
Amelioration of Alzheimer’s Disease by Gut-Pancreas-Liver-Brain Interaction in an App Knock-In Mouse Model

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mice
2.3. Investigation of the Effects of L-Arginine and Limonoid Administration on Mouse Models
2.4. Fecal Samples and DNA Extraction
2.5. Mouse Tissue Samples and DNA Extraction
2.6. Gut Microbiota Analysis: 16SrRNA Amplicon Sequencing Library Preparation
2.7. Microbiota Analysis of Sequencing Data
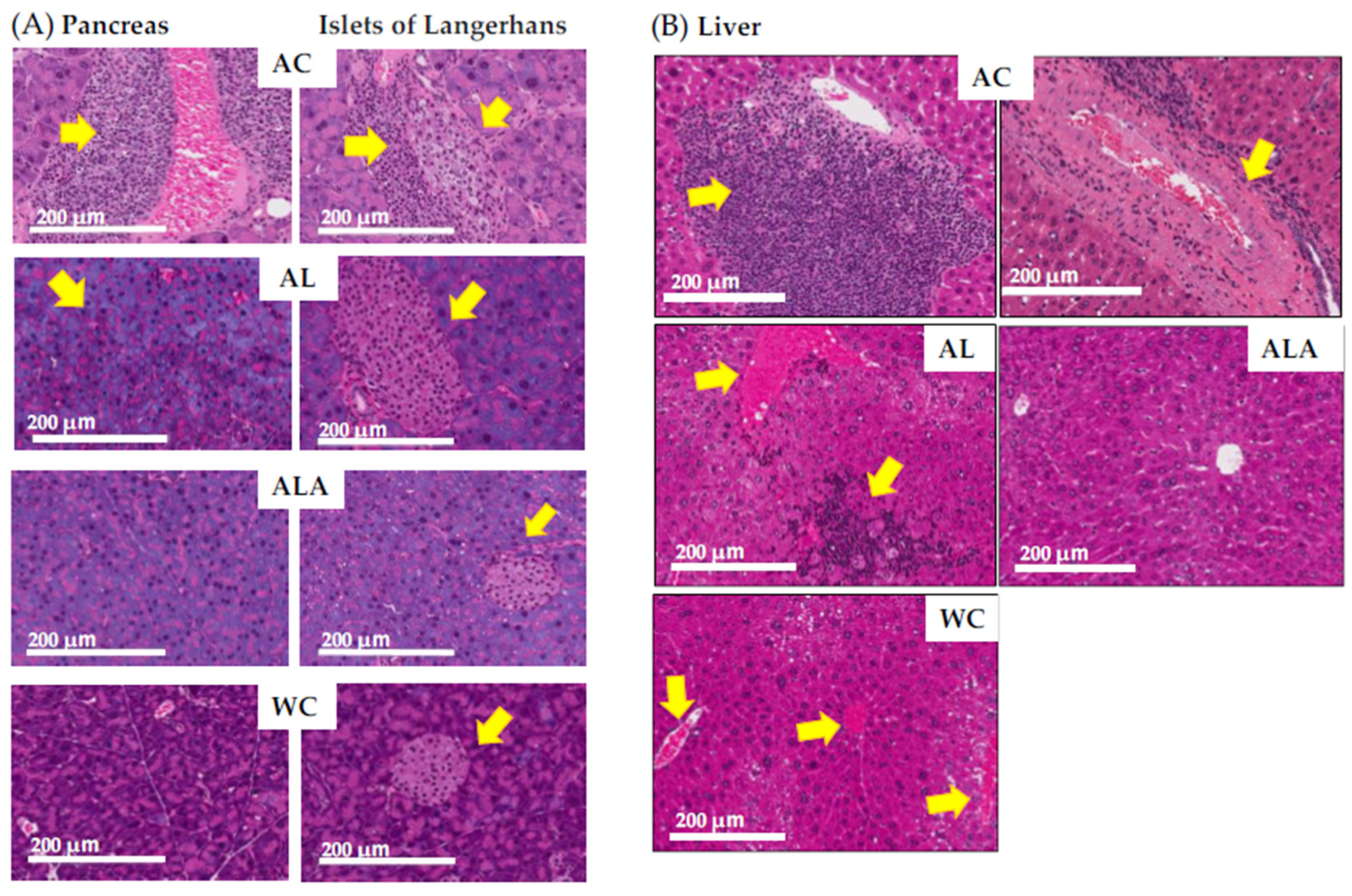
2.8. Histopathological Analysis of Mice Tissues
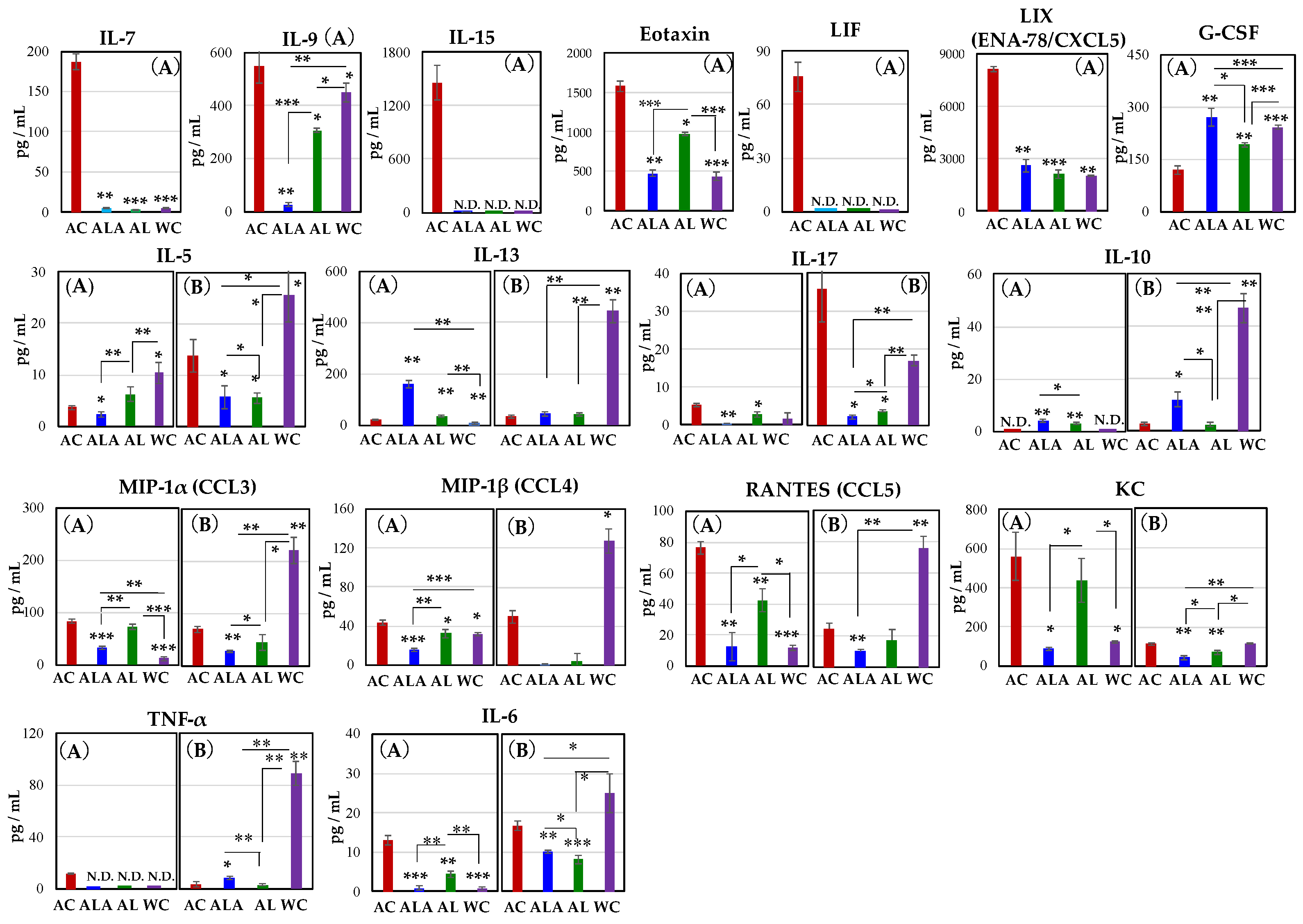
2.9. Measurement of Cytokine/Chemokine Concentration in Mouse Blood
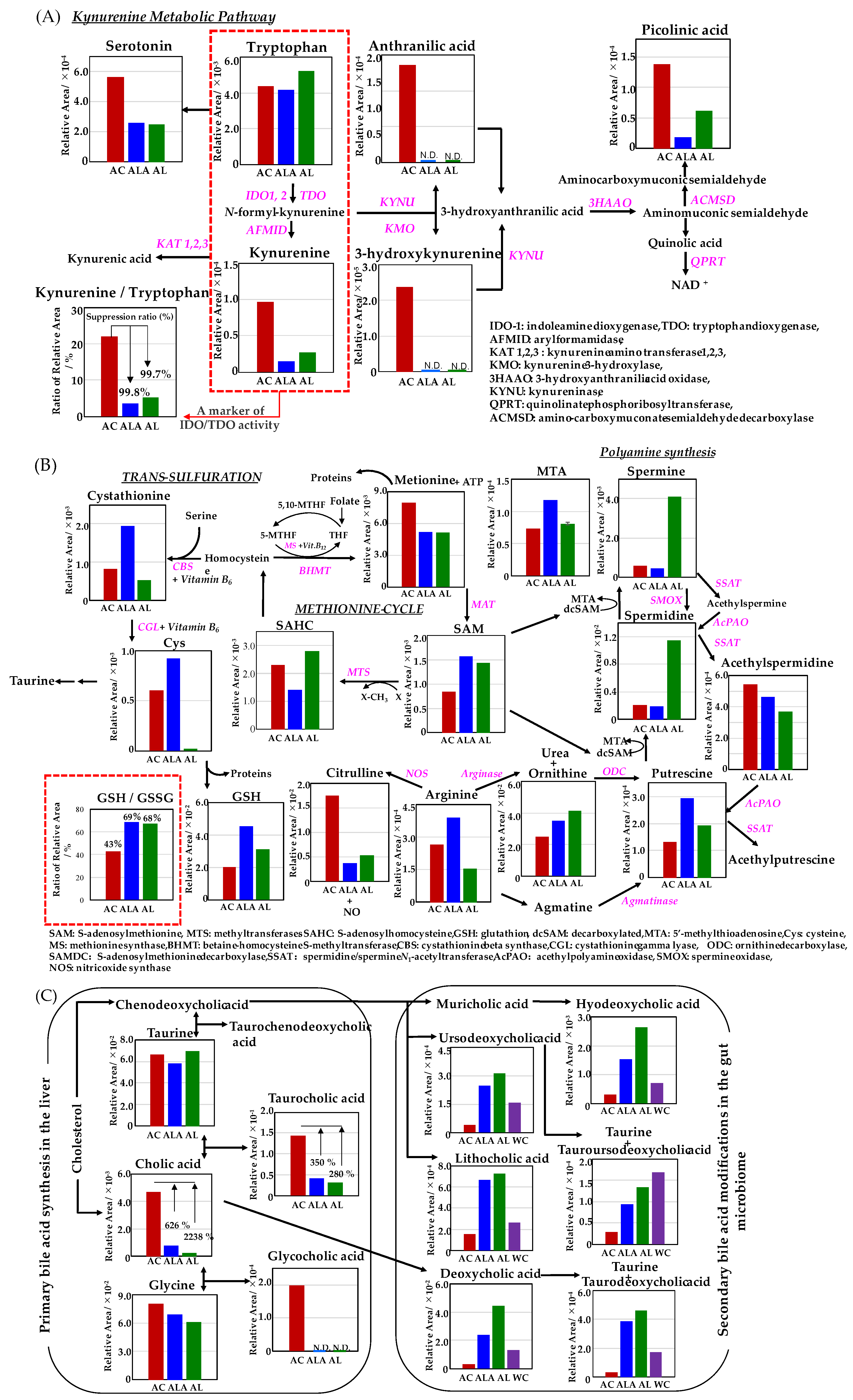
2.10. Metabolome Analysis of Mouse Liver by Capillary Electrophoresis–Time-of-Flight Mass Spectrometry
2.11. Metabolome Analysis of Mouse Feces by Liquid Chromatography/Time-of-Flight Mass Spectrometer
2.12. Processing and Analysis of Metabolome Analysis Data
2.13. Statistical Analysis
3. Results
3.1. Changes in the Gut Microbiota of Mice Treated with Limonoids and L-Arginine in a Mouse Model of AD
3.2. Observation of Bacterial Translocation in AD Mouse Model
3.3. Histopathological Analysis of Pancreas, Liver, and Brain Tissue
3.4. Metabolomic Analysis of Mouse Liver and Feces
3.5. Production of Proinflammatory Cytokines and Chemokines in Serum of AD Model Mice and WT Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- G-Science Academies Statement 2016, Understanding, Protecting, and Developing Global Brain Resources Developing Global Brain Resources. Available online: https://www.academie-sciences.fr/pdf/rapport/2016_G7_Brain.pdf (accessed on 15 August 2021).
