
Sphingosine-1-Phosphate Induces ATP Release via Volume-Regulated Anion Channels in Breast Cell Lines


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Experimental Setup
2.3. Real-Time Imaging of the ATP Release
2.4. Quantitative Reverse Transcription Polymerase Chain Reaction (RTqPCR)
2.5. LRRC8A Knockdown with shRNA
3. Results
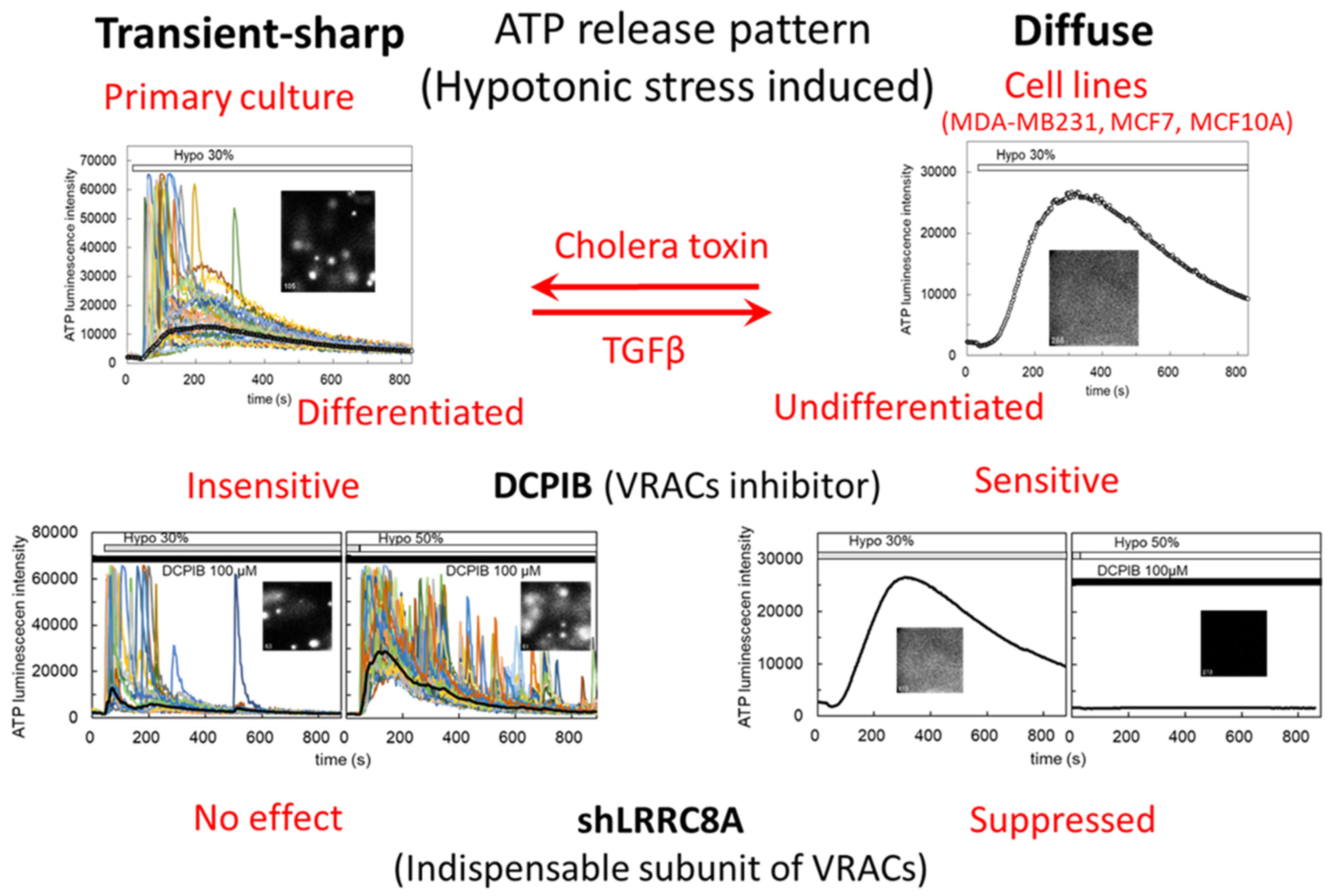
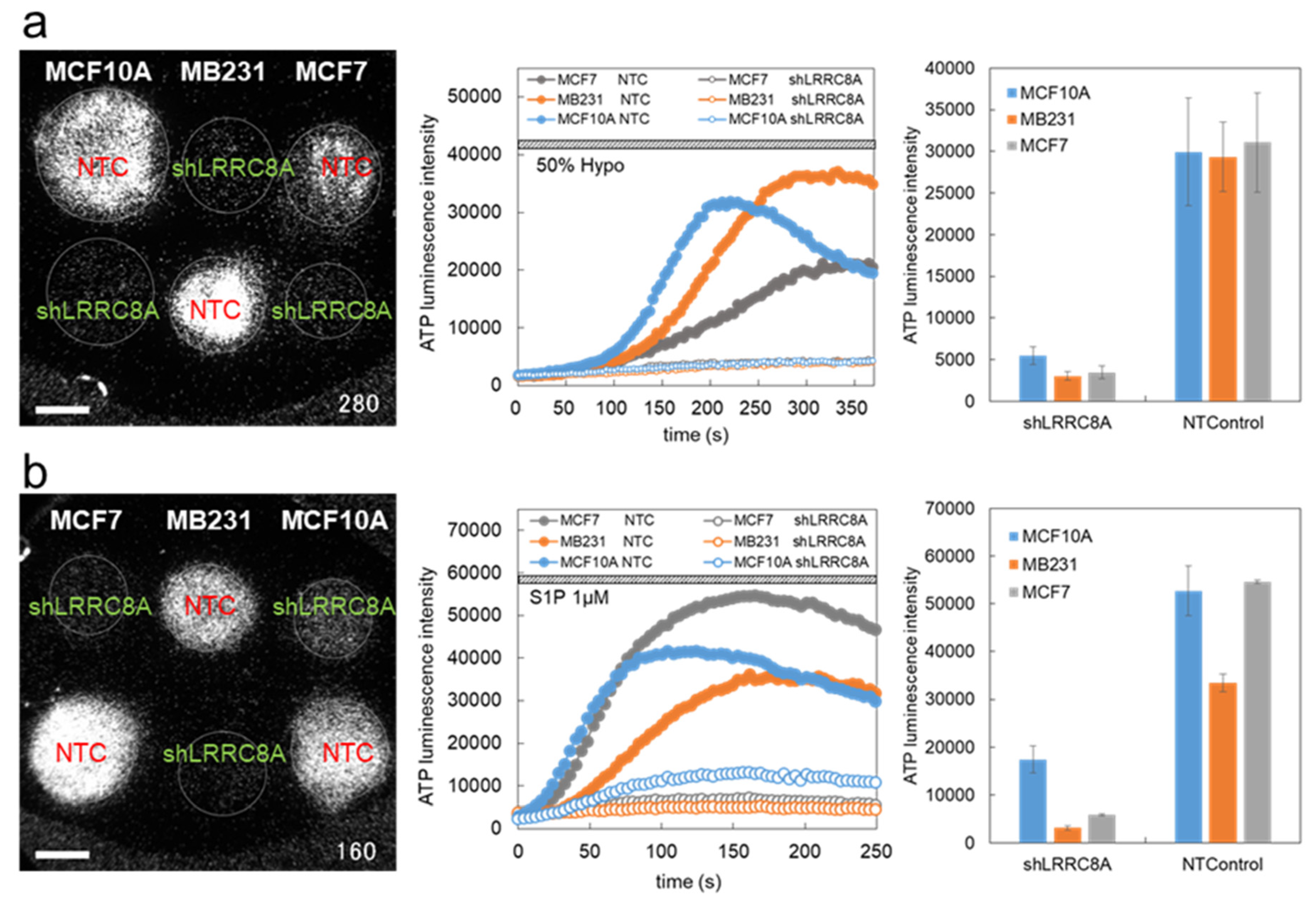
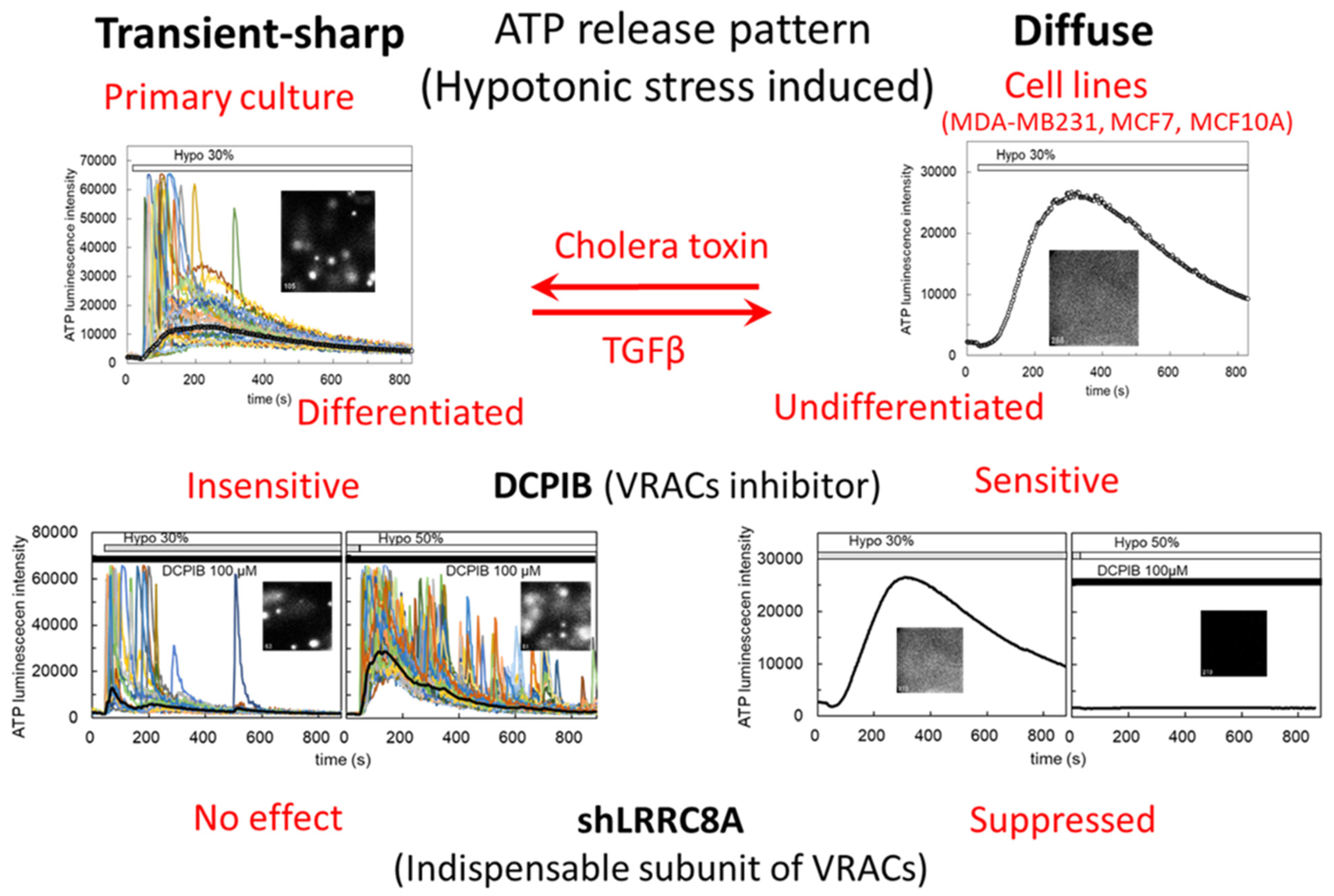
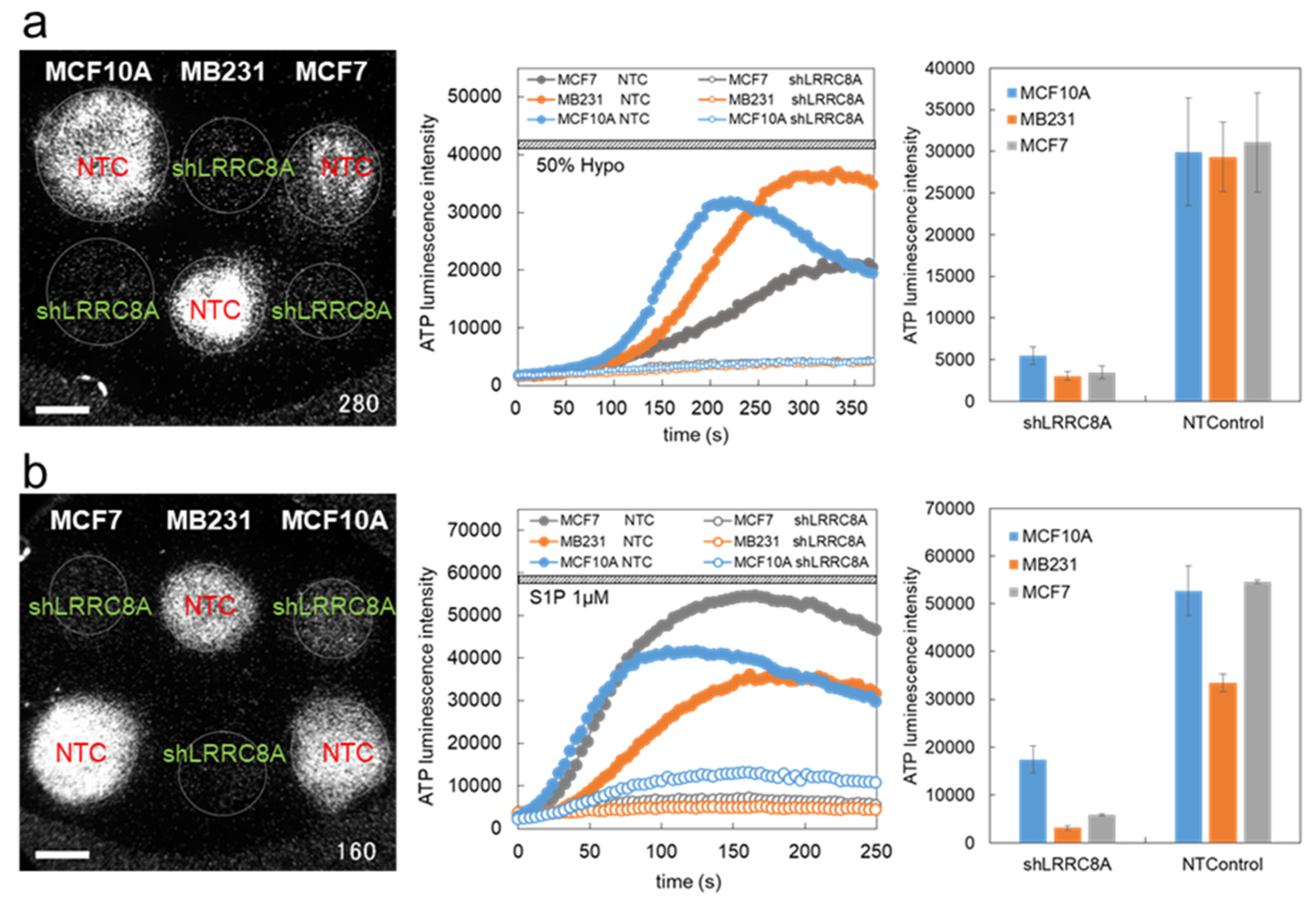
3.1. Hypotonic Stress-Induced ATP Release via VRACs
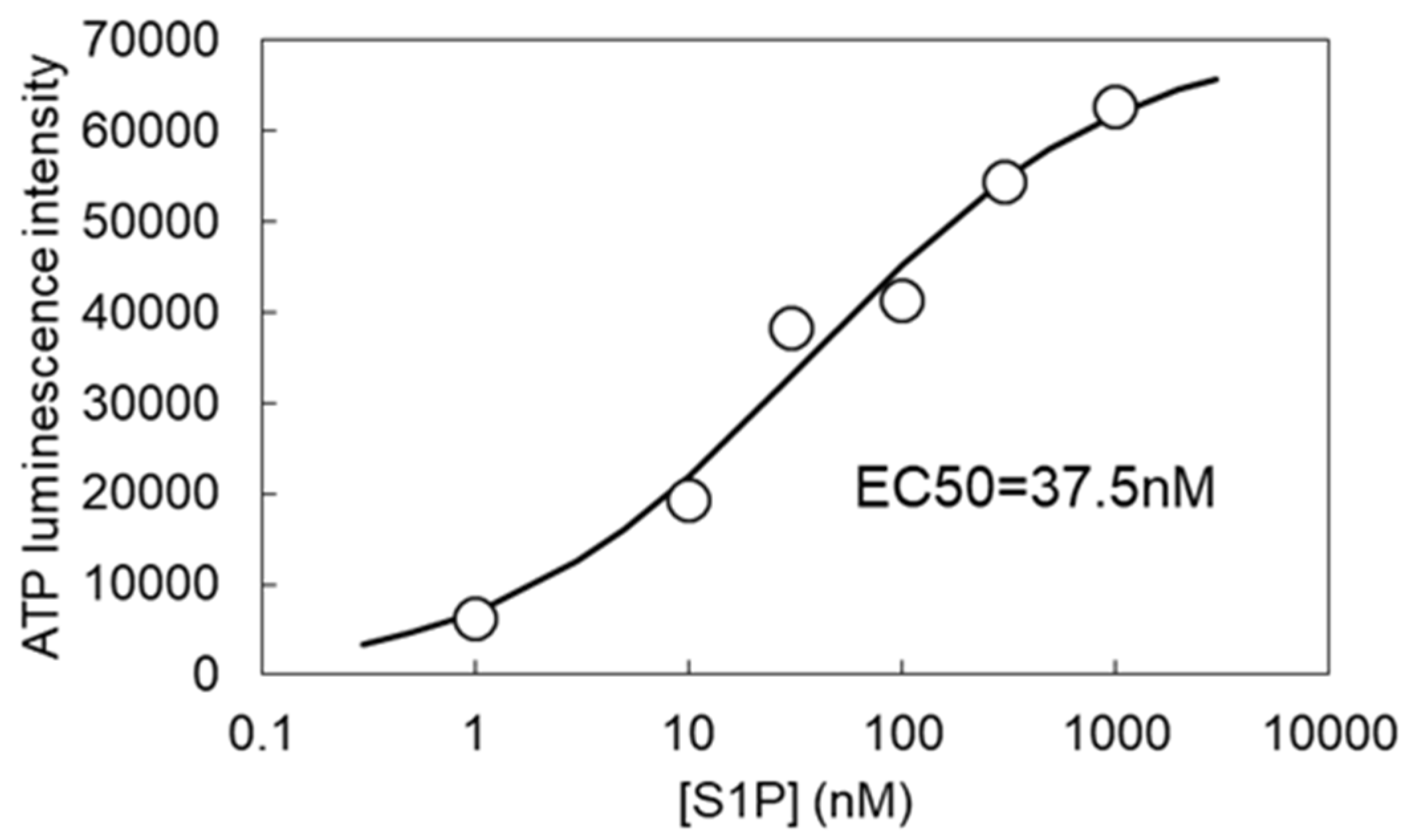
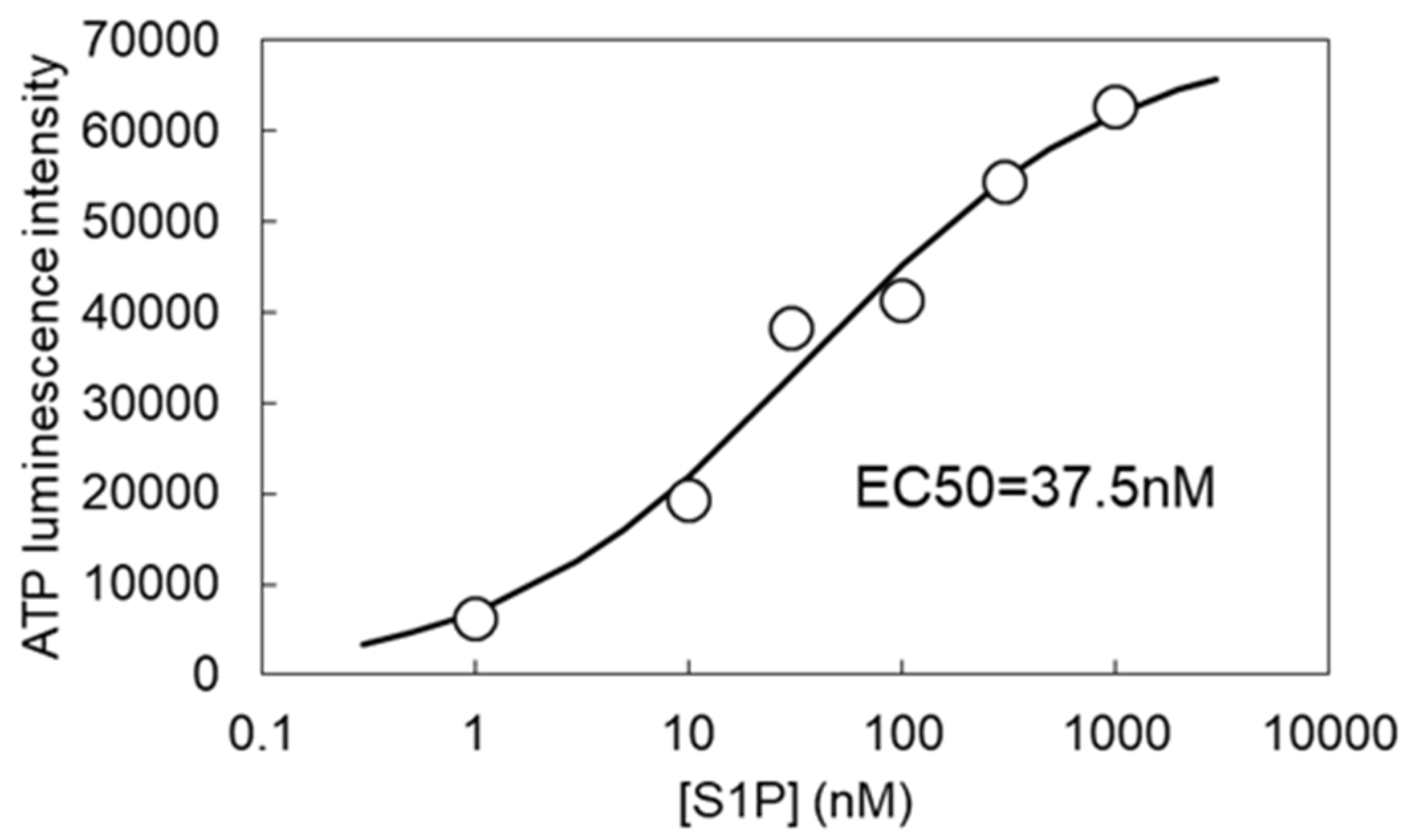
3.2. S1P-Induced ATP Release via VRACs
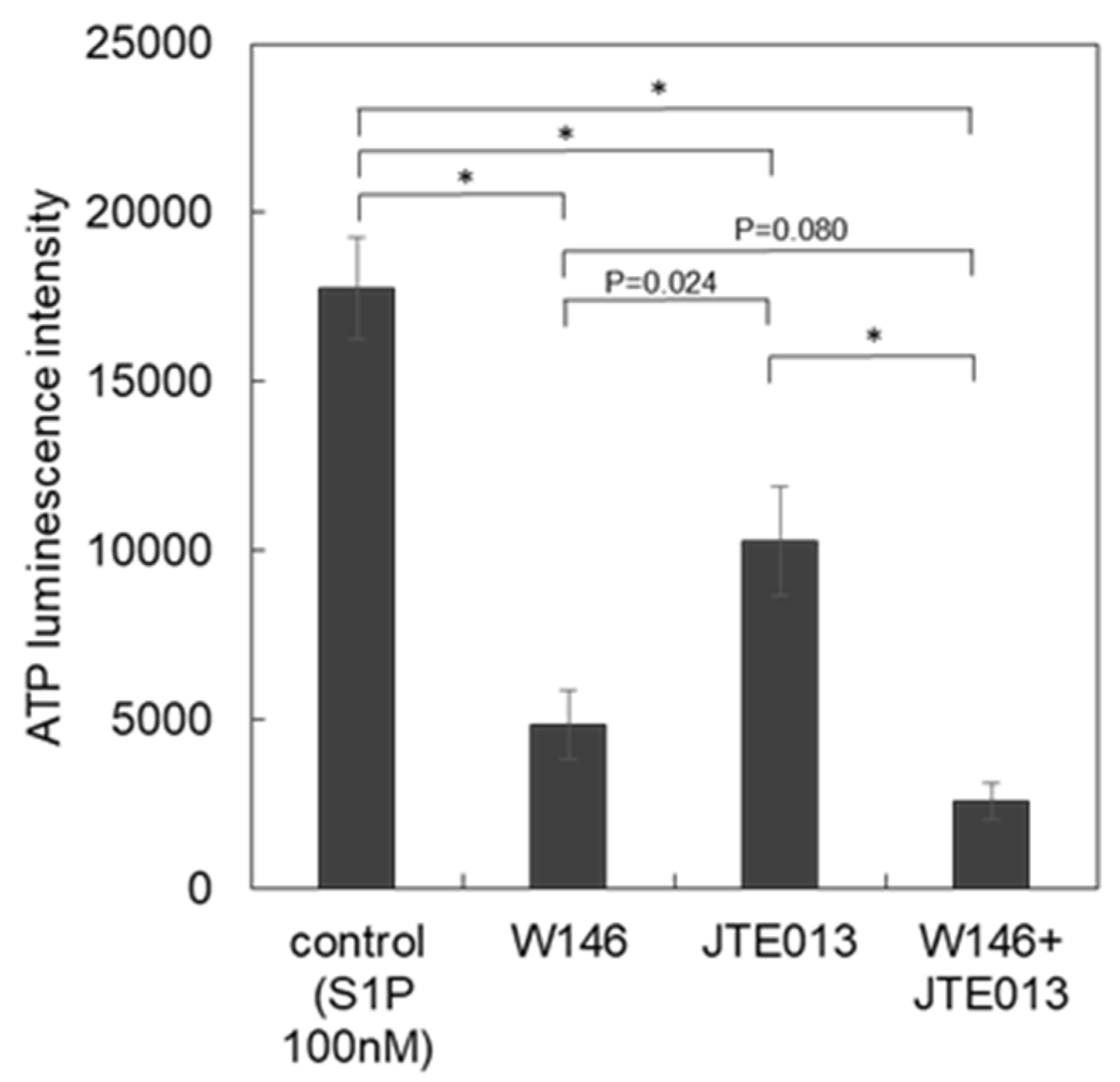
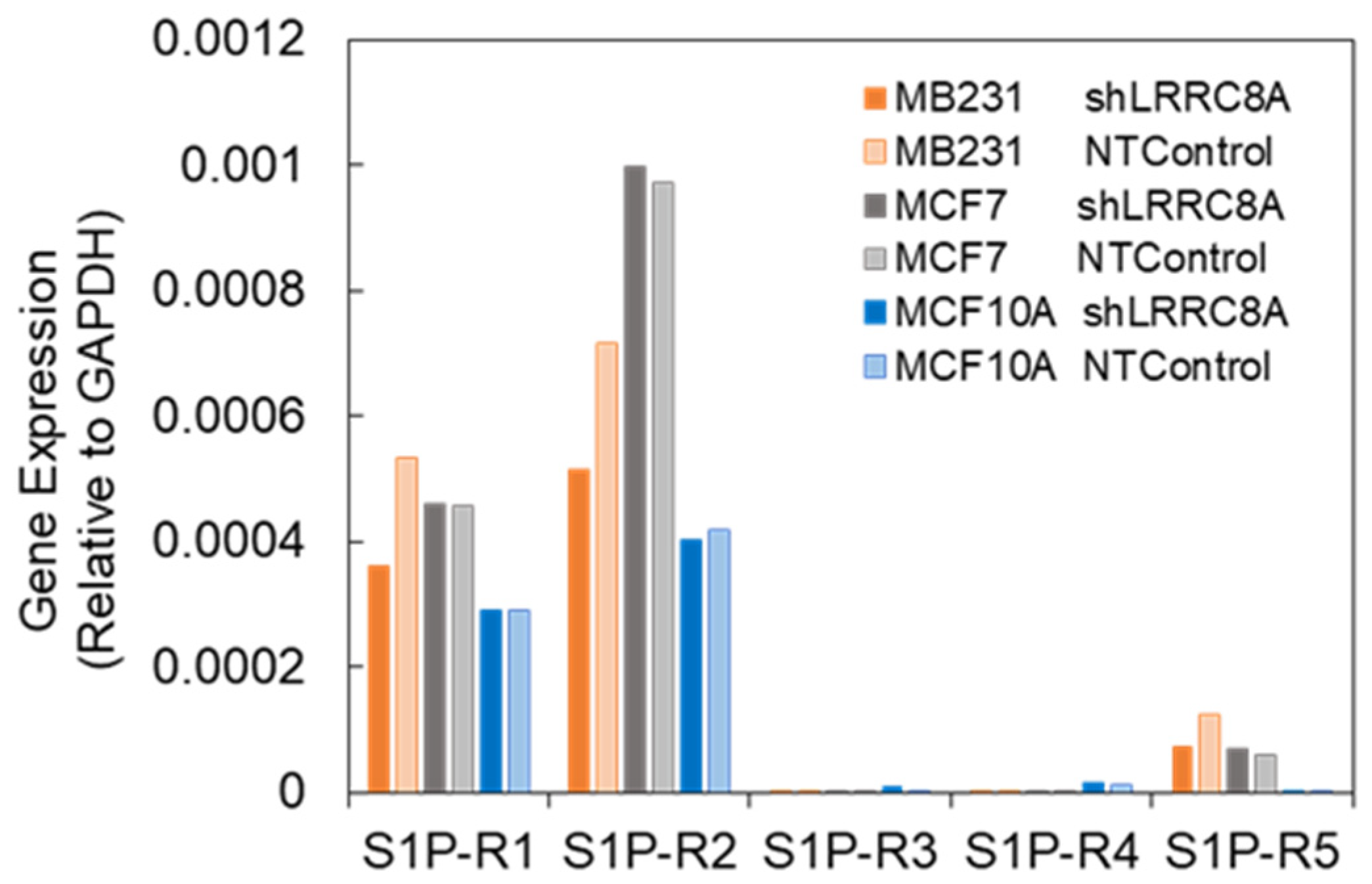
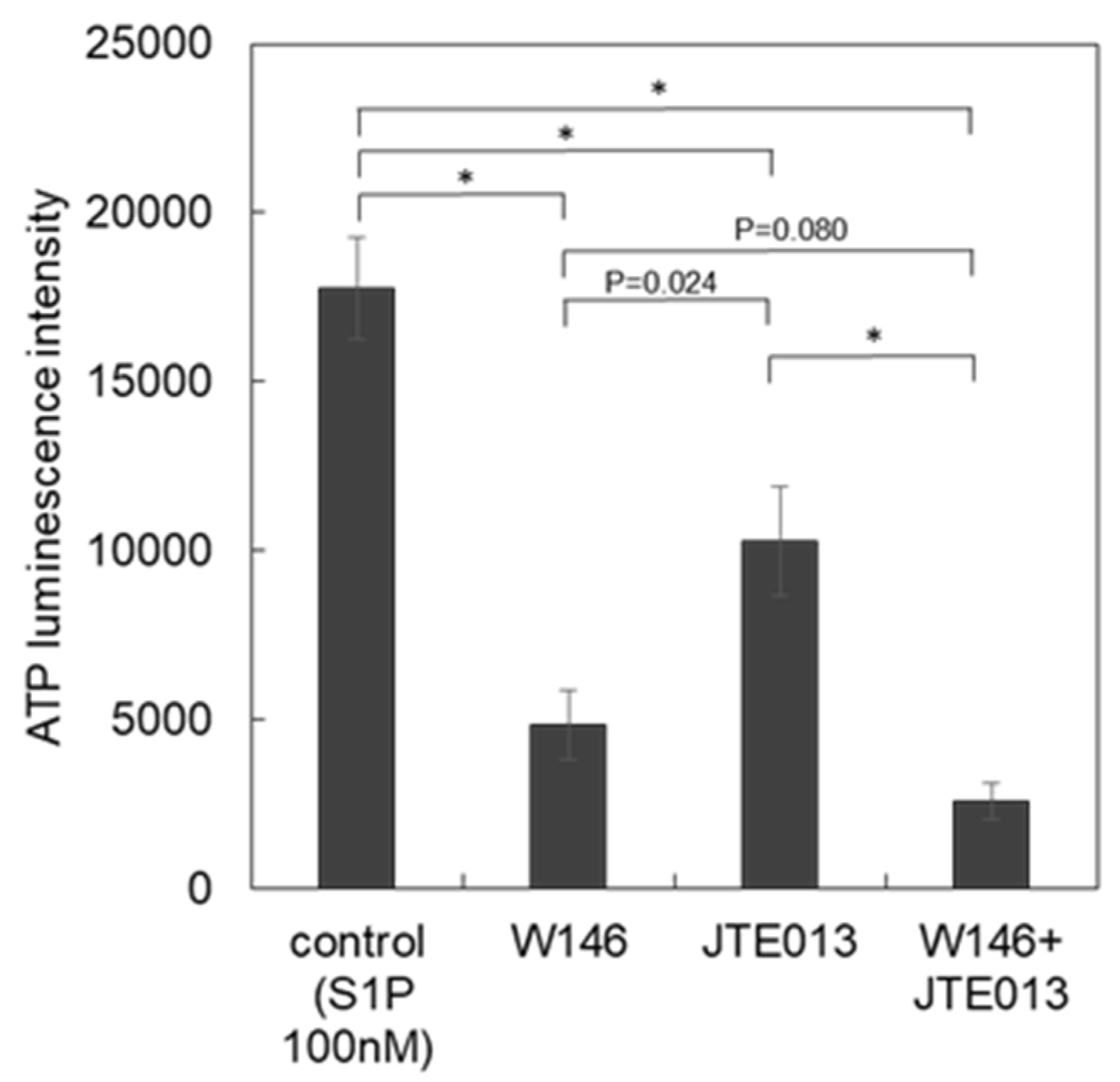
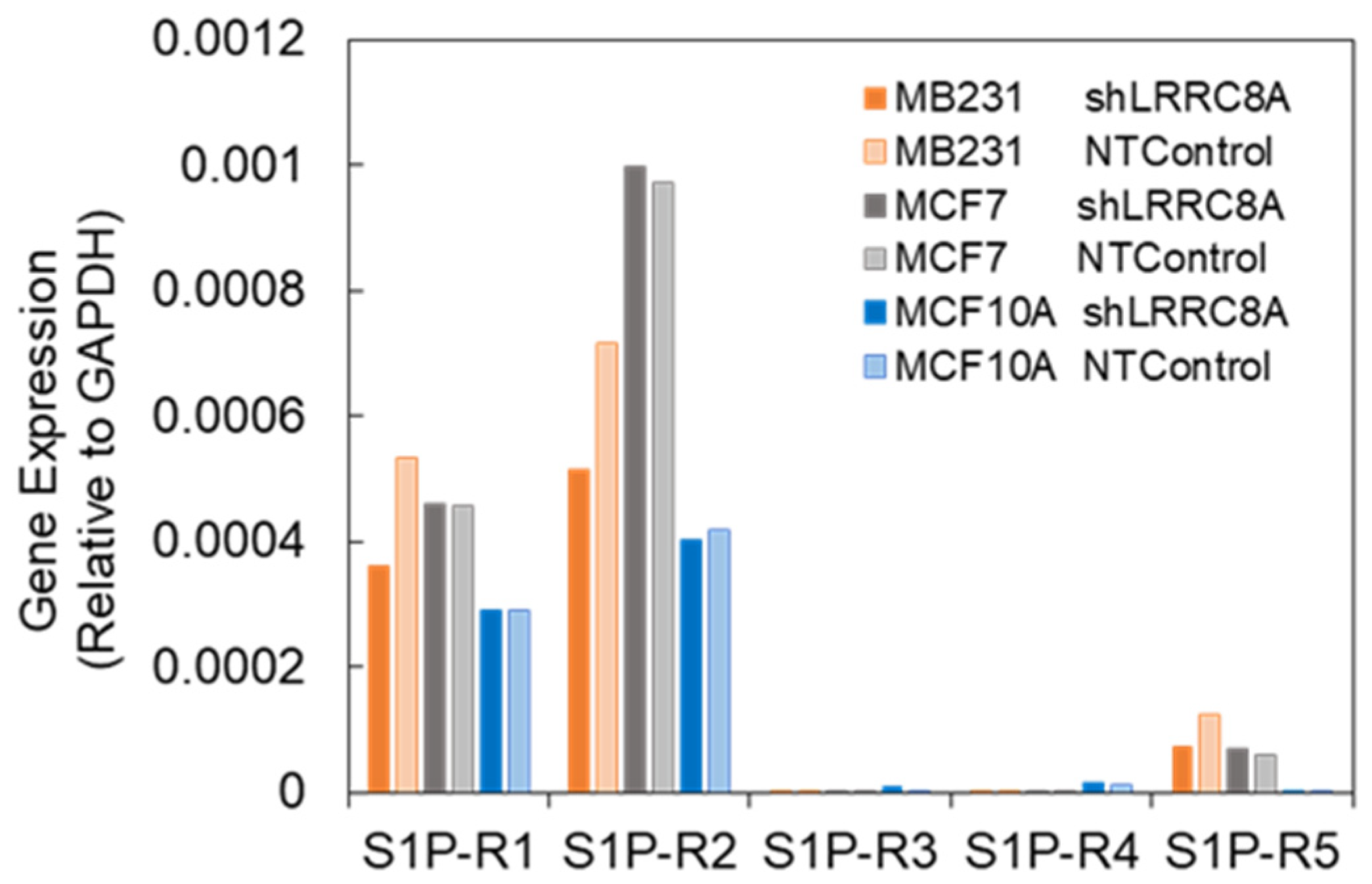
3.3. Subtypes of S1P Receptors
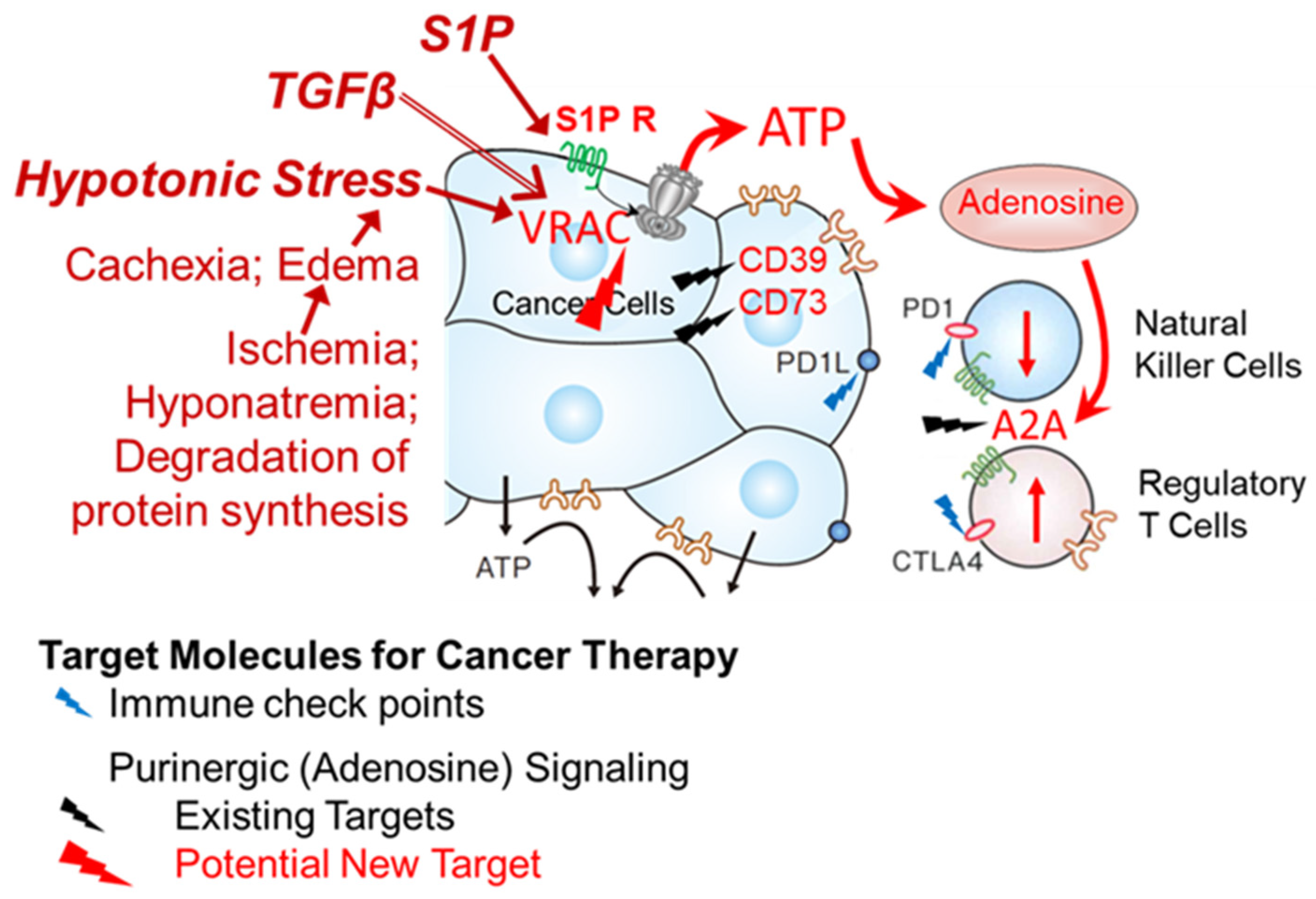
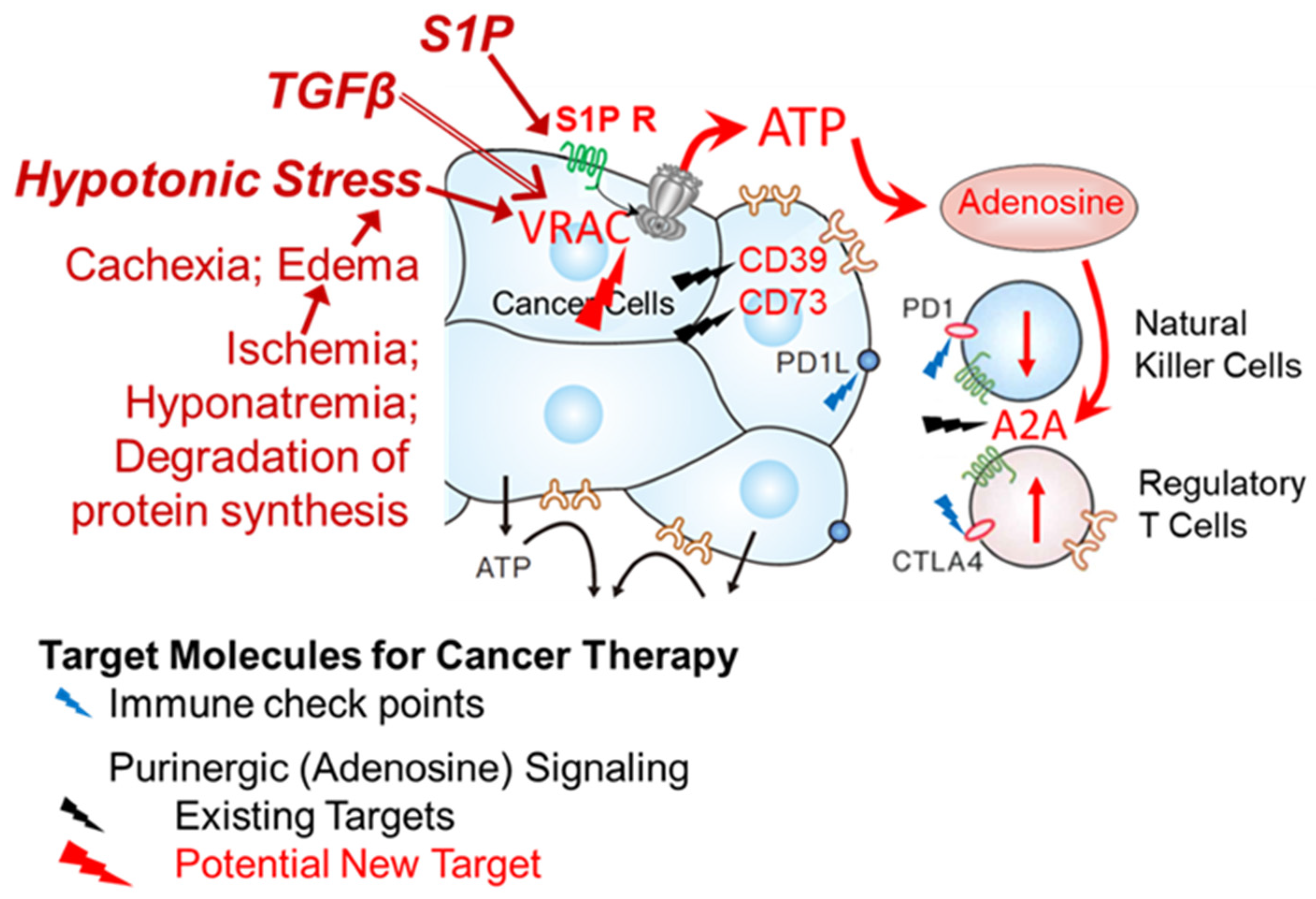
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased Level of Extracellular ATP at Tumor Sites: In Vivo Imaging with Plasma Membrane Luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef]
- Di Virgilio, F. Purines, Purinergic Receptors, and Cancer. Cancer Res. 2012, 72, 5441–5447. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef]
