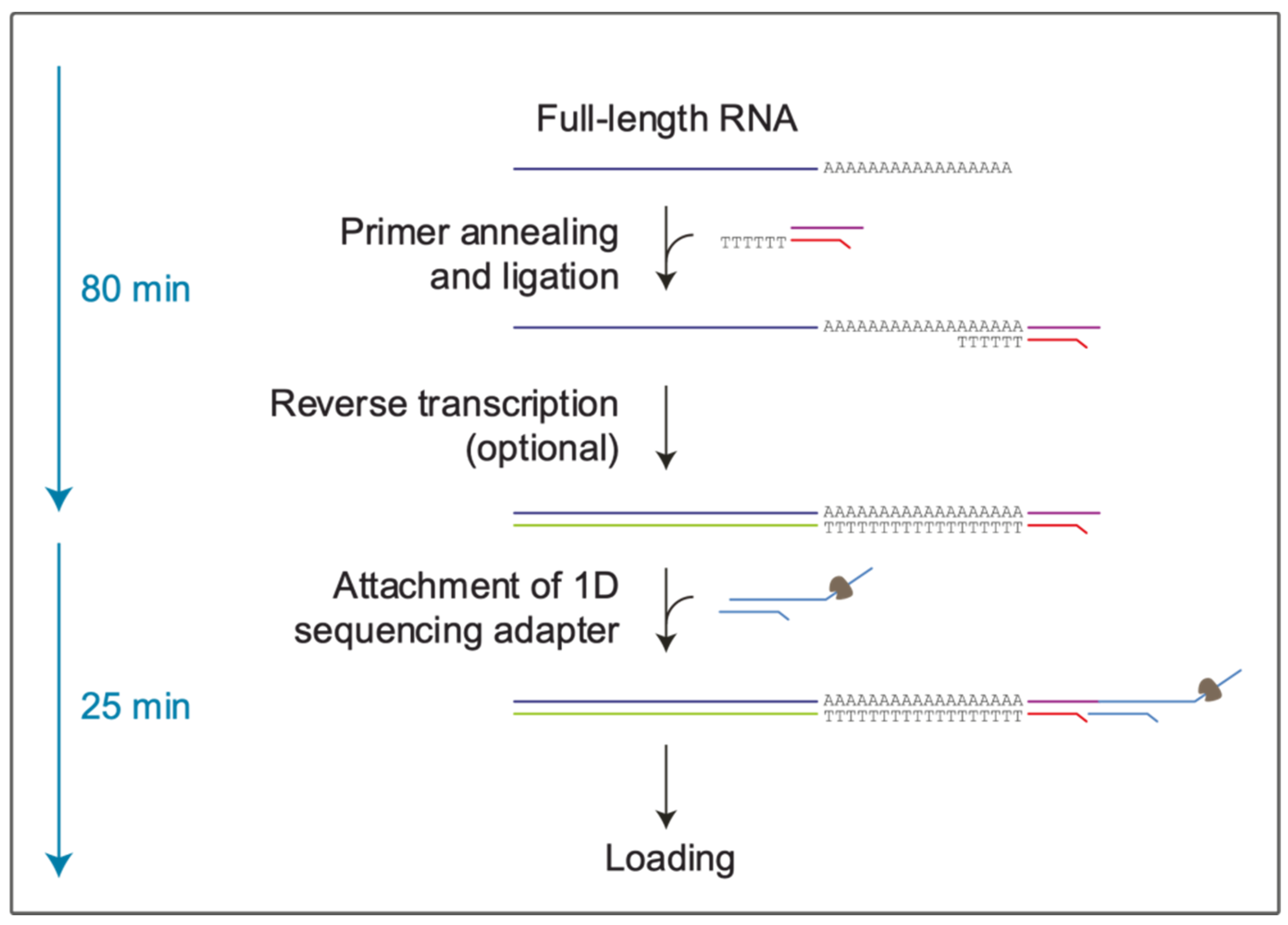
2.2. Preparation of the Libraries and the Computational Pipeline
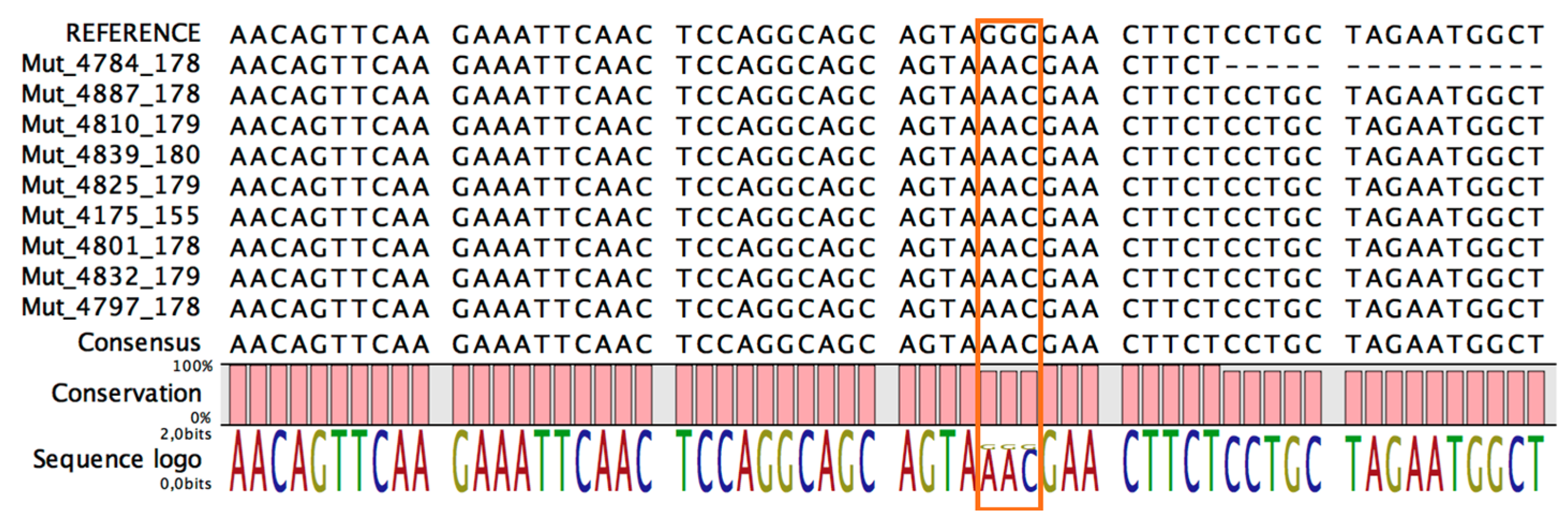
2.3. Calculation of Mutation Frequency
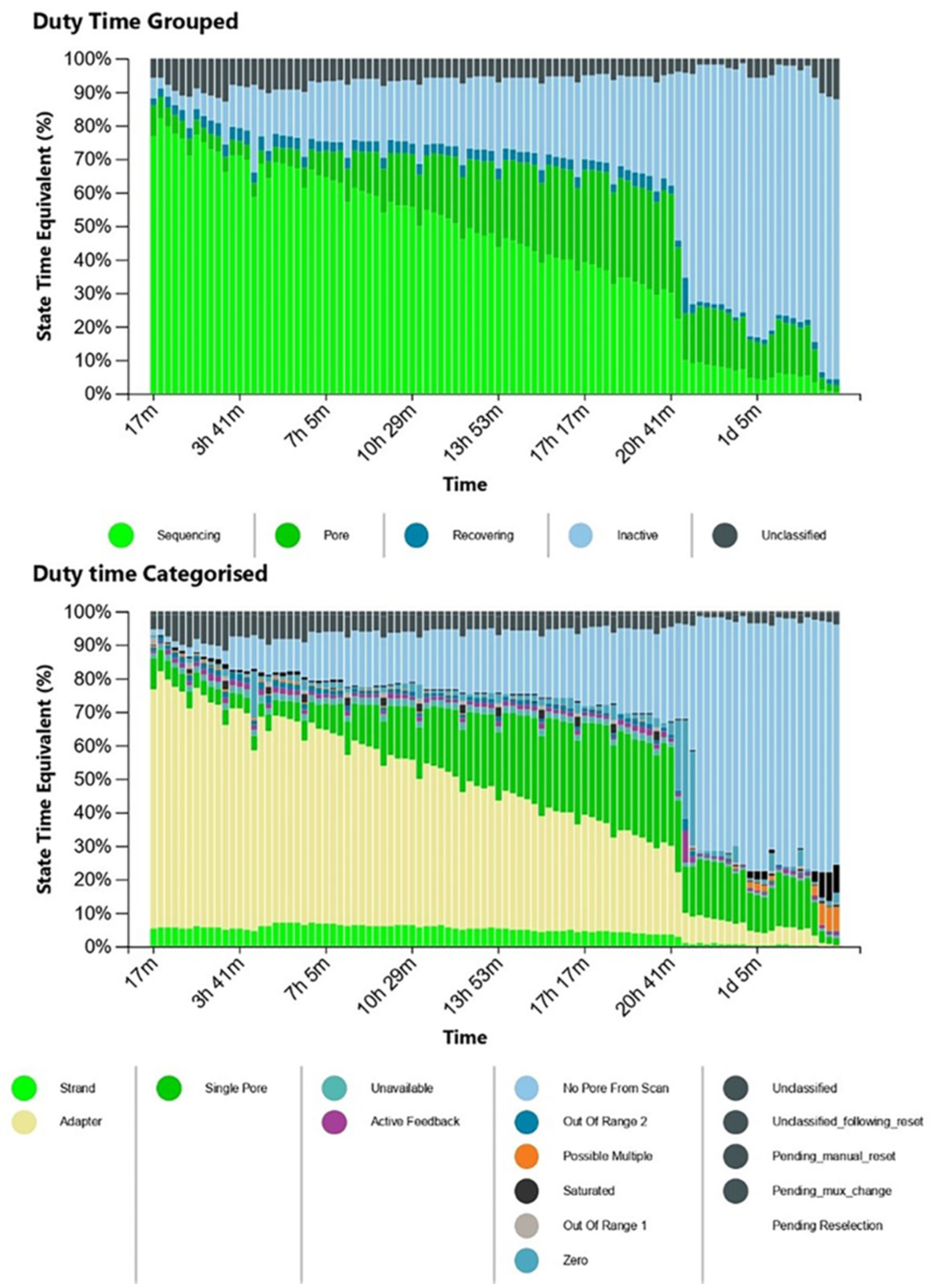
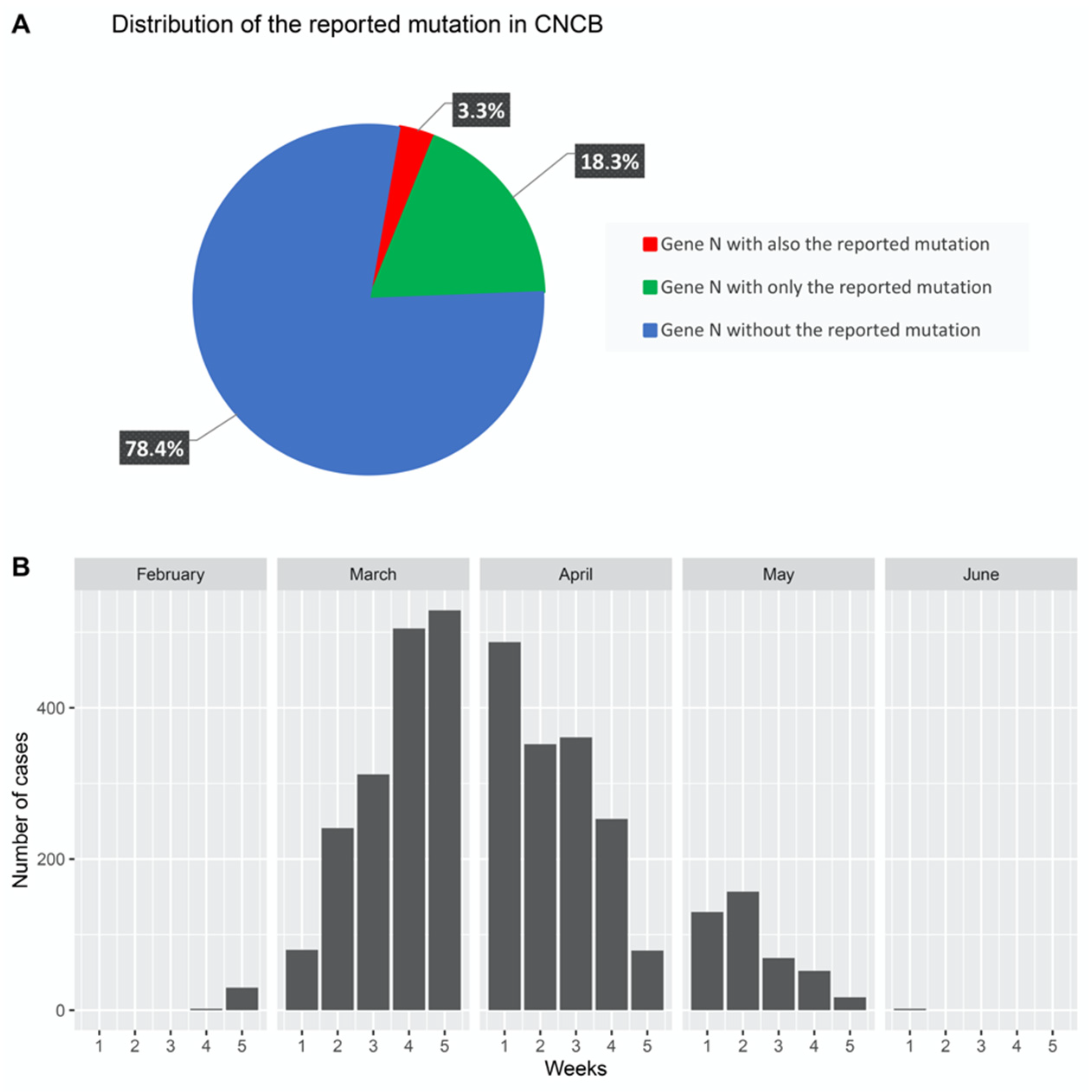
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bafna, K.; Krug, R.M.; Montelione, G.T. Structural Similarity of SARS-CoV-2 