- Walker, E.R.; McGee, R.E.; Druss, B.G. Mortality in mental disorders and global disease burden implications: A systematic review and meta-analysis. JAMA Psychiatry 2015, 72, 334–341. [Google Scholar] [CrossRef]
- Silberberg, D.; Anand, N.P.; Michels, K.; Kalaria, R.N. Brain and other nervous system disorders across the lifespan—Global challenges and opportunities. Nature 2015, 527, S151–S154. [Google Scholar] [CrossRef]
- Patel, V.; Chisholm, D.; Parikh, R.; Charlson, F.J.; Degenhardt, L.; Dua, T.; Ferrari, A.; Hyman, S.; Laxminarayan, R.; Levin, C.; et al. Addressing the Burden of Mental, Neurological, and Substance Use Disorders: Key Messages from Disease Control Priorities, 3rd edition. Lancet 2016, 387, 1672–1685. [Google Scholar] [CrossRef]
- Meguro, K.; Ishii, H.; Yamaguchi, S.; Ishizaki, J.; Shimada, M.; Sato, M.; Hashimoto, R.; Shimada, Y.; Meguro, M.; Yamadori, A.; et al. Prevalence of Dementia and Dementing Diseases in Japan. Arch. Neurol. 2002, 59, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N.; Ishihara, Y.; Ishida, K.; Tada, H.; Funaki-Kato, Y.; Hagiwara, M.; Ferdous, T.; Abdullah, M.; Mitani, A.; Michikawa, M.; et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech. Dis. 2017, 3, 15. [Google Scholar] [CrossRef]
- Boutajangout, A.; Wisniewski, T. Tau-Based Therapeutic Approaches for Alzheimer’s Disease—A Mini-Review. Gerontology 2014, 60, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef]
- Borroni, E.; Bohrmann, B.; Grueninger, F.; Prinssen, E.; Nave, S.; Loetscher, H.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Siddiqui, A.; et al. Sembragiline: A Novel, Selective Monoamine Oxidase Type B Inhibitor for the Treatment of Alzheimer’s Disease. J. Pharmacol. Exp. Ther. 2017, 362, 413–423. [Google Scholar] [CrossRef]
- Oka, S.; Leon, J.; Sakumi, K.; Ide, T.; Kang, N.; LaFerla, F.M.; Nakabeppu, Y. Human mitochondrial transcriptional factor A breaks the mitochondria-mediated vicious cycle in Alzheimer’s disease. Sci. Rep. 2016, 6, 37889. [Google Scholar] [CrossRef]
- Suda, M.; Shimizu, I.; Katsuumi, G.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Matsumoto, N.; Yoshida, Y.; Mikawa, R.; Katayama, A.; et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat. Aging 2021, 1, 1117–1126. [Google Scholar] [CrossRef]
- Umeda, T.; Ono, K.; Sakai, A.; Yamashita, M.; Mizuguchi, M.; Klein, W.L.; Yamada, M.; Mori, H.; Tomiyama, T. Rifampicin is a candidate preventive medicine against amyloid-β and tau oligomers. Brain 2016, 139, 1568–1586. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-G.; Udayanga, K.G.S.; Totsuka, N.; Weinberg, J.B.; Núñez, G.; Shibuya, A. Gut Dysbiosis Promotes M2 Macrophage Polarization and Allergic Airway Inflammation via Fungi-Induced PGE2. Cell Host Microbe 2014, 15, 95–102. [Google Scholar] [CrossRef]
- Okumura, R.; Kurakawa, T.; Nakano, T.; Kayama, H.; Kinoshita, M.; Motooka, D.; Gotoh, K.; Kimura, T.; Kamiyama, N.; Kusu, T.; et al. Lypd8 promotes the segregation of flagellated microbiota and colonic epithelia. Nature 2016, 532, 117–121. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Sharon, G.; Sampson, T.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Godinho-Silva, C.; Cardoso, F.; Veiga-Fernandes, H. Neuro–Immune Cell Units: A New Paradigm in Physiology. Annu. Rev. Immunol. 2019, 37, 19–46. [Google Scholar] [CrossRef] [PubMed]
- Minamisawa, M.; Suzumura, T.; Bose, S.; Taniai, T.; Kawai, G.; Suzuki, K.; Yamaguchi, A.; Yamanaka, S. Effect of Yuzu (Citrus junos) Seed Limonoids and Spermine on Intestinal Microbiota and Hypothalamic Tissue in the Sandhoff Disease Mouse Model. Med. Sci. 2021, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, H.; Guo, Y.; Du, X.; Qin, C. Gut microbiota regulate cognitive deficits and amyloid deposition in a model of Alzheimer’s disease. J. Neurochem. 2020, 155, 448–461. [Google Scholar] [CrossRef]
- Minamisawa, M.; Suzuki, K.; Kawai, G.; Yamaguchi, A.; Yamanaka, S. Functional evaluation of yuzu (Citrus junos) extracts con taining limonoids and polyamine for life extension. J. Funct. Foods 2017, 38, 591–600. [Google Scholar] [CrossRef]
- Kan, M.J.; Lee, J.E.; Wilson, J.G.; Everhart, A.L.; Brown, C.M.; Hoofnagle, A.N.; Jansen, M.; Vitek, M.P.; Gunn, M.D.; Colton, C.A. Arginine Deprivation and Immune Suppression in a Mouse Model of Alzheimer’s disease. J. Neurosci. 2015, 35, 5969–5982. [Google Scholar] [CrossRef]
- Walter, R.; Mark, M.; Reinhart, W.H. Pharmacological Concentrations of Arginine Influence Human Whole Blood Viscosity Independent of Nitric Oxide Synthase Activity in Vitro. Biochem. Biophys. Res. Commun. 2000, 269, 687–691. [Google Scholar] [CrossRef]