- Furuya, K.; Takahashi, Y.; Hirata, H.; Kobayashi, T.; Samsonov, M.; Sokabe, M. Hypo-osmotic Stress Induces ATP Release via Volume-regulated Anion Channels in Undifferentiated Mammary Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Doulberis, M.; Angelopoulou, K.; Kaldrymidou, E.; Tsingotjidou, A.; Abas, Z.; Erdman, S.E.; Poutahidis, T. Cholera-toxin suppresses carcinogenesis in a mouse model of inflammation-driven sporadic colon cancer. Carcinogenesis 2015, 36, 280–290. [Google Scholar] [CrossRef] [Green Version]
- Bharati, K.; Ganguly, N.K. Cholera toxin: A paradigm of a multifunctional protein. Indian J. Med. Res. 2011, 133, 179–187. [Google Scholar]
- Moses, H.; Barcellos-Hoff, M.H. TGF-β Biology in Mammary Development and Breast Cancer. Cold Spring Harb. Perspect. Biol. 2011, 3, a003277. [Google Scholar] [CrossRef] [Green Version]
- Syed, V. TGF-β Signaling in Cancer—A double-edged sword. J. Cell Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef]
- Lutter, D.; Ullrich, F.; Lueck, J.C.; Kempa, S.; Jentsch, T.J. Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J. Cell Sci. 2017, 130, 1122–1133. [Google Scholar] [CrossRef] [Green Version]
- Schober, A.L.; Wilson, C.S.; Mong, A.A. Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J. Physiol. 2017, 595, 6939–6951. [Google Scholar] [CrossRef]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef]
