
RiboNT: A Noise-Tolerant Predictor of Open Reading Frames from Ribosome-Protected Footprints


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Design of RiboNT
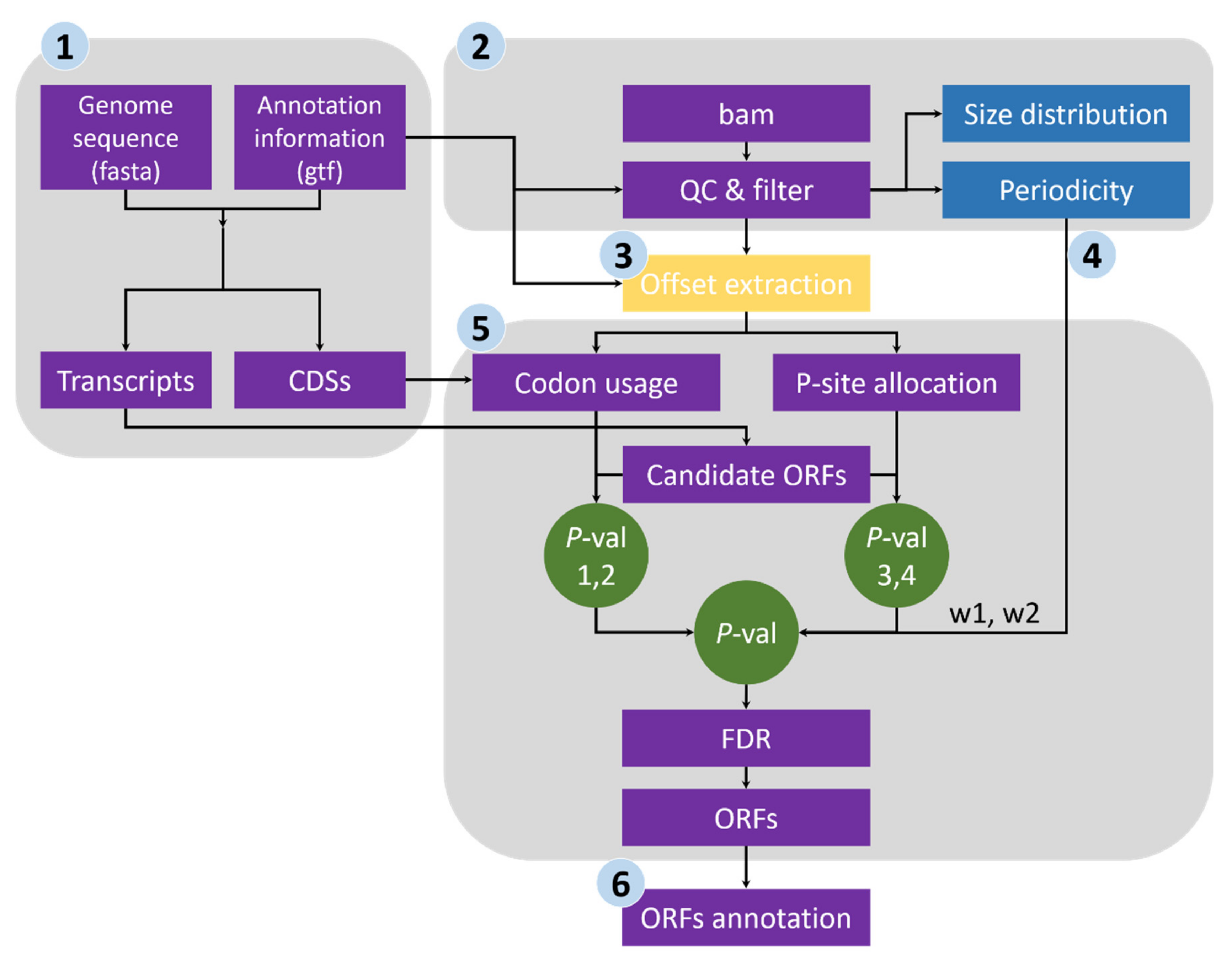
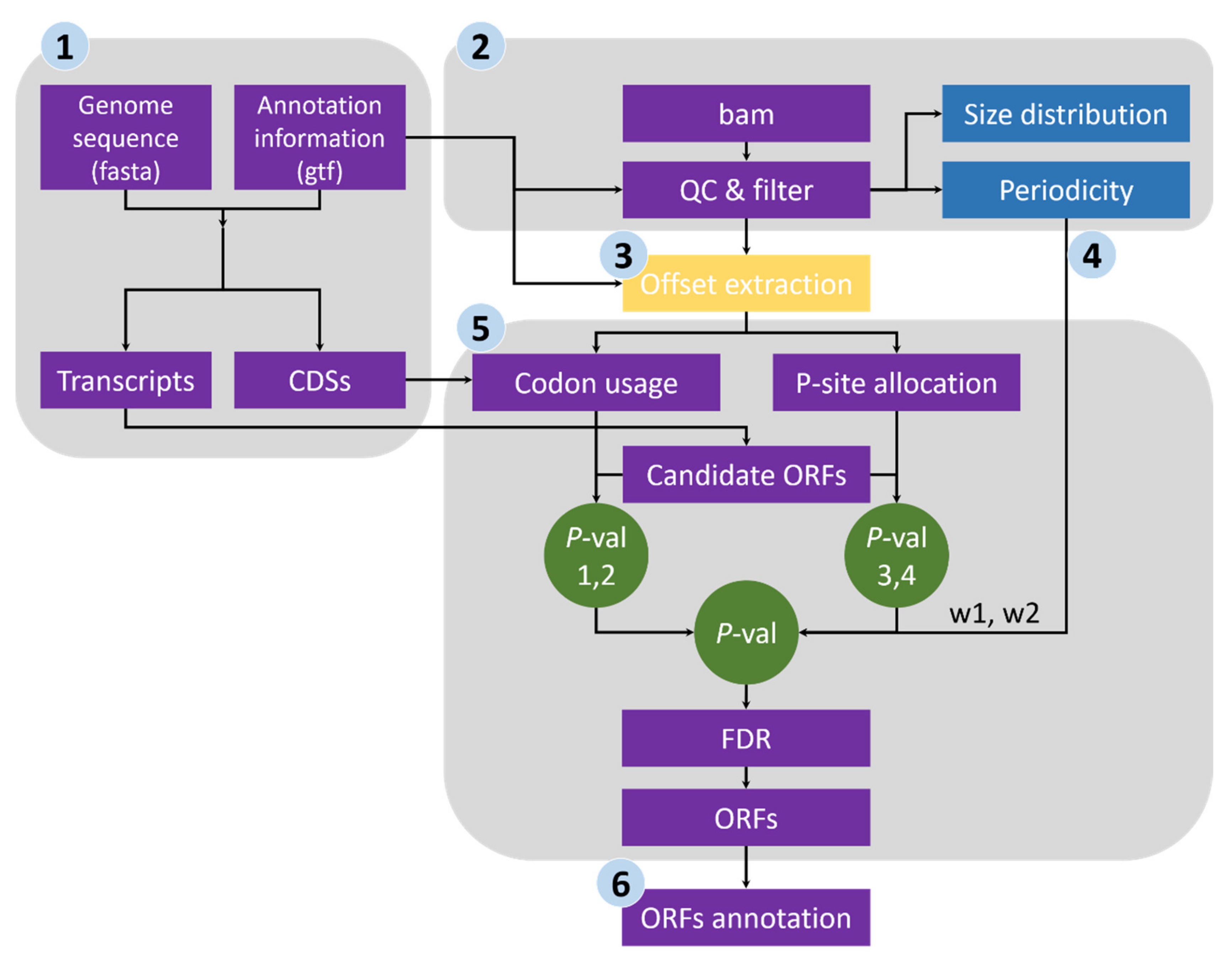
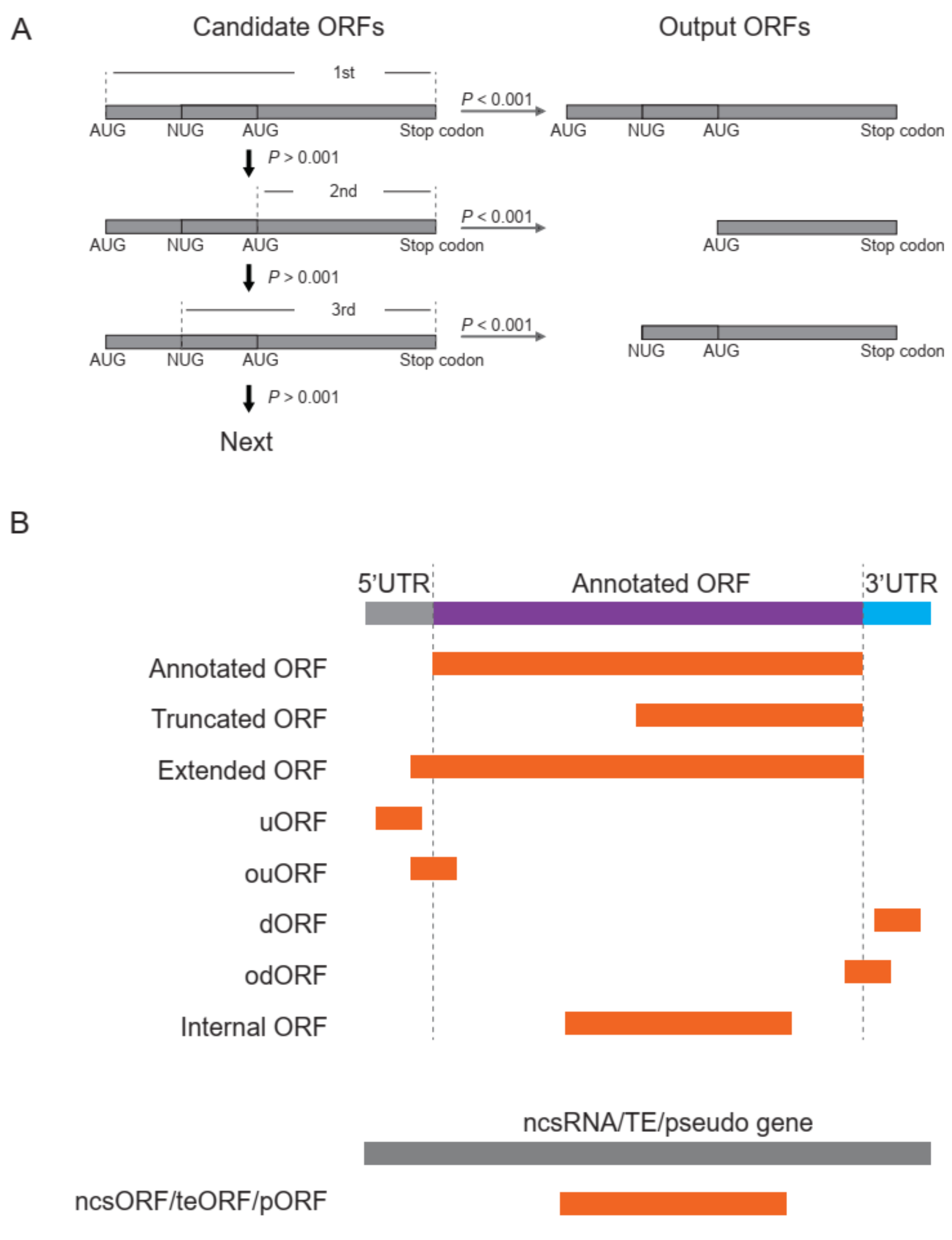
- Sequence extraction of annotated CDSs and ORF candidates. We first extracted the CDS sequences from the reference genome file according to the annotation data and subsequently calculated the