- Siasos, G.; Tousoulis, D.; Antoniades, C.; Stefanadi, E.; Stefanadis, C. L-arginine, the substrate for NO synthesis: Anan alternative treatment for premature atherosclerosis? Int. J. Cardiol. 2007, 116, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Esh, C.; Kokjohn, T.A.; Kalback, W.; Luehrs, D.C.; Seward, J.D.; Sue, L.I.; Beach, T.G. Circle of Willis Atherosclerosis Is a Risk Factor for Sporadic Alzheimer’s Disease. Arter. Thromb. Vasc. Biol. 2003, 23, 2055–2062. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; He, Y.; Ma, J.; Huang, P.; Du, J.; Cao, L.; Wang, Y.; Xiao, Q.; Tang, H.; Chen, S. Mild cognitive impairment has similar alterations as Alzheimer’s disease in gut microbiota. Alzheimer’s Dement. 2019, 15, 1357–1366. [Google Scholar] [CrossRef]
- Goumas, G.; Tentolouris, C.; Tousoulis, D.; Stefanadis, C.; Toutouzas, P. Therapeutic modification of the L-arginine–eNOS path way in cardiovascular diseases. Atherosclerosis 2001, 154, 255–267. [Google Scholar] [CrossRef]
- Böger, R.H.; Bode-Böger, S.M.; Szuba, A.; Tsao, P.S.; Chan, J.R.; Tangphao, O.; Blaschke, T.F.; Cooke, J.P. Asymmetric dimethylarginine (ADMA): A novel risk factor for endothelial dysfunction: Its role in hypercholesterolemia. Circulation 1998, 98, 1842–1847. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kurihara, S.; Kibe, R.; Ashida, H.; Benno, Y. Longevity in Mice is promoted by Probiotic-Induced suppression of Colonic Senescence Dependent senescence dependent on Upregulation of Gut Bacterial Polyamine Production. PLoS ONE 2011, 6, e23652. [Google Scholar] [CrossRef]
- Soda, K.; Kano, Y.; Chiba, F.; Koizumi, K.; Miyaki, Y. Increased polyamine intake inhibits age-associated alteration in global DNA methylation and 1, 2-dimethylhydrazineinduced tumorigenesis. PLoS ONE 2013, 8, e64357. [Google Scholar] [CrossRef]
- Hunt, J.B., Jr.; Placides, D.; Ratnasamy, K.; Selenica, M.L.; Nash, K.; Sandusky, L.A.; Abuqalbeen, F.; Lee, D.C. Argi nine metabolism and higher-order polyamines impact tau aggregation, microtubule assembly and autophagy in models of tauopathies. Alzheimer’s Dement. 2015, 11, 636–637. [Google Scholar] [CrossRef]
- Minamisawa, M.; Yoshida, S.; Uzawa, A. The functional evaluation of waste yuzu (Citrus junos) seeds. Food Funct. 2013, 5, 330–336. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Kawai, S.; Takagi, Y.; Kaneko, S.; Kurosawa, T. Effect of Three Types of Mixed Anesthetic Agents Alternate to Ketamine in Mice. Exp. Anim. 2011, 60, 481–487. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene data base project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome. Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Honda, K. Clostridia and the mucosal immune system. J. Intest. Microbiol. 2013, 27, 187–196. [Google Scholar]
- Lee, A.S.; Hermínia de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [Google Scholar] [CrossRef]
- Freier, T.A.; Beitz, D.C.; Li, L.; Hartman, P.A. Characterization of Eubacterium coprostanoligenes sp. nov., a Cholesterol-Reducing Anaerobe. Int. J. Syst. Bacteriol. 1994, 44, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Fang, B.; Pang, G.F.; Zhang, M.; Ren, F.Z. Age- and diet-specific effects of chronic exposure to chlorpyrifos on hormones, inflammation, and gut microbiota in rats. Pestic. Biochem. Physiol. 2019, 159, 68–79. [Google Scholar] [CrossRef]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Fosso, B.; Melocchi, L.; Nizzoli, G.; Troisi, J.; et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef]
- Kerry, R.G.; Patra, J.K.; Gouda, S.; Park, Y.; Shin, H.-S.; Das, G. Benefaction of probiotics for human health: A review. J. Food Drug Anal. 2018, 26, 927–939. [Google Scholar] [CrossRef]
- Mangifesta, M.; Mancabelli, L.; Milani, C.; Gaiani, F.; De’Angelis, N.; De’Angelis, G.L.; Van Sinderen, D.; Ventura, M.; Turroni, F. Mucosal microbiota of intestinal polyps reveals putative biomarkers of colorectal cancer. Sci. Rep. 2018, 8, 13974. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial role of Human gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Kanda, T.; Ueda, N.; Ikebuchi, Y.; Hashiguchi, K.; Nakao, K.; Isomoto, H. IL-8 and LYPD8 expression levels are associated with the inflammatory response in the colon of patients with ulcerative colitis. Biomed. Rep. 2020, 12, 193–198. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Lee, Y.-S.; Kim, Y.; Lee, S.-H.; Ryu, S.; Fukuda, S.; Hase, K.; Yang, C.-S.; Lim, H.S.; Kim, M.-S.; et al. Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal Immunol. 2016, 10, 104–116. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, S.; Zhong, R.; Wan, F.; Chen, L.; Liu, L.; Yi, B.; Zhang, H. Olive Fruit Extracts Supplement Improve Antioxidant Capacity via Altering Colonic Microbiota Composition in Mice. Front. Nutr. 2021, 8, 645099. [Google Scholar] [CrossRef]