- Gaitán-Peñas, H.; Gradogna, A.; Laparra-Cuervo, L.; Solsona, C.; Fernández-Dueñas, V.; Barrallo-Gimeno, A.; Ciruela, F.; Lakadamyali, M.; Pusch, M.; Estévez, R. Investigation of LRRC8-Mediated Volume-Regulated Anion Currents in Xenopus Oocytes. Biophys. J. 2016, 111, 1429–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, T.J.; Lutter, D.; Planells-Cases, R.; Ullrich, F.; Voss, F.K. VRAC: Molecular identification as LRRC8 heteromers with differential functions. Pflügers Arch. Eur. J. Physiol. 2016, 468, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Islam, M.R.; Tsiferova, N.A.; Okada, Y.; Sabirov, R.Z. Specific and essential but not sufficient roles of LRRC8A in the activity of volume-sensitive outwardly rectifying anion channel (VSOR). Channels 2017, 11, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Sato, K.; Numata, T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. 2009, 587, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Okada, Y.; Nilius, B. Biophysics and Physiology of the Volume-Regulated Anion Channel (VRAC)/Volume-Sensitive Outwardly Rectifying Anion Channel (VSOR). Pflügers Arch. Eur. J. Physiol. 2016, 468, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Ginzberg, M.B.; Kafri, R.; Kirschner, M. On being the right (cell) size. Science 2015, 348, 1245075. [Google Scholar] [CrossRef] [Green Version]