genome-wide usage of codons in the annotated CDSs. Transcript sequences were also extracted, and all potential ORFs (beginning with start codon AUG or NUG, ending with a stop codon and having a multiple-of-three length) were retained (Figure 1).
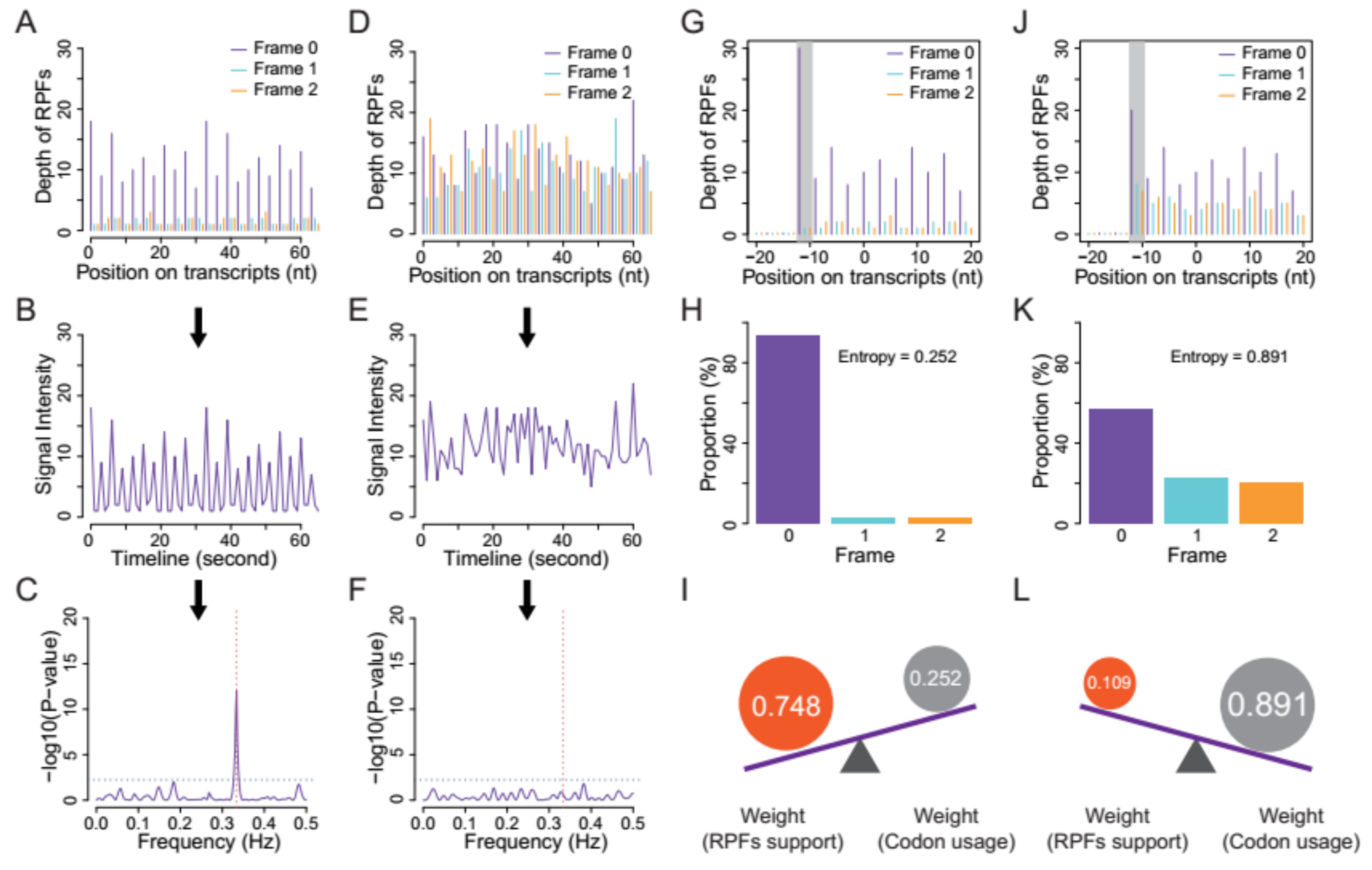
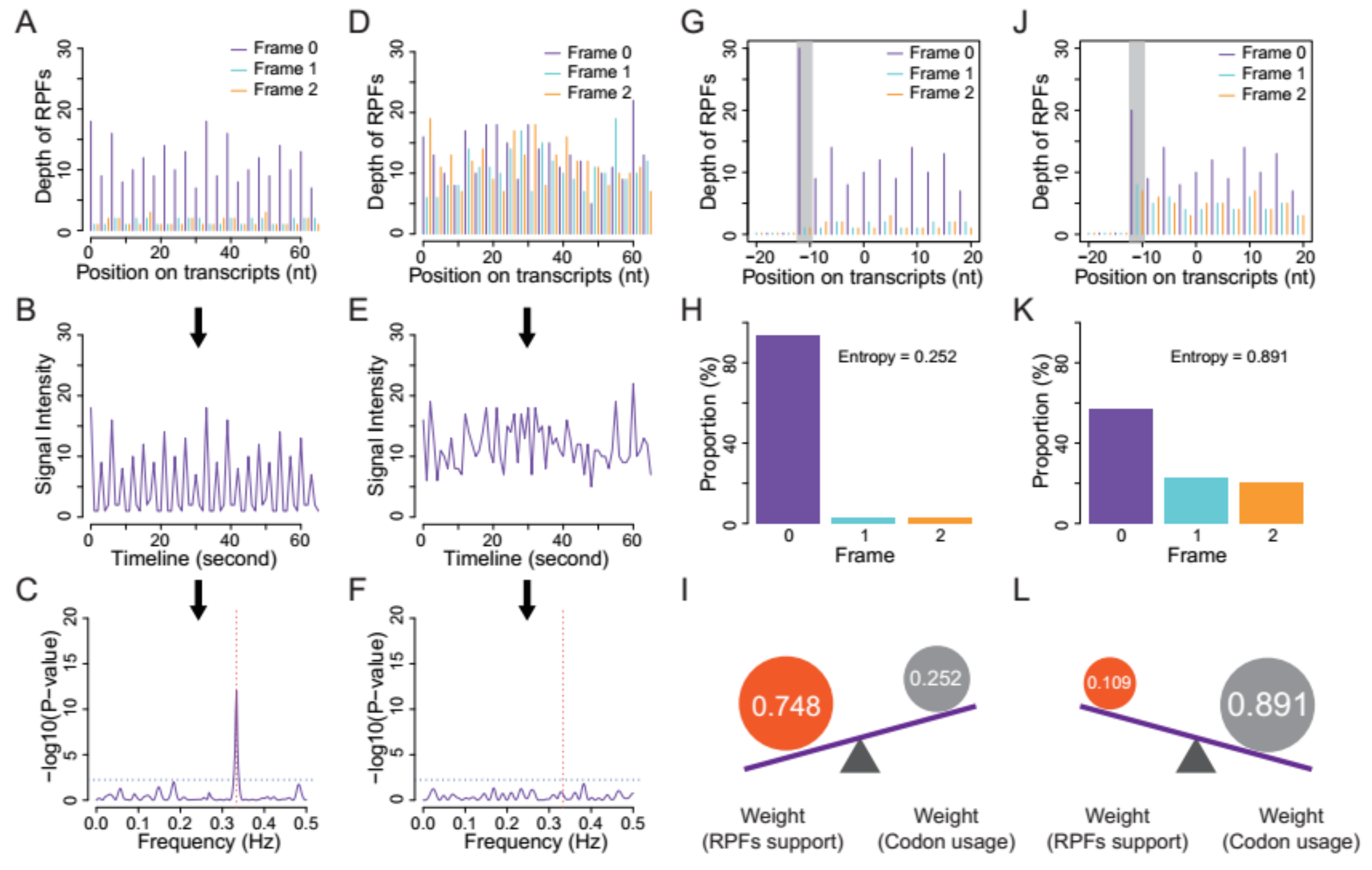
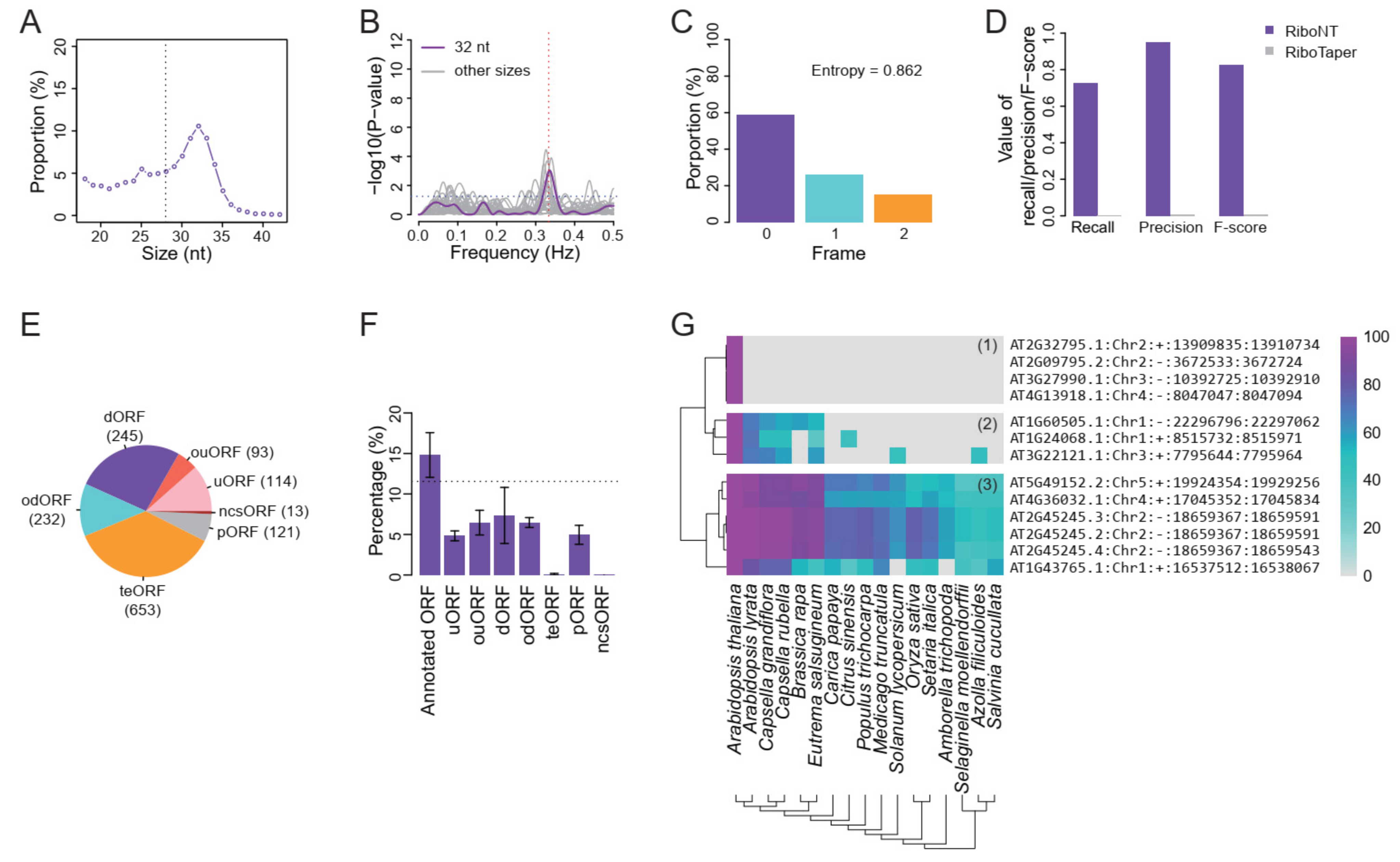
- Quality evaluation of RPFs. RPFs mapped to the first 60 bp of a CDS were used to evaluate RPF periodicity (Figure 