- Gupta, R.S.; Chen, W.J.; Adeolu, M.; Chai, Y. Molecular signatures for the class Coriobacteriia and its different clades; proposal for division of the class Coriobacteriia into the emended order Coriobacteriales, containing the emended family Coriobacteriaceae and Atopobiaceae fam. nov., and Eggerthellales ord. nov., containing the family Eggerthellaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2013, 63, 3379–3397. [Google Scholar] [PubMed]
- Harrison, C.A.; Laubitz, D.; Ohland, C.L.; Midura-Kiela, M.T.; Patil, K.; Besselsen, D.G.; Jamwal, D.R.; Jobin, C.; Ghishan, F.K.; Kiela, P.R. Microbial dysbiosis associated with impaired intestinal Na+/H+ exchange accelerates and exacerbates colitis in exgerm free mice. Mucosal Immunol. 2018, 11, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.; Lamas, B.; Pham, H.P.; Michel, M.-L.; Rainteau, D.; Bridonneau, C.; DA Costa, G.; Van Hylckama Vlieg, J.E.; Sovran, B.; Chamignon, C.; et al. Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat. Commun. 2018, 9, 2802. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A. Gut microbiota and internal diseases: Update Information. Editorial: Unexpected functions of the Gut microbiota as a Virtual Organ. Nihon. Naika. Gakkai. Zasshi. 2015, 104, 29–34. [Google Scholar] [CrossRef][Green Version]
- Uehara, M.; Plank, L.D.; Hill, G.L. Components of energy expenditure in patients with severe sepsis and major trauma: A basis for clinical care. Crit. Care Med. 1999, 27, 1295–1302. [Google Scholar] [CrossRef]
- Ammori, B.J. Gut barrier dysfunction in patients with acute pancreatitis. J. Hepato-Biliary-Pancreat. Surg. 2002, 9, 411–412. [Google Scholar] [CrossRef]
- Wu, L.M.; Sankaran, S.J.; Plank, L.; Windsor, J.A.; Petrov, M.S. Meta-analysis of gut barrier dysfunction in patients with acute pancreatitis. BJS 2014, 101, 1644–1656. [Google Scholar] [CrossRef]
- Tan, C.; Ling, Z.; Huang, Y.; Cao, Y.; Liu, Q.; Cai, T.; Yuan, H.; Liu, C.; Li, Y.; Xu, K. Dysbiosis of intestinal microbiota associated with inflammation involved in the progression of acute pancreatitis. Pancreas 2015, 44, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Leblhuber, F.; Walli, J.; Tilz, G.P.; Demel, U.; Fuchs, D. Tryptophan degradation and immune activation in Alzheimer’s disease. J. Neural Transm. 2000, 107, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Greilberger, J.; Fuchs, D.; Leblhuber, F.; Greilberger, M.; Wintersteiger, R.; Tafeit, E. Carbonyl proteins as a clinical marker in Alzheimer’s disease and its relation to tryptophan degradation and immune activation. Clin. Lab. 2010, 56, 441–448. [Google Scholar]
- Colín-González, A.L.; Maldonado, P.D.; Santamaría, A. 3-Hydroxykynurenine: Anhydroxykynurenine: An intriguing molecule exerting dual actions in the Central Nervous System. Neurotoxicology 2013, 34, 189–204. [Google Scholar] [CrossRef]
- Chouraki, V.; Preis, S.R.; Yang, Q.; Beiser, A.; Li, S.; Larson, M.G.; Weinstein, G.; Wang, T.J.; Gerszten, R.E.; Vasan, R.S.; et al. Association of amine biomarkers with incident dementia and Alzheimer’s disease in the Framingham Study. Alzheimer’s Dement. 2017, 13, 1327–1336. [Google Scholar] [CrossRef]
- Chitapanarux, T.; Tienboon, P.; Pojchamarnwiputh, S.; Leelarungrayub, D. Open-labeled pilot study of cysteine-rich whey protein isolate supplementation for nonalcoholic steatohepatitis patients. J. Gastroenterol. Hepatol. 2009, 24, 1045–1050. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. Bile Acid Metabolism acid metabolism and lntestinal Flora. Bifidus 1992, 5, 157–172. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Zhu, H.; Sharma, S.; Bogdanov, M.; Rozen, S.G.; Matson, W.; Oki, N.O.; Motsinger-Reif, A.A.; Churchill, E.; Lei, Z.; et al. Alterations in metabolic pathways and networks in Alzheimer’s disease. Transl. Psychiatry 2013, 3, e244. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Jana, M.; Mondal, S.; Jana, A.; Pahan, K. Interleukin-12 (IL-12), but not IL-23, induces the expression of IL-7 in microglia and macrophages: Implications for multiple sclerosis. Immunology 2013, 141, 549–563. [Google Scholar] [CrossRef]
- Sawa, Y.; Arima, Y.; Ogura, H.; Kitabayashi, C.; Jiang, J.J.; Fukushima, T.; Kamimura, D.; Hirano, T.; Murakami, M. Hepatic Interleukininterleukin-7 Expression Regulates T Cell Responses. Immunity 2009, 30, 447–457. [Google Scholar] [CrossRef]
