
Differential Mucosal Microbiome Profiles across Stages of Human Colorectal Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Library Preparation
2.2. OTU Picking and Analysis of 16S rRNA Amplicons
2.3. Analyses of Differential Abundances of Gut Mucosal Microbiota during CRC Development
2.4. Co-Occurrence and Co-Excluding Correlation Network Analysis
2.5. Statistical Analysis
3. Results
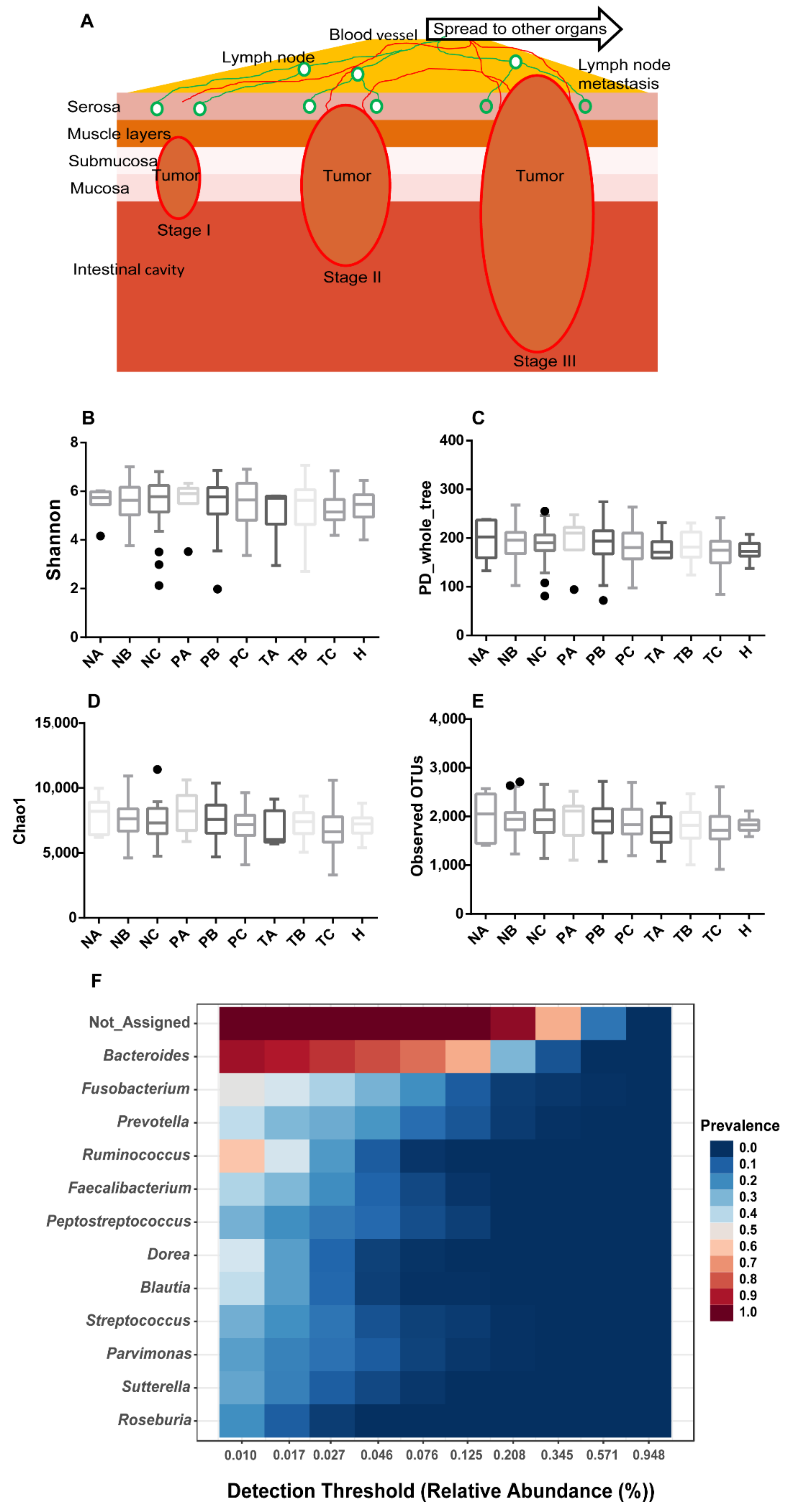
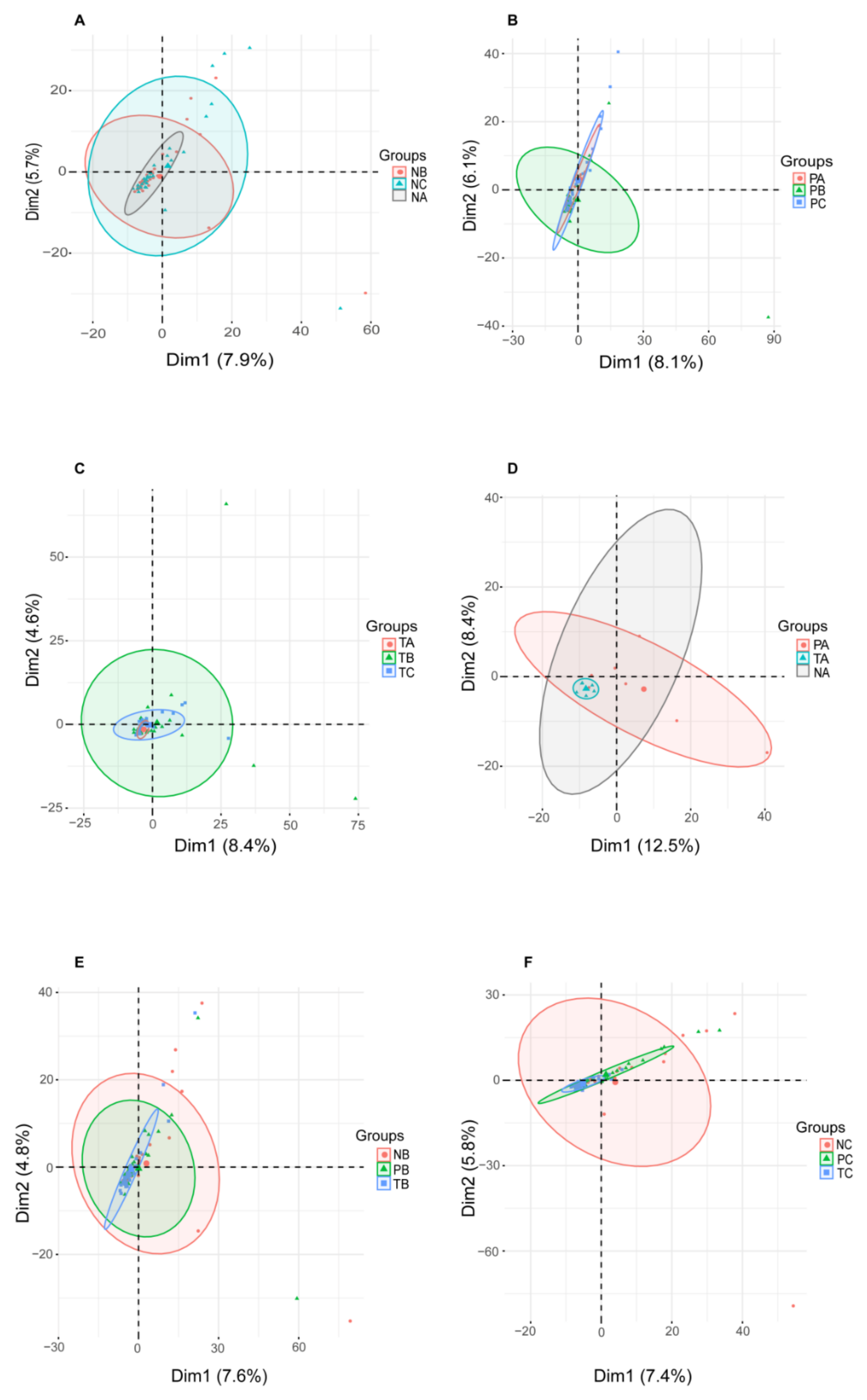
3.1. Gut Mucosal Microbiome Profiles during CRC Evolvement
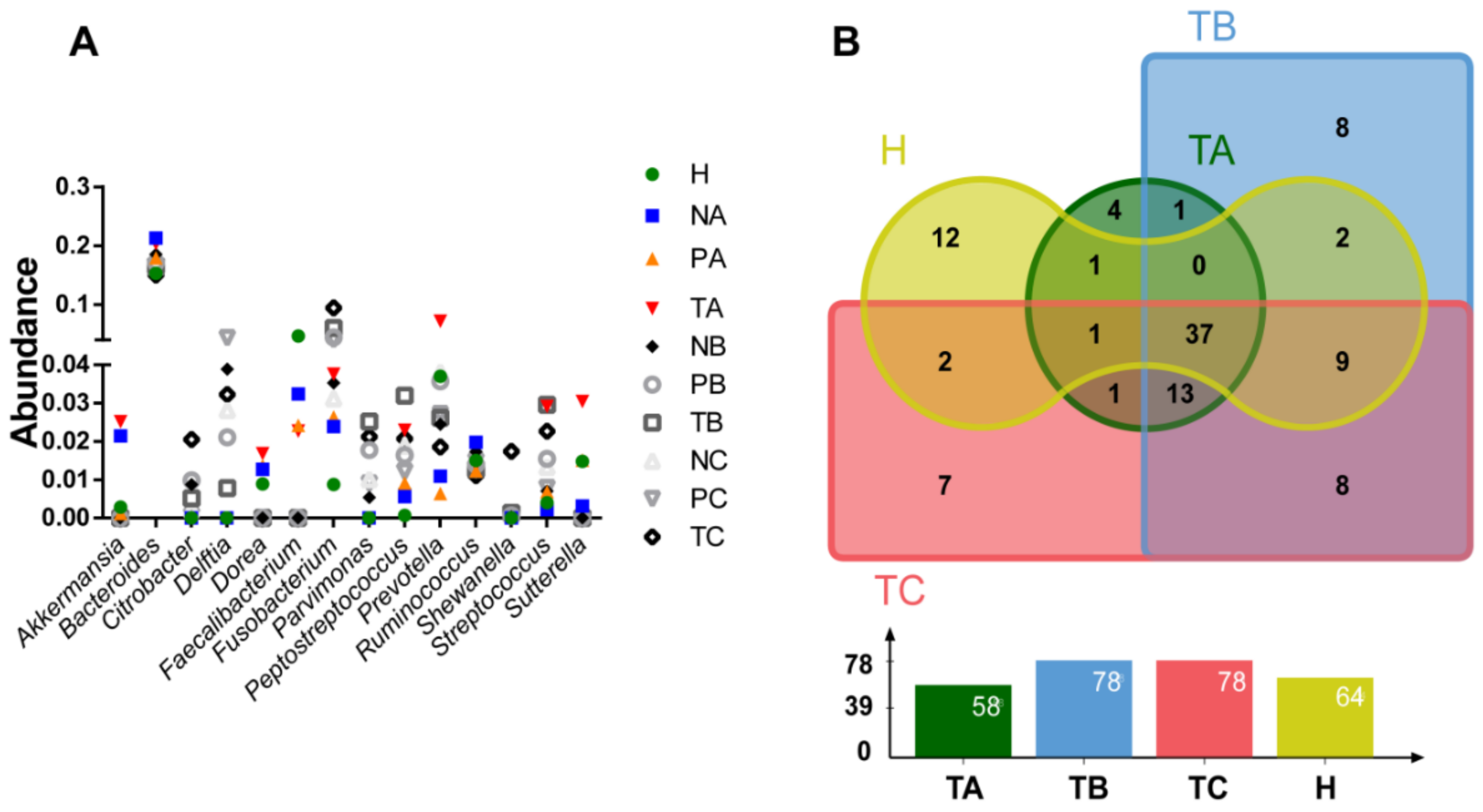
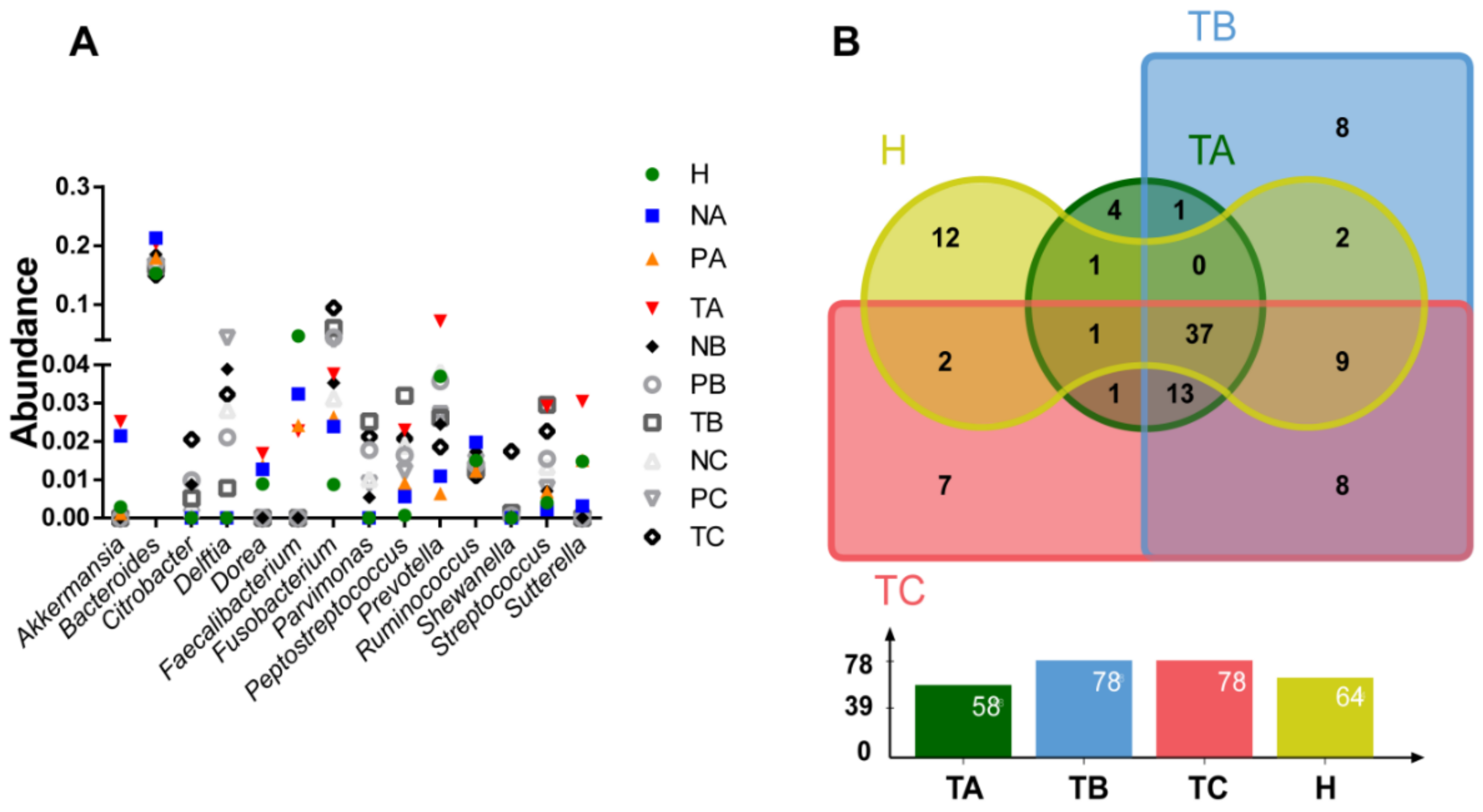
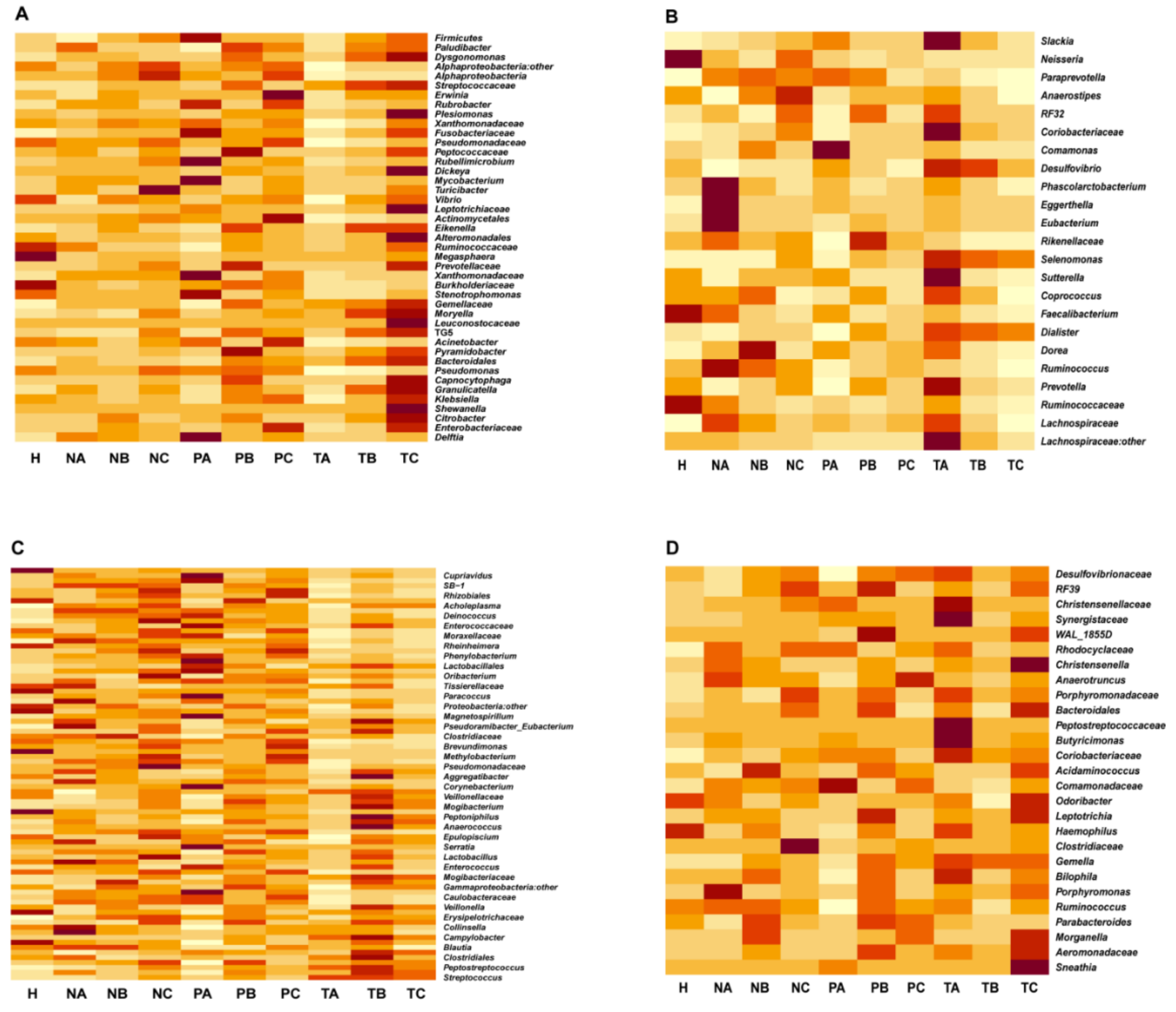
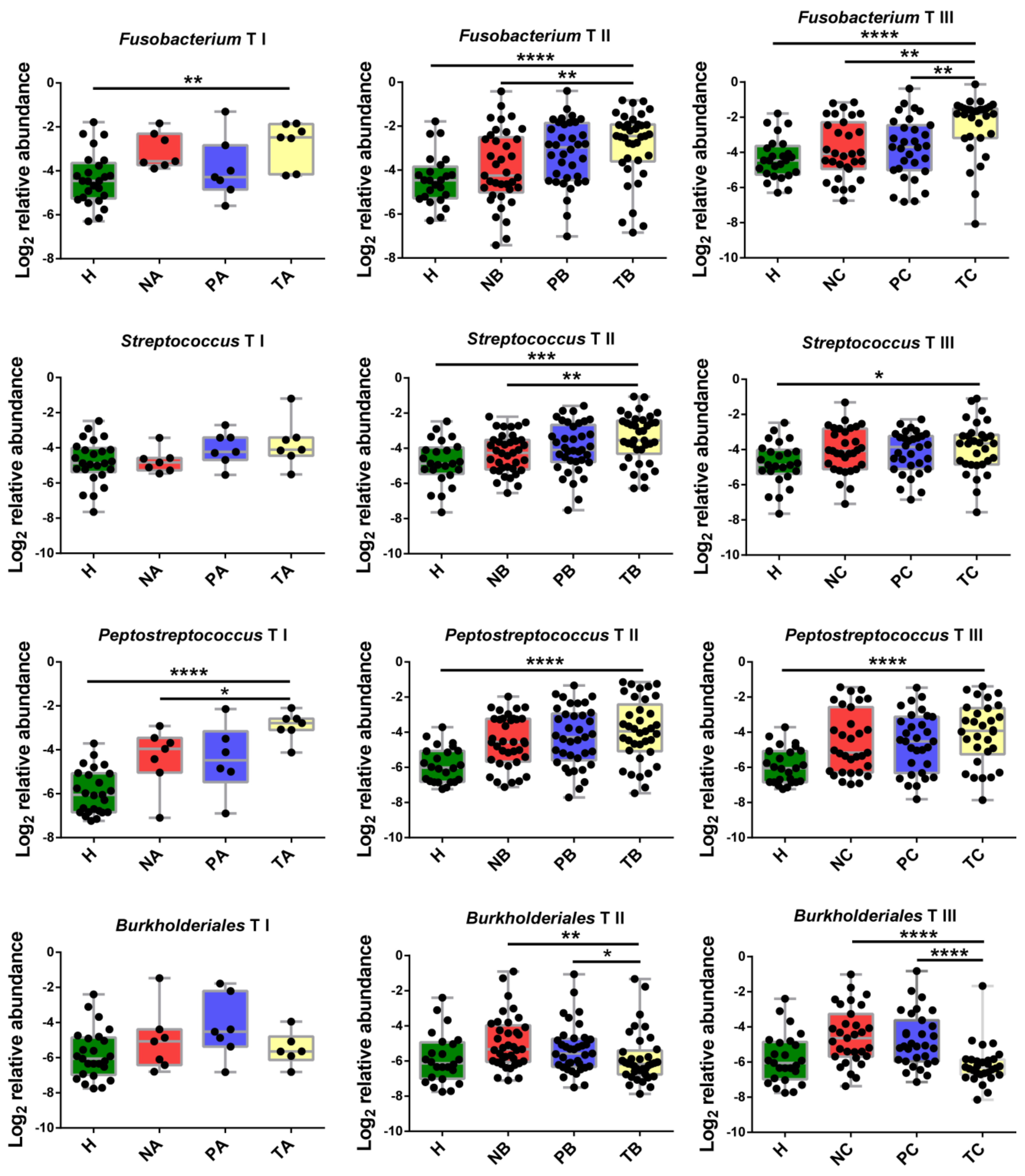
3.2. Genera with Top Abundances Showing Differential Abundances during CRC Progression
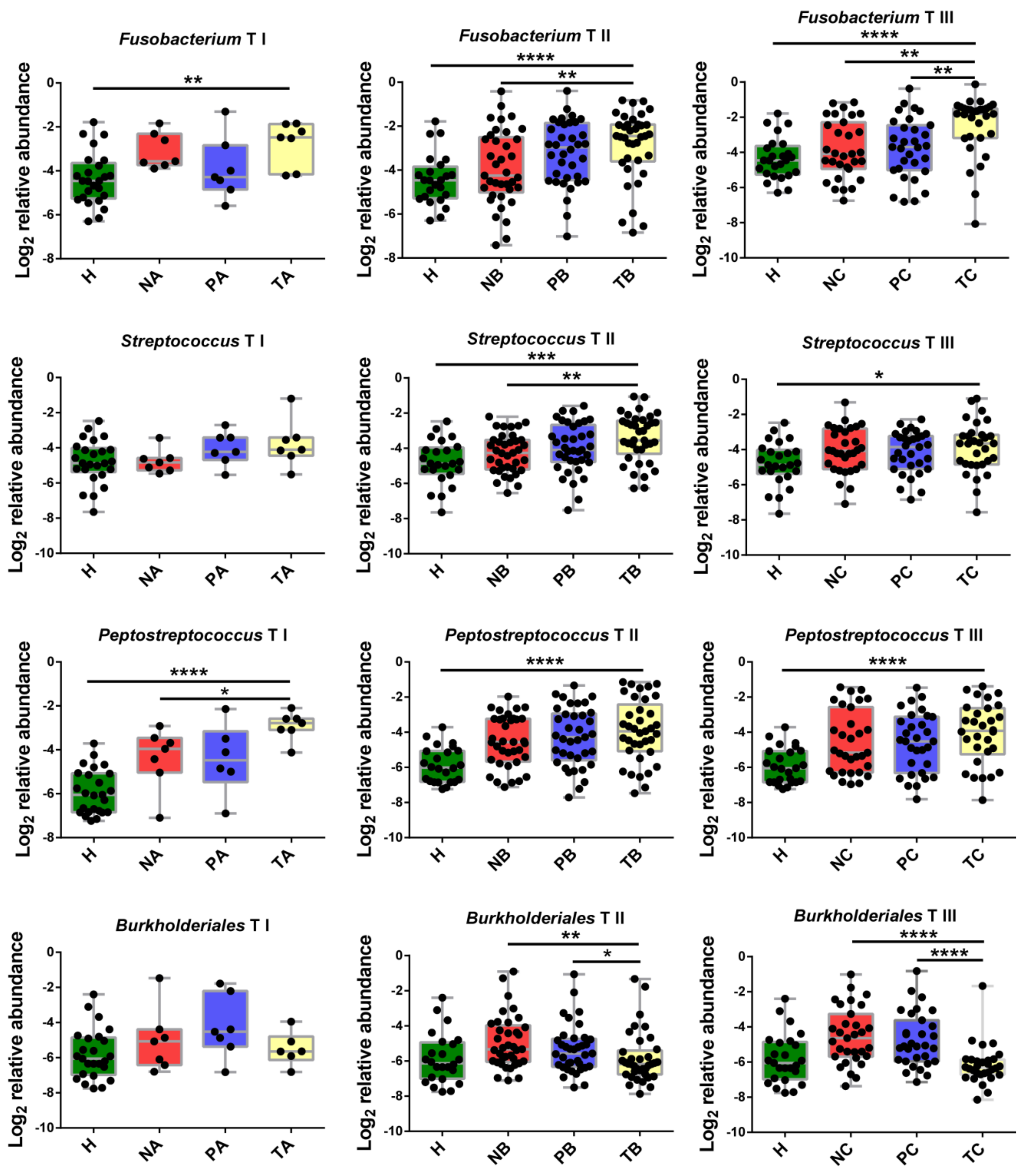
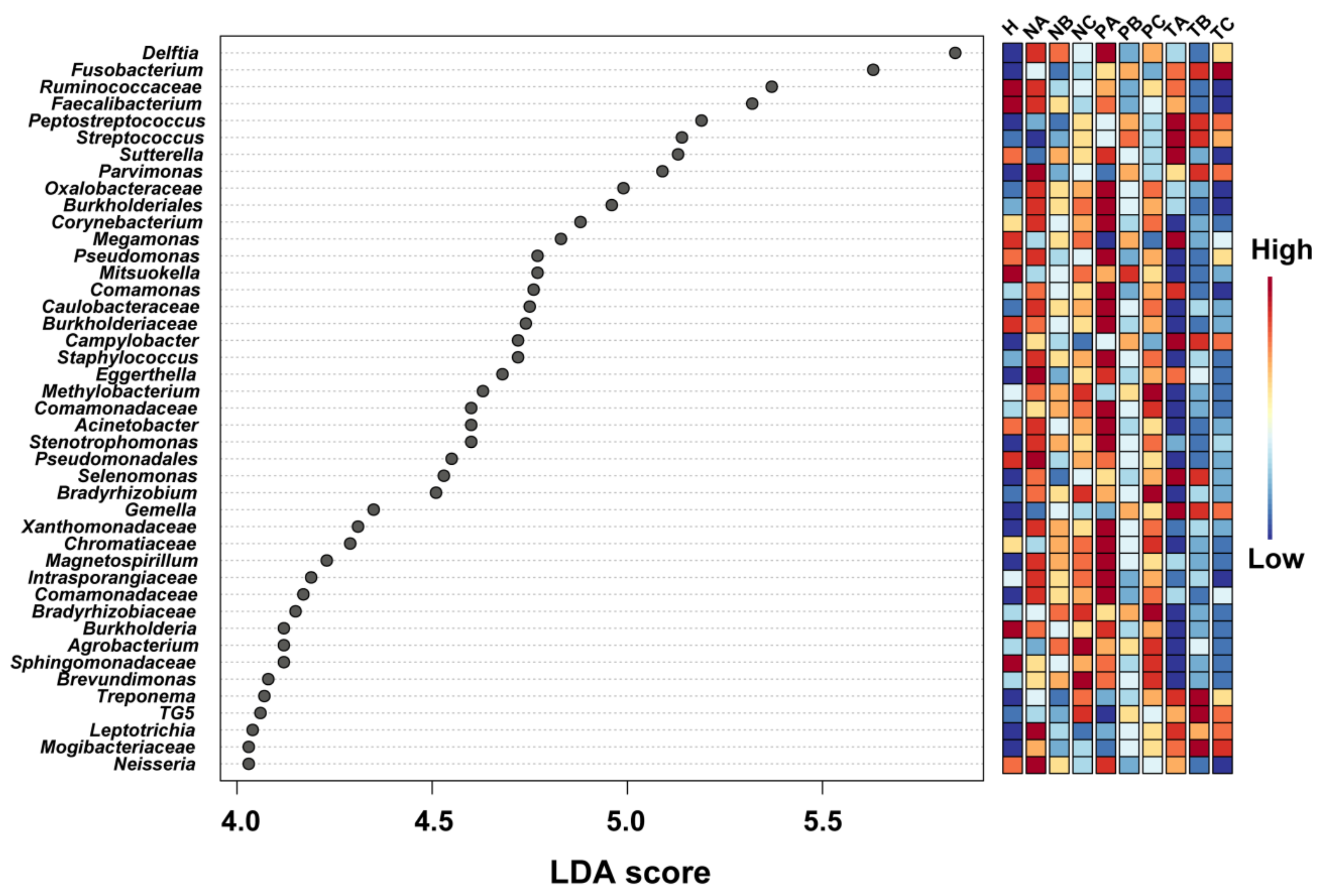
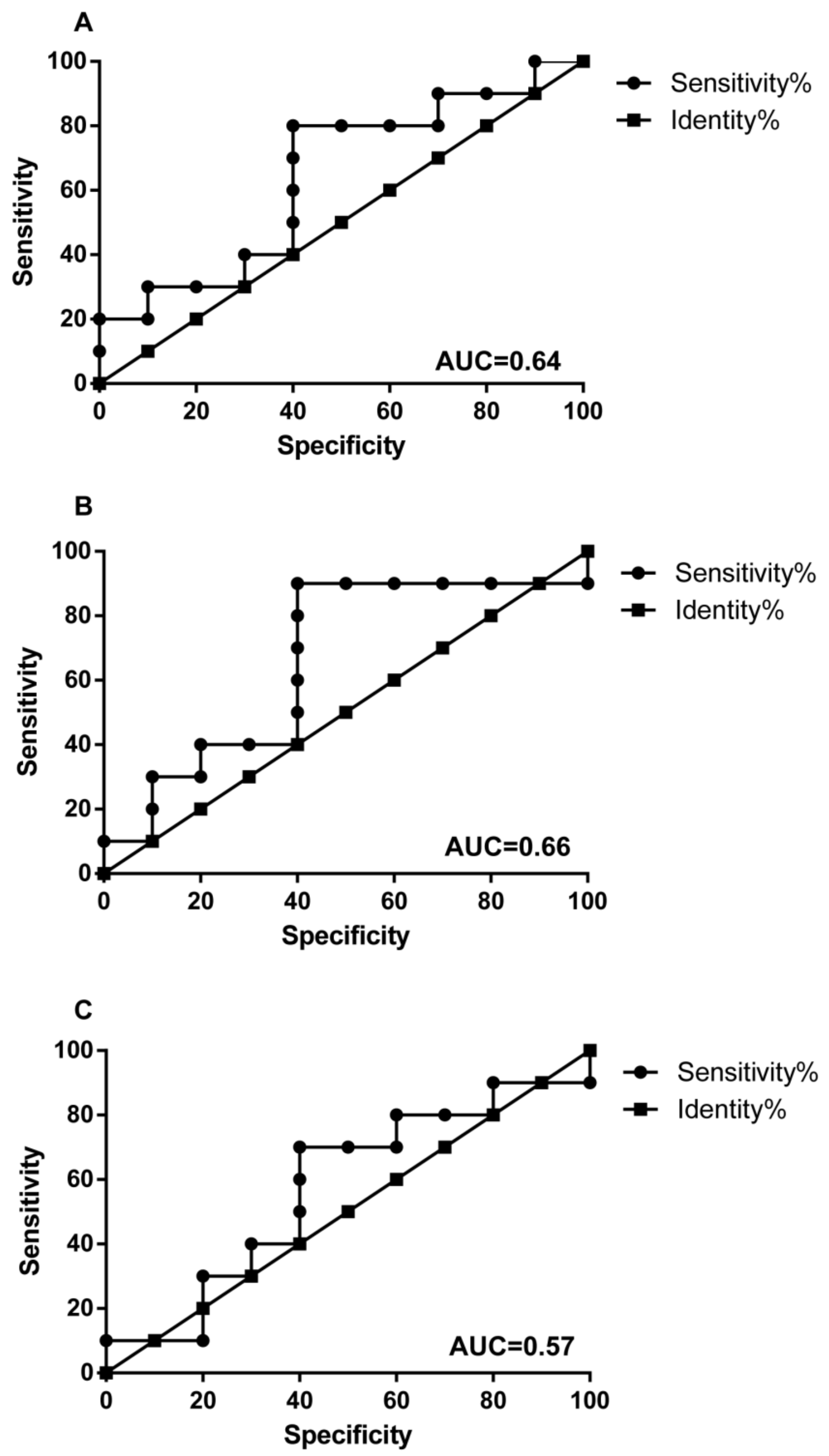
3.3. Altered Microbial Signatures in Different CRC Stages
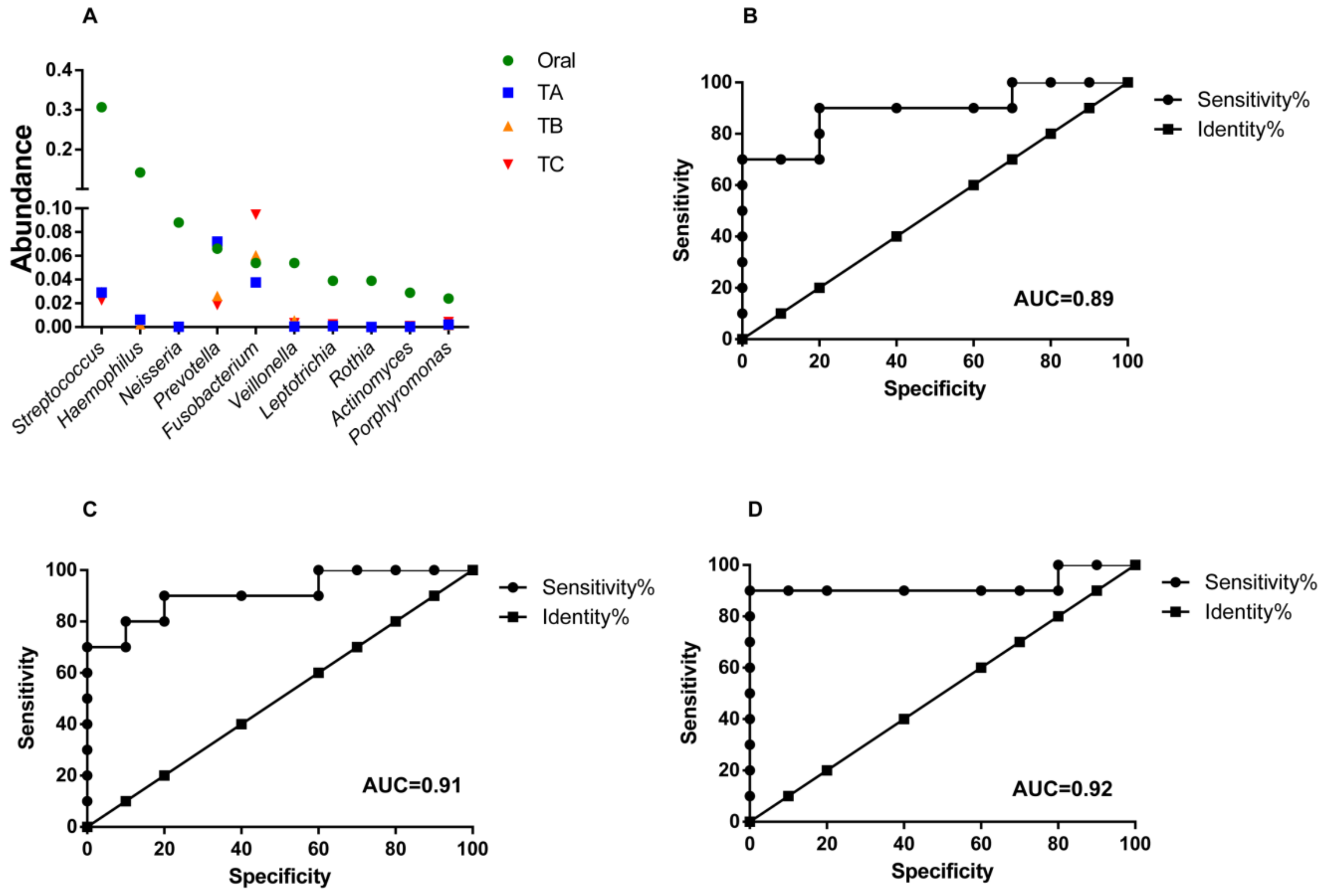
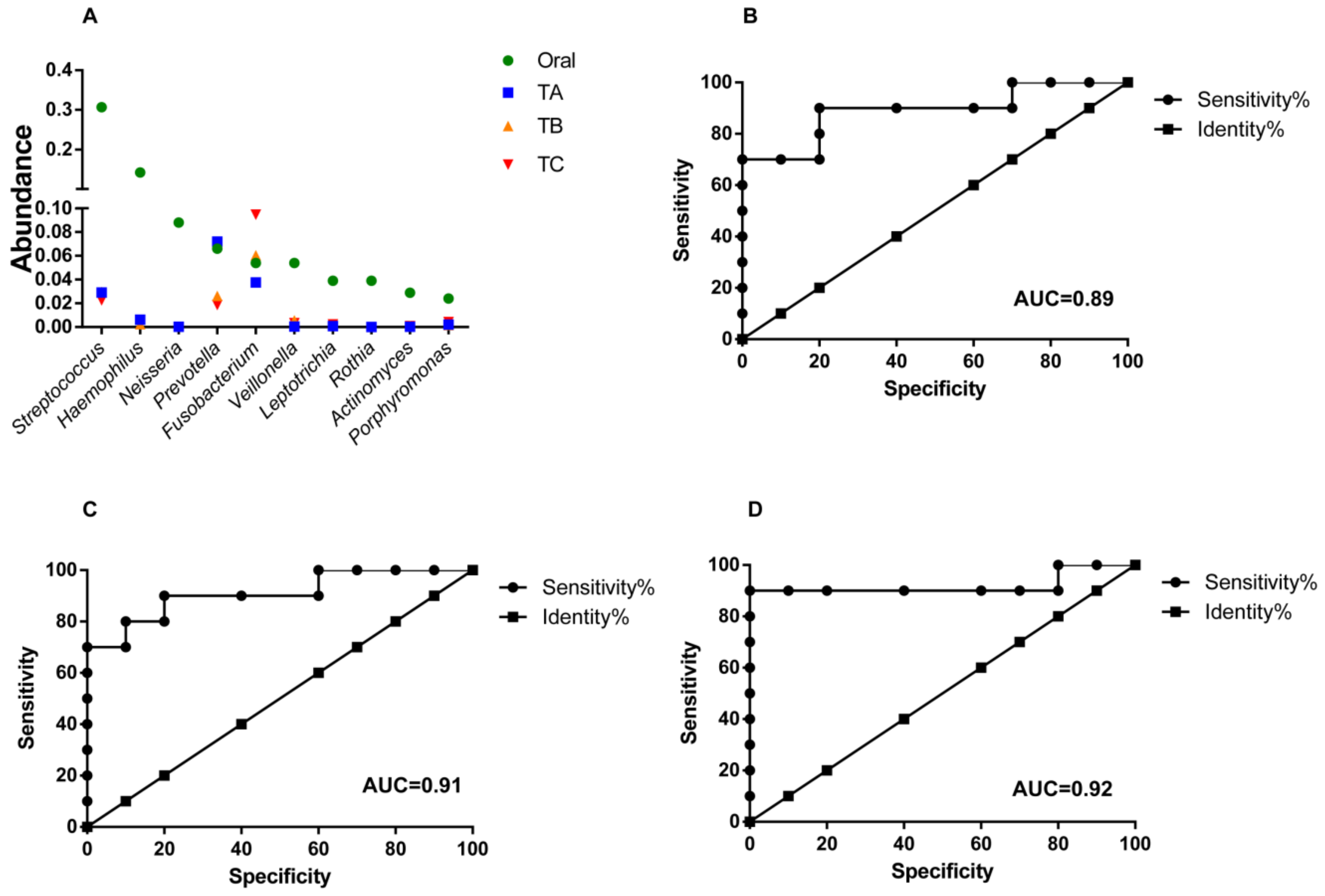
3.4. Oral Microbes Compared to Gut Mucosal Microbes in CRC Stages
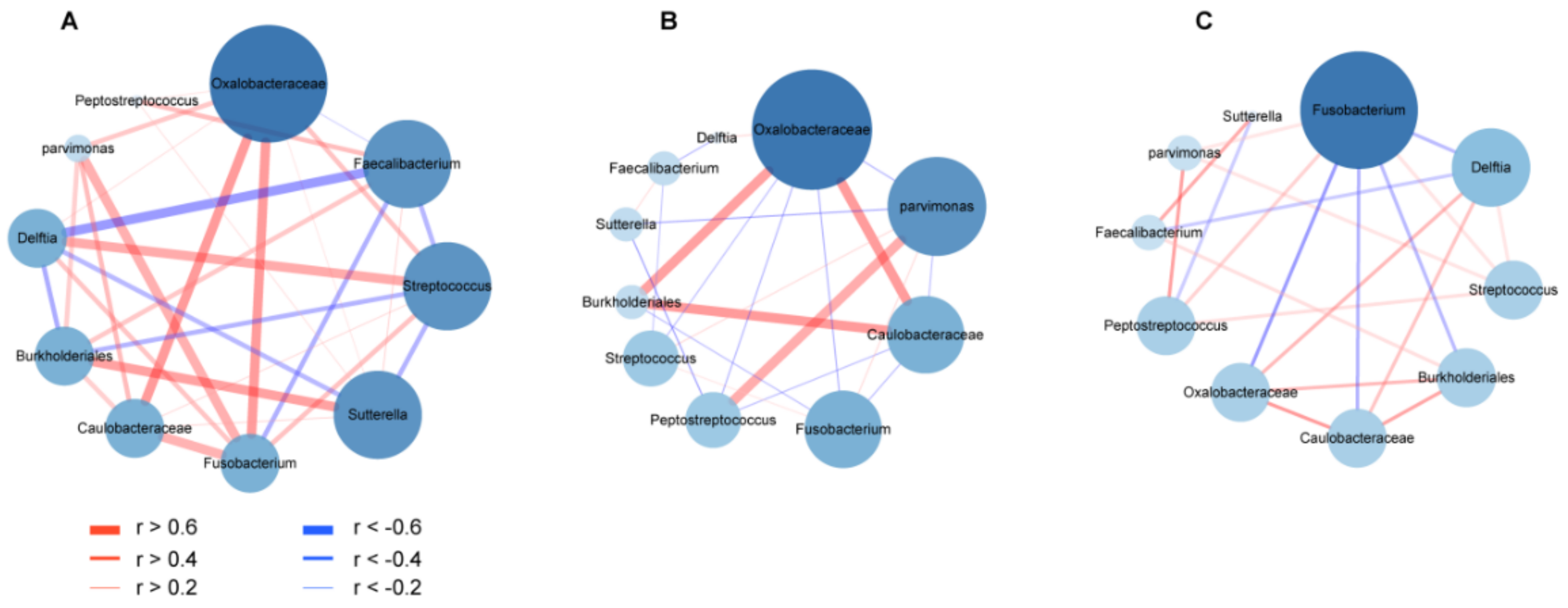
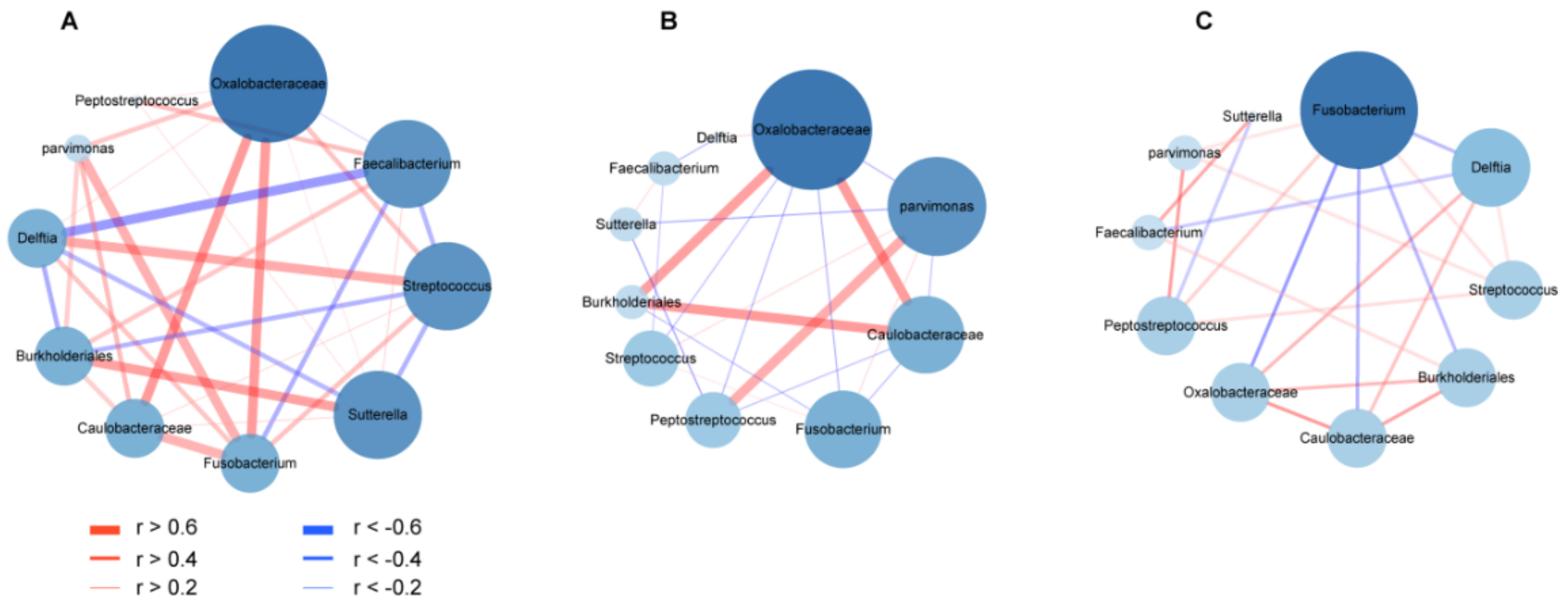
3.5. Co-Occurrence and Co-Excluding Alteration of Gut Microbes during CRC Development