- Rosen, H.; Gonzalez-Cabrera, P.J.; Sanna, M.G.; Brown, S. Sphingosine 1-phosphate receptor signaling. Ann. Rev. Biochem. 2009, 78, 743–768. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Takuwa, Y.; Okamoto, Y.; Yoshioka, K.; Takuwa, N. Sphingosine-1-phosphate signaling in physiology and diseases. BioFactors 2012, 38, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, P.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-Phosphate Produced by Sphingosine Kinase 1 Promotes Breast Cancer Progression by Stimulating Angiogenesis and Lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef] [Green Version]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci. 2018, 109, 3671–3678. [Google Scholar] [CrossRef] [Green Version]
- Furuya, K.; Sokabe, M.; Grygorczyk, R. Real-time Luminescence Imaging of Cellular ATP Release. Methods 2014, 66, 330–344. [Google Scholar] [CrossRef]
- Hirata, H.; Samsonov, M.; Sokabe, M. Actomyosin contractility provokes contact inhibition in E-cadherin-ligated keratinocytes. Sci. Rep. 2017, 7, 46326. [Google Scholar] [CrossRef]
- Burow, P.; Klapperstück, M.; Markwardt, F. Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflügers Arch. Eur. J. Physiol. 2015, 467, 1215–1226. [Google Scholar] [CrossRef]