Mpro and Hcv Ns3/4a Proteases Suggests New Approaches for Identifying Existing Drugs Useful as COVID-19 Therapeutics. ChemRxiv Prepr. Serv. Chem. 2020. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef]
- Shanker, S.; Paulson, A.; Edenberg, H.J.; Peak, A.; Perera, A.; Alekseyev, Y.O.; Beckloff, N.; Bivens, N.J.; Donnelly, R.; Gillaspy, A.F.; et al. Evaluation of Commercially Available RNA Amplification Kits for RNA Sequencing Using Very Low Input Amounts of Total RNA. J. Biomol. Tech. JBT 2015, 26, 4–18. [Google Scholar] [CrossRef][Green Version]
- Brandariz-Fontes, C.; Camacho-Sanchez, M.; Vilà, C.; Vega-Pla, J.L.; Rico, C.; Leonard, J.A. Effect of the enzyme and PCR conditions on the quality of high-throughput DNA sequencing results. Sci. Rep. 2015, 5, 8056. [Google Scholar] [CrossRef]
- Depledge, D.P.; Wilson, A.C. Using Direct RNA Nanopore Sequencing to Deconvolute Viral Transcriptomes. Curr. Protoc. Microbiol. 2020, 57, e99. [Google Scholar] [CrossRef]
- PCR Tiling of COVID-19 Virus Version: PTC_9096_v109_revF_06Feb2020. Available online: https://community.nanoporetech.com/protocols/classic-pcr-tiling-SARS-CoV-2/v (accessed on 6 February 2020).