- Mattei, F.; Schiavoni, G.; Belardelli, F.; Tough, D.F. IL-15 Is Expressed by Dendritic Cells in Response to Type I IFN, Double-Stranded RNA, or Lipopolysaccharide and Promotes Dendritic Cell Activation. J. Immunol. 2001, 167, 1179–1187. [Google Scholar] [CrossRef]
- Zdrenghea, M.; Telcian, A.G.; Laza-Stanca, V.; Bellettato, C.M.; Edwards, M.R.; Nikonova, A.; Khaitov, M.; Azimi, N.; Groh, V.; Mallia, P.; et al. RSV infection modulates IL-15 production and MICA levels in respiratory epithelial cells. Eur. Respir. J. 2011, 39, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Hara, T.; Simmons, S.; Wagatsuma, K.; Abe, A.; Miyachi, H.; Kitano, S.; Ishii, M.; Tani-Ichi, S.; Ikuta, K. Characterization of the IL-15 niche in primary and secondary lymphoid organs in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 1915–1920. [Google Scholar] [CrossRef]
- Liu, Z.; Geboes, K.; Colpaert, S.; D’Haens, G.R.; Rutgeerts, P.; Ceuppens, J.L. IL-15 is highly expressed in inflammatory bowel disease and regulates local T cell-dependent cytokine production. J. Immunol. 2000, 164, 3608–3615. [Google Scholar] [CrossRef]
- Cacci, E.; Ajmone-Cat, M.A.; Anelli, T.; Biagioni, S.; Minghetti, L. In vitro neuronal and glial differentiation from embryonic or adult neural precursor cells are differently affected by chronic or acute activation of microglia. Glia 2008, 56, 412–425. [Google Scholar] [CrossRef]
- Butovsky, O.; Bukshpan, S.; Kunis, G.; Jung, S.; Schwartz, M. Microglia can be induced by IFN-γ or IL-4 to express neural or dendritic-like markers. Mol. Cell. Neurosci. 2007, 35, 490–500. [Google Scholar] [CrossRef]
- Lacy, P. Eosinophil Cytokines in Allergy. In Cytokine Effector Functions in Tissues; Academic Press: Cambridge, MA, USA, 2017; pp. 173–218. [Google Scholar] [CrossRef]
- Li, L.; Xia, Y.; Nguyen, A.; Lai, Y.H.; Feng, L.; Mosmann, T.R.; Lo, D. Effects of Th2 cytokines on chemokine expression in the lung: IL-13 potently induces eotaxin expression by airway epithelial cells. J. Immunol. 1999, 162, 2477–2487. [Google Scholar]
- Gleich, G.J. Mechanisms of eosinophil-associated inflammation. J. Allergy Clin. Immunol. 2000, 105, 651–663. [Google Scholar] [CrossRef]
- Hogan, S.P.; Rosenberg, H.F.; Moqbel, R.; Phipps, S.; Foster, P.S.; Lacy, P.; Kay, A.B.; Rothenberg, M.E. Eosinophils: Biological Properties and Role in Health and Disease. In Allergy and Allergic Diseases, 2nd ed.; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2008; pp. 258–294. [Google Scholar] [CrossRef]
- Ackerman, S.J.; Bochner, B.S. Mechanisms of Eosinophilia in the Pathogenesis of Hypereosinophilic Disorders. Immunol. Allergy Clin. N. Am. 2007, 27, 357–375. [Google Scholar] [CrossRef]
- Stefanovic, L.; Stefanovic, B. Mechanism of Direct Hepatotoxic Effect of KC Chemokine: Sequential Activation of Gene Expression and Progression from Inflammation to Necrosis. J. Interf. Cytokine Res. 2006, 26, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Terao, S.; Rantes, C. Chemokine RANTES in brain infarction, Cerebral. J. Cereb. Blood. Flow Metab. 2017, 28, 327–331. [Google Scholar] [CrossRef]
- Appay, V.; Brown, A.; Cribbes, S.; Randle, E.; Czaplewski, L.G. Aggregation of RANTES Is Responsible for Its Inflammatory Properties. J. Biol. Chem. 1999, 274, 27505–27512. [Google Scholar] [CrossRef] [PubMed]
- Suffee, N.; Richard, B.; Hlawaty, H.; Oudar, O.; Charnaux, N.; Sutton, A. Angiogenic properties of the chemokine RANTES/CCL5. Biochem. Soc. Trans. 2011, 39, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-J.; Tsai, Y.-C.; Shen, C.-K.J. G-CSF rescues the memory impairment of animal models of Alzheimer’s disease. J. Exp. Med. 2007, 204, 1273–1280. [Google Scholar] [CrossRef]
- Schneider, A.; Krüger, C.; Steigleder, T.; Weber, D.; Pitzer, C.; Laage, R.; Aronowski, J.; Maurer, M.H.; Gassler, N.; Mier, W.; et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Investig. 2005, 115, 2083–2098. [Google Scholar] [CrossRef]
- Kawada, H.; Takizawa, S.; Takanashi, T.; Morita, Y.; Fujita, J.; Fukuda, K.; Takagi, S.; Okano, H.; Ando, K.; Hotta, T. Administration of Hematopoietic Cytokines in the Subacute Phase After Cerebral Infarction Is Effective for Functional Recovery Facilitating Proliferation of Intrinsic Neural Stem/Progenitor Cells and Transition of Bone Marrow-Derived Neuronal Cells. Circulation 2006, 113, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Tang, C.; Kakuta, S.; Shimizu, K.; Kadoki, M.; Kamiya, T.; Shimazu, T.; Kubo, S.; Saijo, S.; Ishigame, H.; Nakae, S.; et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Nat. Immunol. 2018, 19, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Hurtado, I.; Santacruz, A.; Peiró, G.; Zapater, P.; Gutiérrez, A.; Pérez-Mateo, M.; Sanz, Y.; Francés, R. Gut Microbiota Dysbiosis Is Associated with Inflammation and Bacterial Translocation in Mice with CCl4-Induced Fibrosis. PLoS ONE 2011, 6, e23037. [Google Scholar] [CrossRef]