- Zahiri, D.; Burow, P.; Großmann, C.; Müller, C.E.; Klapperstück, M.; Markwardt, F. Sphingosine-1-phosphate induces migration of microglial cells via activation of volume-sensitive anion channels, ATP secretion and activation of purinergic receptors. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2021, 1868, 118915. [Google Scholar] [CrossRef]
- Okada, Y.; Okada, T.; Sato-Numata, K.; Islam, M.R.; Ando-Akatsuka, Y.; Numata, T.; Kubo, M.; Shimizu, T.; Kurbannazarova, R.S.; Marunaka, Y.; et al. Cell Volume-Activated and Volume-Correlated Anion Channels in Mammalian Cells: Their Biophysical, Molecular, and Pharmacological Properties. Pharmacol. Rev. 2019, 71, 49–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, S.; Remigante, A.; Zuccolini, P.; Barbieri, R.; Ferrera, L.; Picco, C.; Gavazzo, P.; Pusch, M. Mechanisms of Activation of LRRC8 Volume Regulated Anion Channels. Cell Physiol. Biochem. 2021, 55, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Strange, K.; Yamada, T.; Denton, J.S. A 30-year journey from volume-regulated anion currents to molecular structure of the LRRC8 channel. J. Gen. Physiol. 2019, 151, 100–117. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y. Volume expansion-sensing outward-rectifier Cl− channel: Fresh start to the molecular identity and volume sensor. Am. J. Physiol.-Cell Physiol. 1997, 273, C755–C789. [Google Scholar] [CrossRef]