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irrazabal, T.; Belcheva, A.; Girardin, S.E.; Martin, A.; Philpott, D.J. The multifaceted role of the intestinal microbiota in colon cancer. Mol. Cell 2014, 54, 309–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef]
- Zhang, S.; Cai, S.; Ma, Y. Association between Fusobacterium nucleatum and colorectal cancer: Progress and future directions. J. Cancer 2018, 9, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Wong, C.C.; Tong, L.; Chu, E.S.H.; Szeto, C.H.; Go, M.Y.Y.; Coker, O.O.; Chan, A.W.H.; Chan, F.K.; Sung, J.J.Y.; et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat. Microbiol. 2019, 4, 12. [Google Scholar] [CrossRef]
- Rhee, K.-J.; Wu, S.; Wu, X.; Huso, D.L.; Karim, B.; Franco, A.A.; Rabizadeh, S.; Golub, J.E.; Mathews, L.E.; Shin, J.; et al. Induction of Persistent Colitis by a Human Commensal, Enterotoxigenic Bacteroides fragilis, in Wild-Type C57BL/6 Mice. Infect. Immun. 2009, 77, 1708–1718. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wan, X.; Wu, X.; Zhang, C.; Liu, J.; Hou, S. Eubacterium rectale contributes to colorectal cancer initiation via promoting colitis. Gut Pathog. 2021, 13, 1–11. [Google Scholar] [CrossRef]