- Livingston, D.H.; Mosenthal, A.C.; Deitch, E.A. Sepsis and multiple organ dysfunction syndrome: A clinical-mechanistic overview. New Horiz. 1995, 3, 257–266. [Google Scholar]
- Tripathy, D.; Thirumangalakudi, L.; Grammas, P. RANTES upregulation in the Alzheimer’s disease brain: A possible neuroprot ective role. Neurobiol. Aging 2010, 31, 8–16. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef]
- An, C.; Shi, Y.; Li, P.; Hu, X.; Gan, Y.; Stetler, R.A.; Leak, R.; Gao, Y.; Sun, B.-L.; Zheng, P.; et al. Molecular dialogs between the ischemic brain and the peripheral immune system: Dualistic roles in injury and repair. Prog. Neurobiol. 2013, 115, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, Y.; Kigoshi, T.; Murasaki, S.; Arai, F.; Shimada, K.; Seki, N.; Kim, Y.G.; Hase, K.; Ohno, H.; Kawano, K.; et al. Pancreatic glycoprotein 2 is a first line of defense for mucosal protection in intestinal inflammation. Nat. Commun. 2021, 12, 1067. [Google Scholar] [CrossRef]
- Graefe, K.H.; Bossle, F.; Wölfel, R.; Burger, A.; Souladaki, M.; Bier, D.; Dutschka, K.; Farahati, J.; Bönisch, H. Sympathomimetic effects of MIBG: Comparison with tyramine. J. Nucl. Med. 1999, 40, 1342–1351. [Google Scholar]
- Hirose, H.; Maruyama, H.; Kido, K.; Ito, K.; Koyama, K.; Tashiro, Y.; Saruta, T. α- and β-Cell Function in Obese Zucker (fa/fa) Rats: A Study with the Isolated Perfused Pancreas. Clin. Sci. 1994, 86, 311–316. [Google Scholar] [CrossRef]
- Gilliam, L.K.; Palmer, J.P.; Taborsky, G.J. Tyramine-Mediated Activation of Sympathetic Nerves Inhibits Insulin Secretion in Humans. J. Clin. Endocrinol. Metab. 2007, 92, 4035–4038. [Google Scholar] [CrossRef]
- Valoti, M.; Morón, J.; Benocci, A.; Sgaragli, G.; Unzeta, M. Evidence of a coupled mechanism between monoamine oxidase and peroxidase in the metabolism of tyramine by rat intestinal mitochondria. Biochem. Pharmacol. 1998, 55, 37–43. [Google Scholar] [CrossRef]
- Dhakal, S.; Macreadie, I. Tyramine and Amyloid Beta 42: A Toxic Synergy. Biomedicines 2020, 8, 145. [Google Scholar] [CrossRef]
- Cohen, G.; Farooqui, R.; Kesler, N. Parkinson disease: A new link between monoamine oxidase and mitochondrial electron flow. Proc. Natl. Acad. Sci. USA 1997, 94, 4890–4894. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace amines and their receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, N.; Grimsby, J.; Shih, J.C.; Cadenas, E. The metabolism of tyramine by monoamine oxidase A/B causes oxidative damage to mitochondrial DNA. Arch. Biochem. Biophys. 1996, 335, 295–304. [Google Scholar] [CrossRef]
- Sherwani, S.I.; Khan, H.A. Trace Amines in Neuropsychiatric Disorders. In Trace Amines and Neurological Disorders: Potential Mechanisms and Risk Factors; Academic Press: San Diego, CA, USA, 2016; p. 269. [Google Scholar]
- Revel, F.G.; Moreau, J.L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef] [PubMed]
- Vallim, T.Q.D.A.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.P.K.; Sanyal, A.J. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef]
- Wiest, R.; Albillos, A.; Trauner, M.; Bajaj, J.S.; Jalan, R. Targeting the gut-liver axis in liver disease. J. Hepatol. 2017, 67, 1084–1103. [Google Scholar] [CrossRef]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 2012, 28, 159–165. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Guo, G.L. Role of FXR in liver inflammation during nonalcoholic steatohepatitis. Curr. Pharmacol. Rep. 2017, 3, 92–100. [Google Scholar] [CrossRef]
- Ferrell, M.; Bazeley, P.; Wang, Z.; Levison, B.S.; Li, X.S.; Jia, X.; Krauss, R.M.; Knight, R.; Lusis, A.J.; Garcia-Garcia, J.C.; et al. Fecal Microbiome Composition Does Not Predict Diet-Induced TMAO Production in Healthy Adults. J. Am. Heart Assoc. 2021, 10, e021934. [Google Scholar] [CrossRef]
- Giil, L.M.; Midttun, Ø.; Refsum, H.; Ulvik, A.; Advani, R.; Smith, A.D.; Ueland, P.M. Kynurenine Pathway Metabolites in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gerbarg, P.; Bottiglieri, T.; Massoumi, L.; Carpenter, L.L.; Lavretsky, H.; Muskin, P.R.; Brown, R.P.; Mischoulon, D.; Work Group of the American Psychiatric Association Council on Research. S-adenosylmethionine (SAMe) for neuropsychiatric disorders: A clinician-oriented review of research. J. Clin. Psychiatry 2017, 78, e656–e667. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Z.; Ma, S.; Kong, F.; He, Y.; Yang, X.; Wang, Y.; Xu, H.; Tian, J.; Cao, J.; et al. Ratio of S-adenosylmethionine to S-adenosylhomocysteine as a sensitive indicator of atherosclerosis. Mol. Med. Rep. 2016, 14, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Murín, R.; Vidomanová, E.; Kowtharapu, B.S.; Hatok, J.; Dobrota, D. Role of S-adenosylmethionine cycle in carcinogenesis. Gen. Physiol. Biophys. 2017, 36, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Sarris, J.; Byrne, G.J.; Bousman, C.; Stough, C.; Murphy, J.; MacDonald, P.; Adams, L.; Nazareth, S.; Mischoulon, D. Adjunctive S-adenosylmethionine (SAMe) in treating non-remittent major depressive disorder: An 8-week double-blind, randomized, controlled trial. Eur. Neuropsychopharmacol. 2018, 28, 1126–1136. [Google Scholar] [CrossRef]