- Benedetto, R.; Sirianant, L.; Pankonien, I.; Wanitchakool, P.; Ousingsawat, J.; Cabrita, I.; Schreiber, R.; Amaral, M.; Kunzelmann, K. Relationship between TMEM16A/anoctamin 1 and LRRC8A. Pflügers Arch. Eur. J. Physiol. 2016, 468, 1751–1763. [Google Scholar] [CrossRef]
- Gunasekar, S.K.; Xie, L.; Sah, R. SWELL signalling in adipocytes: Can fat ‘feel’ fat? Adipocyte 2019, 8, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Dunn, P.J.; Salm, E.J.; Tomita, S. ABC transporters control ATP release through cholesterol-dependent volume-regulated anion channel activity. J. Biol. Chem. 2020, 295, 5192–5203. [Google Scholar] [CrossRef] [Green Version]
- Cailleau, R.; Young, R.; Olive, M.; Reeves, W.J., Jr. Breast Tumor Cell Lines from Pleural Effusions. J. Natl. Cancer Inst. 1974, 53, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Soule, H.D.; Vazquez, J.; Long, A.; Albert, S.; Brennan, M. A Human Cell Line From a Pleural Effusion Derived From a Breast Carcinoma. J. Natl. Cancer Inst. 1973, 51, 1409–1416. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Monville, F.; Finetti, P.; Adélaïde, J.; Cervera, N.; Fekairi, S.; Xerri, L.; Jacquemier, J.; Birnbaum, D.; et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006, 25, 2273–2284. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, P.; Sukocheva, O.A.; Wang, T.; Mayne, G.C.; Watson, D.I.; Hussey, D.J. Effects of chemotherapy agents on Sphingosine-1-Phosphate receptors expression in MCF-7 mammary cancer cells. Biomed. Pharmacother. 2016, 81, 218–224. [Google Scholar] [CrossRef]