- Dong, X.; Pan, P.; Zheng, D.-W.; Bao, P.; Zeng, X.; Zhang, X.-Z. Bioinorganic hybrid bacteriophage for modulation of intestinal microbiota to remodel tumor-immune microenvironment against colorectal cancer. Sci. Adv. 2020, 6, eaba1590. [Google Scholar] [CrossRef]
- Boyaval, F.; Van Zeijl, R.; Dalebout, H.; Holst, S.; Van Pelt, G.W.; Fariña-Sarasqueta, A.; Mesker, W.E.; Tollenaar, R.; Morreau, H.; Wuhrer, M.; et al. N-glycomic signature of stage II colorectal cancer and its association with the tumor microenvironment. Mol. Cell. Proteom. 2021, 20, 100057. [Google Scholar] [CrossRef]
- Kasprzak, A. The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia. Int. J. Mol. Sci. 2021, 22, 1565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chang, Y.; Zheng, Q.; Hu, C.; Jia, W. Altered intestinal microbiota associated with colorectal cancer. Front. Med. 2019, 13, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Pascual, L.M.; Cabrera-Rubio, R.; Ocon, S.; Costales, P.; Parra, A.; Suarez, A.; Moris, F.; Rodrigo, L.; Mira, A.; Collado, M.C. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J. Gastroenterol. 2015, 50, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Pasquereau-Kotula, E.; Martins, M.; Aymeric, L.; Dramsi, S. Significance of Streptococcus gallolyticus subsp. gallolyticus association with colorectal cancer. Front. Microbiol. 2018, 9, 614. [Google Scholar] [CrossRef] [PubMed]