2A). RPF filtering was performed using an F-test implemented in the ‘multitaper’ R package [11], which was also used for ORF prediction in RiboTaper [12]. Briefly, we first converted the RPF position and depth along the CDS into a time axis (in seconds) and signal intensities, respectively (Figure 2B). The ‘multitaper’ R package (version 1.0-14) [11] was applied to extract the spectrum and frequency of this ‘signal’. A frequency of 0.33 Hz indicates that the peak of the ‘signal’ appears every three seconds (nucleotides in CDS). An F-test implemented in ‘multitaper’ was performed to calculate the p values for all the frequencies extracted from this ‘signal’. In this pipeline, RPFs with a p value less than 0.01 at a frequency of 0.33 were selected as periodic RPFs for downstream steps (Figure 2C); those that did not satisfy these criteria (Figure 2D–F) were discarded.
- Offset extraction of RPFs. The offsets to the P-site were counted for RPFs in each size class using the RPFs that overlapped with the start (P-site) or stop codon (A-site). Translation initiates from the start codon, so the largest distance from the RPF 5′ terminus to the start codon is the offset to the P-site (Figure 2G–L). As noisy RPFs may show different offsets, instead of a unique offset for each size (Figure 2J), three offsets with corresponding probabilities were calculated for each RPF size using the RPF depths at the first three positions (Figure 2H,K).