- Calis, I.U.; Cosan, D.T.; Mutlu, F. Effects of S1P1 and S1P3 in ER+ and ER− Breast Cancer Cells. Anticancer Res. 2017, 37, 5469–5475. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Haleem, A.M.; Lewis, N.E.; Jamshidi, N.; Mineta, K.; Gao, X.; Gojobori, T. The Emerging Facets of Non-Cancerous Warburg Effect. Front. Endocrinol. 2017, 8, 279. [Google Scholar] [CrossRef] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, V.J.; Brown, J.K.; Maybin, J.; Saunders, P.T.K.; Duncan, W.C.; Horne, A.W. Transforming Growth Factor-β Induced Warburg-Like Metabolic Reprogramming May Underpin the Development of Peritoneal Endometriosis. J. Clin. Endocrinol. Metab. 2014, 99, 3450–3459. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1α Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017, 18, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti–PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RT-qPCR Primers | |||
| Target | Sequence(5′-3′) | Oligo Name (TaKaRa) | |
| S1PR1 | F | GGCTATGTTGAGTACGTAGGCTGTG | HA258013-F |
| R | TCCCGCTTACATGGAAACTTTG | HA258013-R | |
| S1PR2 | F | ATGCAAGGCGCAACTTGAGA | HA264697-F |
| R | CTGCAGGTGTGGAGCTGAGAA | HA264697-R | |
| S1PR3 | F | AAAGCCCTAACCTTGAAGTTTGGAA | HA178037-F |