- Boleij, A.; Muytjens, C.M.J.; Bukhari, S.I.; Cayet, N.; Glaser, P.; Hermans, P.W.M.; Swinkels, D.W.; Bolhuis, A.; Tjalsma, H. Novel clues on the specific association of Streptococcus gallolyticus subsp gallolyticus with colorectal cancer. J. Infect. Dis. 2011, 203, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Boleij, A.; Dutilh, B.E.; Kortman, G.A.M.; Roelofs, R.; Laarakkers, C.M.; Engelke, U.F.; Tjalsma, H. Bacterial responses to a simulated colon tumor microenvironment. Mol. Cell. Proteom. 2012, 11, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. Microbiome Analyst—A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hibberd, A.A.; Lyra, A.; Ouwehand, A.; Rolny, P.; Lindegren, H.; Cedgård, L.; Wettergren, Y. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017, 4, e000145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.-A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Zheng, J.; Hu, G.; Wang, J.; Huang, C.; Lou, L.; Wang, X.; Zeng, Y. Mucosal adherent bacterial profiles in patients with colorectal adenomas. Sci. Rep. 2016, 6, 26337. [Google Scholar]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef] [PubMed]
- Finotello, F.; Mastrorilli, E.; Camillo, B.D. Measuring the diversity of the human microbiota with targeted next-generation sequencing. Brief. Bioinform. 2018, 19, 679–692. [Google Scholar] [CrossRef]
- Peng, M.; Tabashsum, Z.; Patel, P.; Bernhardt, C.; Biswas, C.; Meng, J.; Biswas, D. Prevention of enteric bacterial infections and modulation of gut microbiota with conjugated linoleic acids producing Lactobacillus in mice. Gut Microbes 2020, 11, 433–452. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. SEER Cancer Statistics Review, 1975–2013; National Cancer Institute: Bethesda, MD, USA, 2016. Available online: https://seer.cancer.gov/archive/csr/1975_2013 (accessed on 18 June 2021).
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.E.C.; Moore, L.V.H. The bacteria of periodontal diseases. Periodontology 1994, 5, 66–77. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mima, K.; Sukawa, Y.; Nishihara, R.; Qian, Z.R.; Yamauchi, M.; Inamura, K.; Kim, S.A.; Masuda, A.; Nowak, J.A.; Nosho, K.; et al. Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA Oncol. 2015, 1, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casasanta, M.A.; Yoo, C.C.; Udayasuryan, B.; Sanders, B.E.; Umaña, A.; Zhang, Y.; Peng, H.; Duncan, A.J.; Wang, Y.; Li, L.; et al. Fusobacterium nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that drives colorectal cancer cell migration. Sci. Signal. 