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Brejová, B.; Boršová, K.; Hodorová, V.; Čabanová, V.; Gafurov, A.; Fričová, D.; Neboháčová, M.; Vinař, T.; Klempa, B.; Nosek, J. Nanopore sequencing of SARS-CoV-2: Comparison of short and long PCR-tiling amplicon protocols. PLoS ONE 2021, 16, e0259277. [Google Scholar] [CrossRef]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. BioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef]
- MinION Mk1B Device Specs. Available online: https://community.nanoporetech.com/requirements_documents/minion-mk1b-spec.pdf (accessed on 23 November 2020).
- Direct RNA Sequencing (SQK-RNA002) Protocol. Available online: https://community.nanoporetech.com/protocols/direct-rna-sequencing-sqk-rna002/v/drs_9080_v2_revp_14aug2019 (accessed on 14 August 2019).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Input DNA/RNA QC. Available online: https://community.nanoporetech.com/protocols/input-dna-rna-qc/v/idi_s1006_v1_revb_18apr2016 (accessed on 16 July 2019).
- MinKNOW Technical Document. Available online: https://community.nanoporetech.com/technical_documents/minknow-tech-doc/v/mitd_5000_v1_revaa_16may2016 (accessed on 17 September 2021).
- Guppy Protocol. Available online: https://community.nanoporetech.com/protocols/Guppy-protocol/v/gpb_2003_v1_revaa_14dec2018 (accessed on 27 October 2021).
- Leger, A.; Leonardi, T. pycoQC, interactive quality control for Oxford Nanopore Sequencing. J. Open Source Softw. 2019, 4, 1236. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinforma. Oxf. Engl. 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinforma. Oxf. Engl. 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Lin, H.-N.; Hsu, W.-L. GSAlign: An efficient sequence alignment tool for intra-species genomes. BMC Genom. 2020, 21, 182. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinforma. Oxf. Engl. 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Zhao, W.-M.; Song, S.-H.; Chen, M.-L.; Zou, D.; Ma, L.-N.; Ma, Y.-K.; Li, R.-J.; Hao, L.-L.; Li, C.-P.; Tian, D.-M.; et al. The 2019 novel coronavirus resource. Yi Chuan Hered. 2020, 42, 212–221. [Google Scholar] [CrossRef]
- Trost, B.; Walker, S.; Haider, S.A.; Sung, W.W.L.; Pereira, S.; Phillips, C.L.; Higginbotham, E.J.; Strug, L.J.; Nguyen, C.; Raajkumar, A.; et al. Impact of DNA source on genetic variant detection from human whole-genome sequencing data. J. Med. Genet. 2019, 56, 809–817. [Google Scholar] [CrossRef]
- Zhu, Q.; Hu, Q.; Shepherd, L.; Wang, J.; Wei, L.; Morrison, C.D.; Conroy, J.M.; Glenn, S.T.; Davis, W.; Kwan, M.L.; et al. The impact of DNA input amount and DNA source on the performance of whole-exome sequencing in cancer epidemiology. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1207–1213. [Google Scholar] [CrossRef]
- Stupnikov, A.; McInerney, C.E.; Savage, K.I.; McIntosh, S.A.; Emmert-Streib, F.; Kennedy, R.; Salto-Tellez, M.; Prise, K.M.; McArt, D.G. Robustness of differential gene expression analysis of RNA-seq. Comput. Struct. Biotechnol. J. 2021, 19, 3470–3481. [Google Scholar] [CrossRef]