| R | CAATCCCATCACATGGACTACGA | HA178037-R | |
| S1PR4 | F | TCGCTCAGCTTTCGGATG | HA139390-F |
| R | ATCCACACGCAAGACTGCAA | HA139390-R | |
| S1PR5 | F | AACCGGCTGCAGACTGACAC | HA203213-F |
| R | TGCACCTTTGGCTGCATTTC | HA203213-R | |
| GAPDH | F | GCACCGTCAAGGCTGAGAAC | HA067812-F |
| R | TGGTGAAGACGCCAGTGGA | HA067812-R | |
| shRNA Target Sequences | |||
| LRRC8A | GAGCGCAGTATTTGGATAA | shA | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furuya, K.; Hirata, H.; Kobayashi, T.; Sokabe, M. Sphingosine-1-Phosphate Induces ATP Release via Volume-Regulated Anion Channels in Breast Cell Lines. Life 2021, 11, 851. https://doi.org/10.3390/life11080851
Furuya K, Hirata H, Kobayashi T, Sokabe M. Sphingosine-1-Phosphate Induces ATP Release via Volume-Regulated Anion Channels in Breast Cell Lines. Life. 2021; 11(8):851. https://doi.org/10.3390/life11080851
Chicago/Turabian StyleFuruya, Kishio, Hiroaki Hirata, Takeshi Kobayashi, and Masahiro Sokabe. 2021. "Sphingosine-1-Phosphate Induces ATP Release via Volume-Regulated Anion Channels in Breast Cell Lines" Life 11, no. 8: 851. https://doi.org/10.3390/life11080851
APA StyleFuruya, K., Hirata, H., Kobayashi, T., & Sokabe, M. (2021). Sphingosine-1-Phosphate Induces ATP Release via Volume-Regulated Anion Channels in Breast Cell Lines. Life, 11(8), 851. https://doi.org/10.3390/life11080851