2020, 13, 641. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, A.; Kokubu, E.; Warita, T.; Ishihara, K. Synergistic biofilm formation by Parvimonas micra and Fusobacterium nucleatum. Anaerobe 2020, 62, 102100. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, L.; Zhao, R.; Long, X.; Coker, O.O.; Sung, J.J. The role of Parvimonas micra in intestinal tumorigenesis in germ-free and conventional APCmin/+ mice. J. Clin. Oncol. 2019, 37, 531. [Google Scholar] [CrossRef]
- Taylor, J.C.; Gao, X.; Xu, J.; Holder, M.; Petrosino, J.; Kumar, R.; Liu, W.; Höök, M.; MacKenzie, C.; Hillhouse, A.; et al. A type VII secretion system of Streptococcus gallolyticus subsp. gallolyticus contributes to gut colonization and the development of colon tumors. PLoS Pathog. 2020, 17, e1009182. [Google Scholar]
- Aymeric, L.; Donnadieu, F.; Mulet, C.; du Merle, L.; Nigro, G.; Saffarian, A.; Bérard, M.; Poyart, C.; Robine, S.; Regnault, B.; et al. Colorectal cancer specific conditions promote Streptococcus gallolyticus gut colonization. Proc. Natl. Acad. Sci. USA 2018, 115, E283–E291. [Google Scholar] [CrossRef] [Green Version]