- Weight balance. We integrated the support from RPFs and codon usage in this pipeline. One underlying principle is that RPFs with greater periodicity are assigned greater weight; if the periodicity is poor, greater weight should be given to the support of codon usage. We used the differences in RPF distribution on frame 0, 1 and 2 to measure the degree of periodicity. RPFs with high periodicity were preferentially distributed on one of these frames with very high proportions. The diversity was calculated using the following formula for entropy:where i denotes the frame (0, 1 and 2), pi denotes the proportion of RPFs distributed on frame i, and n is logarithmic base. To constrain the interval of entropy between 0 and 1, we set n = 3 for the pipeline. From this formula, RPFs with greater periodicity will have lower entropy (Figure 2H,K). With an even distribution of RPFs among the three frames (the lowest periodicity), pi constantly equals 1/3 when i = 0, 1 and 2, resulting in an entropy of 1. By contrast, exclusive accumulation of RPFs on only one of the frames (the greatest periodicity), e.g., frame 0, would result in an entropy of 0. Finally, we used the entropy value to weigh the contributions from codon usage and [1 − Entropy] to weight those from RPF support (Figure 2I,L).
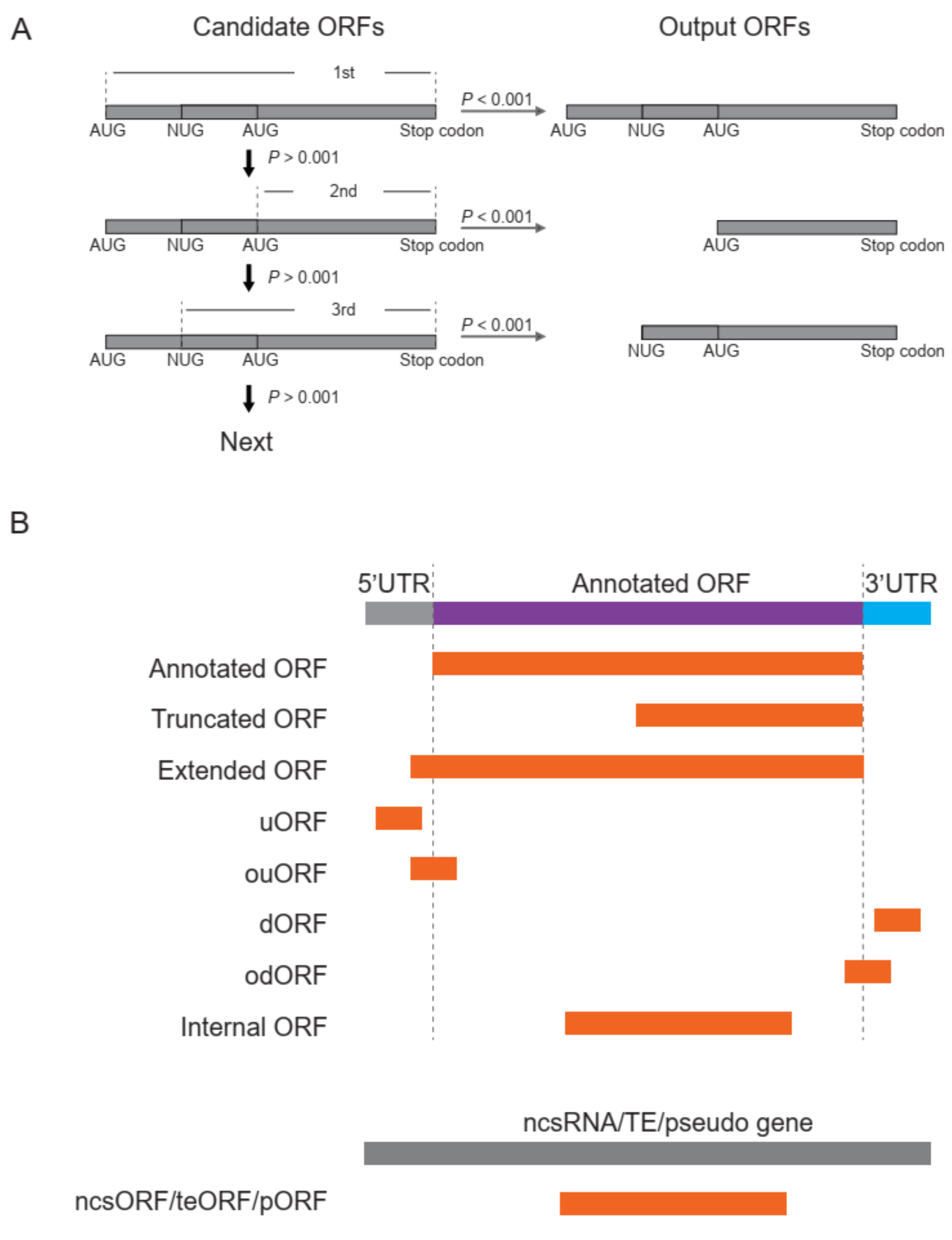
- ORF identification. RPFs were proportionally allocated to their corresponding P-sites according to the offsets extracted in step 3. The RPF depth was transferred to P-site depth, and the values were normalized to a Z-score before two Student’s t-tests were performed to determine whether the depths at frame 0 were significantly greater than those at frame 1 and 2 for a given ORF candidate. Similarly, the codon usage was also assigned to each triplet in the sequence of a given ORF candidate, and two additional Student’s t-tests were performed to determine whether the triplets at frame 0 had greater usage than those at frame 1 and 2. The four p values were weighted according to the RPF periodicity calculated in step 4 and combined using a weighted chi-square method [13] with the following formula:where pi denotes the p values and wi denotes the weight for pi and satisfies = 1. The M value is distributed as 2χ2k/k, where k indicates degrees of freedom, determined according to the weights and correlation between the four tests. According to the algorithm described in [13], k ranges from 2 to 8 in this study. When the four p values are the same, k = 2, suggesting these four tests are identical; when the four tests are independent, k = 8, which is equivalent to that used in Fisher’s method (k = 2n, where n is the number of p values to be combined). We rejected H0, a non-translated candidate ORF, if M > 2 × χ2k, 1-α/k. The value of α was set to 0.001 in this pipeline, candidates with p values less than 0.001 were retained, and the false discovery rate (FDR) was set to 0.0001. For each stop codon, stepwise searching for the longest candidate is performed in RiboNT, which will stop and output the candidate when its p value is smaller than α. The start codon(s) (AUG by default) can be optionally customized; however, considering the higher usage of AUG [2], RiboNT would still output ORFs starting with AUG with higher priority even if other codons were selected (Figure 3A).