- Potapov, V.; Ong, J.L. Correction: Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE 2017, 12, e0181128. [Google Scholar] [CrossRef]
- Moldován, N.; Balázs, Z.; Tombácz, D.; Csabai, Z.; Szűcs, A.; Snyder, M.; Boldogkői, Z. Multi-platform analysis reveals a complex transcriptome architecture of a circovirus. Virus Res. 2017, 237, 37–46. [Google Scholar] [CrossRef][Green Version]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Balázs, Z.; Kis, E.; Molnár, J.; Boldogkői, Z. Third-generation Sequencing Reveals Extensive Polycistronism and Transcriptional Overlapping in a Baculovirus. Sci. Rep. 2018, 8, 8604. [Google Scholar] [CrossRef]
- Tombácz, D.; Csabai, Z.; Szűcs, A.; Balázs, Z.; Moldován, N.; Sharon, D.; Snyder, M.; Boldogkői, Z. Long-Read Isoform Sequencing Reveals a Hidden Complexity of the Transcriptional Landscape of Herpes Simplex Virus Type 1. Front. Microbiol. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T.; DuBois, R.M.; Forsberg, E.C.; Akeson, M.; Vollmers, C. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017, 8, 16027. [Google Scholar] [CrossRef]
- Križanovic, K.; Echchiki, A.; Roux, J.; Šikic, M. Evaluation of tools for long read RNA-seq splice-aware alignment. Bioinforma. Oxf. Engl. 2018, 34, 748–754. [Google Scholar] [CrossRef]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef]
- Depledge, D.P.; Srinivas, K.P.; Sadaoka, T.; Bready, D.; Mori, Y.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 2019, 10, 754. [Google Scholar] [CrossRef]
- Keller, M.W.; Rambo-Martin, B.L.; Wilson, M.M.; Ridenour, C.A.; Shepard, S.S.; Stark, T.J.; Neuhaus, E.B.; Dugan, V.G.; Wentworth, D.E.; Barnes, J.R. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Sci. Rep. 2018, 8, 14408. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vacca, D.; Fiannaca, A.; Tramuto, F.; Cancila, V.; La Paglia, L.; Mazzucco, W.; Gulino, A.; La Rosa, M.; Maida, C.M.; Morello, G.; et al. Direct RNA Nanopore Sequencing of SARS-CoV-2 Extracted from Critical Material from Swabs. Life 2022, 12, 69. https://doi.org/10.3390/life12010069
Vacca D, Fiannaca A, Tramuto F, Cancila V, La Paglia L, Mazzucco W, Gulino A, La Rosa M, Maida CM, Morello G, et al. Direct RNA Nanopore Sequencing of SARS-CoV-2 Extracted from Critical Material from Swabs. Life. 2022; 12(1):69. https://doi.org/10.3390/life12010069
Chicago/Turabian StyleVacca, Davide, Antonino Fiannaca, Fabio Tramuto, Valeria Cancila, Laura La Paglia, Walter Mazzucco, Alessandro Gulino, Massimo La Rosa, Carmelo Massimo Maida, Gaia Morello, and et al. 2022. "Direct RNA Nanopore Sequencing of SARS-CoV-2 Extracted from Critical Material from Swabs" Life 12, no. 1: 69. https://doi.org/10.3390/life12010069
APA StyleVacca, D., Fiannaca, A., Tramuto, F., Cancila, V., La Paglia, L., Mazzucco, W., Gulino, A., La Rosa, M., Maida, C. M., Morello, G., Belmonte, B., Casuccio, A., Maugeri, R., Iacopino, G., Balistreri, C. R., Vitale, F., Tripodo, C., & Urso, A. (2022). Direct RNA Nanopore Sequencing of SARS-CoV-2 Extracted from Critical Material from Swabs. Life, 12(1), 69. https://doi.org/10.3390/life12010069