- Moosavi, S.M.; Sepahi, A.A.; Mousavi, S.F.; Vaziri, F.; Siadat, S.D. The effect of Faecalibacterium prausnitzii and its extracellular vesicles on the permeability of intestinal epithelial cells and expression of PPARs and ANGPTL4 in the Caco-2 cell culture model. J. Diabetes Metab. Disord. 2020, 19, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Kaakoush, N.O. Sutterella Species, IgA-degrading Bacteria in Ulcerative Colitis. Trends Microbiol. 2020, 28, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Borrellio, S.P. The Normal Flora of the Gastrointestinal Tract; Kamm, M.A., Ed.; Martin Dunitz: London, UK, 2002; pp. 1–12. [Google Scholar]
- Kolenbrander, P.E.; Palmer, R.J.; Periasamy, S.; Jakubovics, N. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Al Mamun, A.A.M.; Luong, T.T.; Hu, B.; Gu, J.; Lee, J.H.; D’Amore, M.; Das, A.; Ton-That, H. Forward Genetic Dissection of Biofilm Development by Fusobacterium nucleatum: Novel Functions of Cell Division Proteins FtsX and EnvC. mBio 2018, 9, e00360-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgård, J.E.M.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A.; et al. Colon cancer-associated Fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front. Cell. Infect. Microbiol. 2020, 10, 400. [Google Scholar] [CrossRef]
- Suzuki, H.; Matsuo, K.; Okamoto, M.; Nakata, H.; Sakamoto, H.; Fujita, M. Perioperative changes in oral bacteria number in patients undergoing cardiac valve surgery. J. Oral Sci. 2019, 61, 526–528. [Google Scholar] [CrossRef] [Green Version]