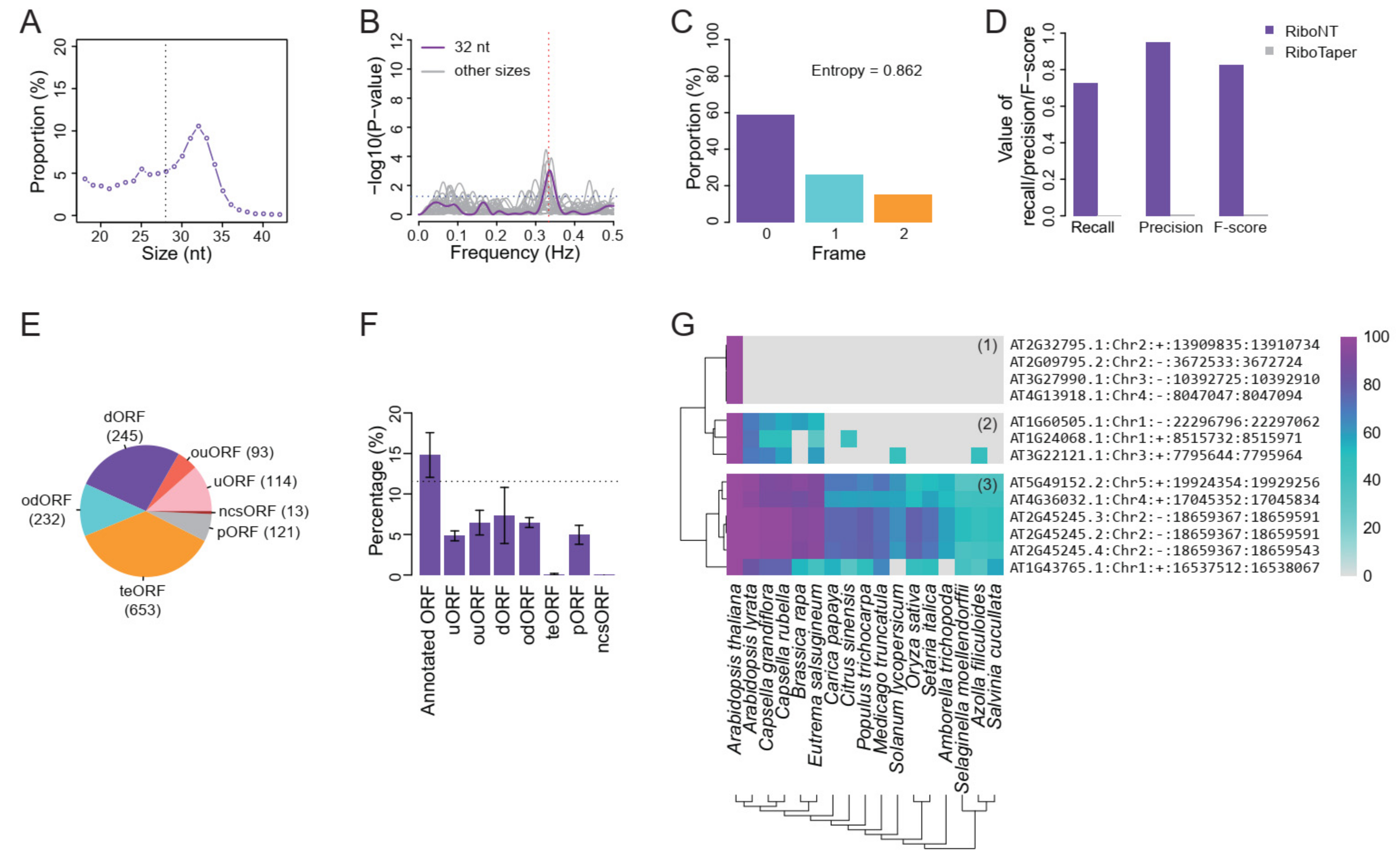
- Classification of predicted ORFs. To ensure consistency with the categories reported in previous works [12,14], several criteria from those studies were incorporated into RiboNT, which classifies the predicted ORFs into 11 categories: (i) annotated ORF, ORFs identical to annotated ORFs; (ii) truncated ORF, ORFs with the same start or stop codon but shorter than the annotated sequence; (iii) extended ORF, ORFs with the same start or stop codon but longer than the annotated sequence; (iv) uORF, upstream ORF, ORFs located in 5′-UTRs; (v) ouORF, overlapped uORF, ORFs located in 5′-UTRs and overlapping an annotated start codon; (vi) dORF, downstream ORF, ORFs located in 3′-UTRs; (vii) odORF, overlapped dORF, ORFs located in 3′UTRs and overlapping an annotated stop codon; (viii) ncsORF, ORFs located in non-coding RNAs, with ORFs predicted from genes without any annotated CDSs also classified as ncsORFs; (ix) internal ORF, ORFs located inside annotated ORFs; (x) teORF, ORFs located in transposable elements; and (xi) pORF, ORFs on pseudogenes (Figure 3B).