- Frostesjö, L.; Holm, I.; Grahn, B.; Page, A.W.; Bestor, T.H.; Heby, O. Interference with DNA methyltransferase activity and genome methylation during F9 teratocarcinoma stem cell differentiation induced by polyamine depletion. J. Biol. Chem. 1997, 272, 4359–4366. [Google Scholar] [CrossRef]
- Shin, H.R.; Kim, H.; Kim, K.I.; Baek, S.H. Epigenetic and transcriptional regulation of autophagy. Autophagy 2016, 12, 2248–2249. [Google Scholar] [CrossRef]
- Mato, J.M.; Chantar, M.L.M.; Lu, S.C. S-adenosylmethionine metabolism and liver disease. Ann. Hepatol. 2013, 12, 183–189. [Google Scholar] [CrossRef]
- Kredich, N.M. Biosynthesis of Cysteine. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Soda, K. Polyamine Metabolism and Gene Methylation in Conjunction with One-Carbon Metabolism. Int. J. Mol. Sci. 2018, 19, 3106. [Google Scholar] [CrossRef]
- Wan, X.; Ma, B.; Wang, X.; Guo, C.; Sun, J.; Cui, J.; Li, L. S-Adenosylmethionine Alleviates Amyloid-β-Induced Neural Injury by Enhancing Trans-Sulfuration Pathway Activity in Astrocytes. J. Alzheimer’s Dis. 2020, 76, 981–995. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Modulation of cellular function by polyamines. Int. J. Biochem. Cell Biol. 2010, 42, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Vanrell, M.C.; Cueto, J.A.; Barclay, J.J.; Carrillo, C.; Colombo, M.I.; Gottlieb, R.A.; Romano, P.S. Polyamine depletion inhibits the autophagic response modulating Trypanosoma cruzi infectivity. Autophagy 2013, 9, 1080–1093. [Google Scholar] [CrossRef]
- Sharmin, S.; Sakata, K.; Kashiwagi, K.; Ueda, S.; Iwasaki, S.; Shirahata, A.; Igarashi, K. Polyamine cytotoxicity in the presence of bovine serum amine oxidase. Biochem. Biophys. Res. Commun. 2001, 282, 228–235. [Google Scholar] [CrossRef]
- Yang, H.; Ko, K.; Xia, M.; Li, T.W.; Oh, P.; Li, J.; Lu, S.C. Induction of avian musculoaponeurotic fibrosarcoma proteins by toxic bile acid inhibits expression of glutathione synthetic enzymes and contributes to cholestatic liver injury in mice. Hepatology 2009, 51, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Tanaka, Y.; Higashi, K.; Miyamori, D.; Takasaka, T.; Nagano, T.; Toida, T.; Yoshimoto, K.; Igarashi, K.; Ikegaya, H. Acetaldehyde-induced cytotoxicity involves induction of spermine oxidase at the transcriptional level. Toxicology 2013, 310, 1–7. [Google Scholar] [CrossRef]
- Imai, J.; Katagiri, H.; Yamada, T.; Ishigaki, Y.; Suzuki, T.; Kudo, H.; Uno, K.; Hasegawa, Y.; Gao, J.; Oka, Y. Regulation of Pancreatic β Cell Mass cell mass by Neuronal Signals from the Liver. Science 2008, 322, 1250–1254. [Google Scholar] [CrossRef] [PubMed]
- Teratani, T.; Mikami, Y.; Nakamoto, N.; Suzuki, T.; Harada, Y.; Okabayashi, K.; Hagihara, Y.; Taniki; Kanai, T. The liver–brain–gut neural arc maintains the Treg cell niche in the gut. Nature 2020, 585, 591–596. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minamisawa, M.; Sato, Y.; Ishiguro, E.; Taniai, T.; Sakamoto, T.; Kawai, G.; Saito, T.; Saido, T.C. Amelioration of Alzheimer’s Disease by Gut-Pancreas-Liver-Brain Interaction in an App Knock-In Mouse Model. Life 2022, 12, 34. https://doi.org/10.3390/life12010034
Minamisawa M, Sato Y, Ishiguro E, Taniai T, Sakamoto T, Kawai G, Saito T, Saido TC. Amelioration of Alzheimer’s Disease by Gut-Pancreas-Liver-Brain Interaction in an App Knock-In Mouse Model. Life. 2022; 12(1):34. https://doi.org/10.3390/life12010034
Chicago/Turabian StyleMinamisawa, Mayumi, Yuma Sato, Eitarou Ishiguro, Tetsuyuki Taniai, Taiichi Sakamoto, Gota Kawai, Takashi Saito, and Takaomi C. Saido. 2022. "Amelioration of Alzheimer’s Disease by Gut-Pancreas-Liver-Brain Interaction in an App Knock-In Mouse Model" Life 12, no. 1: 34. https://doi.org/10.3390/life12010034
APA StyleMinamisawa, M., Sato, Y., Ishiguro, E., Taniai, T., Sakamoto, T., Kawai, G., Saito, T., & Saido, T. C. (2022). Amelioration of Alzheimer’s Disease by Gut-Pancreas-Liver-Brain Interaction in an App Knock-In Mouse Model. Life, 12(1), 34. https://doi.org/10.3390/life12010034