- Kawano, T.; Shigeishi, H.; Fukada, E.; Yanagisawa, T.; Kuroda, N.; Takemoto, T.; Sugiyama, M. Changes in bacterial number at different sites of oral cavity during perioperative oral care management in gastrointestinal cancer patients: Preliminary study. J. Appl. Oral Sci. 2018, 26, e20170516. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Lv, Y.; Hou, S.; Liu, Y.; Wang, Y.; Wan, X. Differential Mucosal Microbiome Profiles across Stages of Human Colorectal Cancer. Life 2021, 11, 831. https://doi.org/10.3390/life11080831
Zhang M, Lv Y, Hou S, Liu Y, Wang Y, Wan X. Differential Mucosal Microbiome Profiles across Stages of Human Colorectal Cancer. Life. 2021; 11(8):831. https://doi.org/10.3390/life11080831
Chicago/Turabian StyleZhang, Mingqing, Yongming Lv, Shaobin Hou, Yanfei Liu, Yijia Wang, and Xuehua Wan. 2021. "Differential Mucosal Microbiome Profiles across Stages of Human Colorectal Cancer" Life 11, no. 8: 831. https://doi.org/10.3390/life11080831
APA StyleZhang, M., Lv, Y., Hou, S., Liu, Y., Wang, Y., & Wan, X. (2021). Differential Mucosal Microbiome Profiles across Stages of Human Colorectal Cancer. Life, 11(8), 831. https://doi.org/10.3390/life11080831