2.2. Comparison between RiboNT and Other Predictors
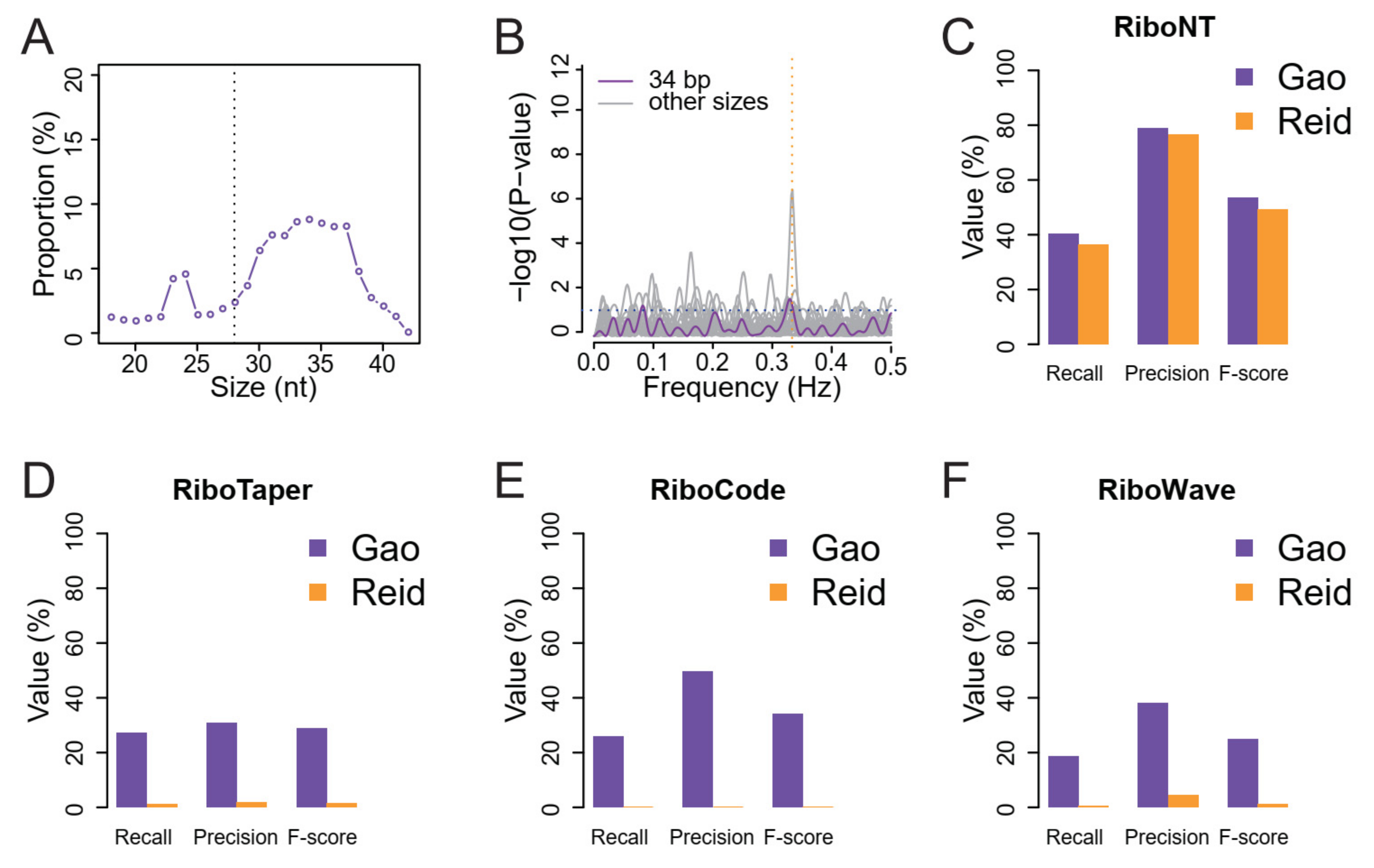
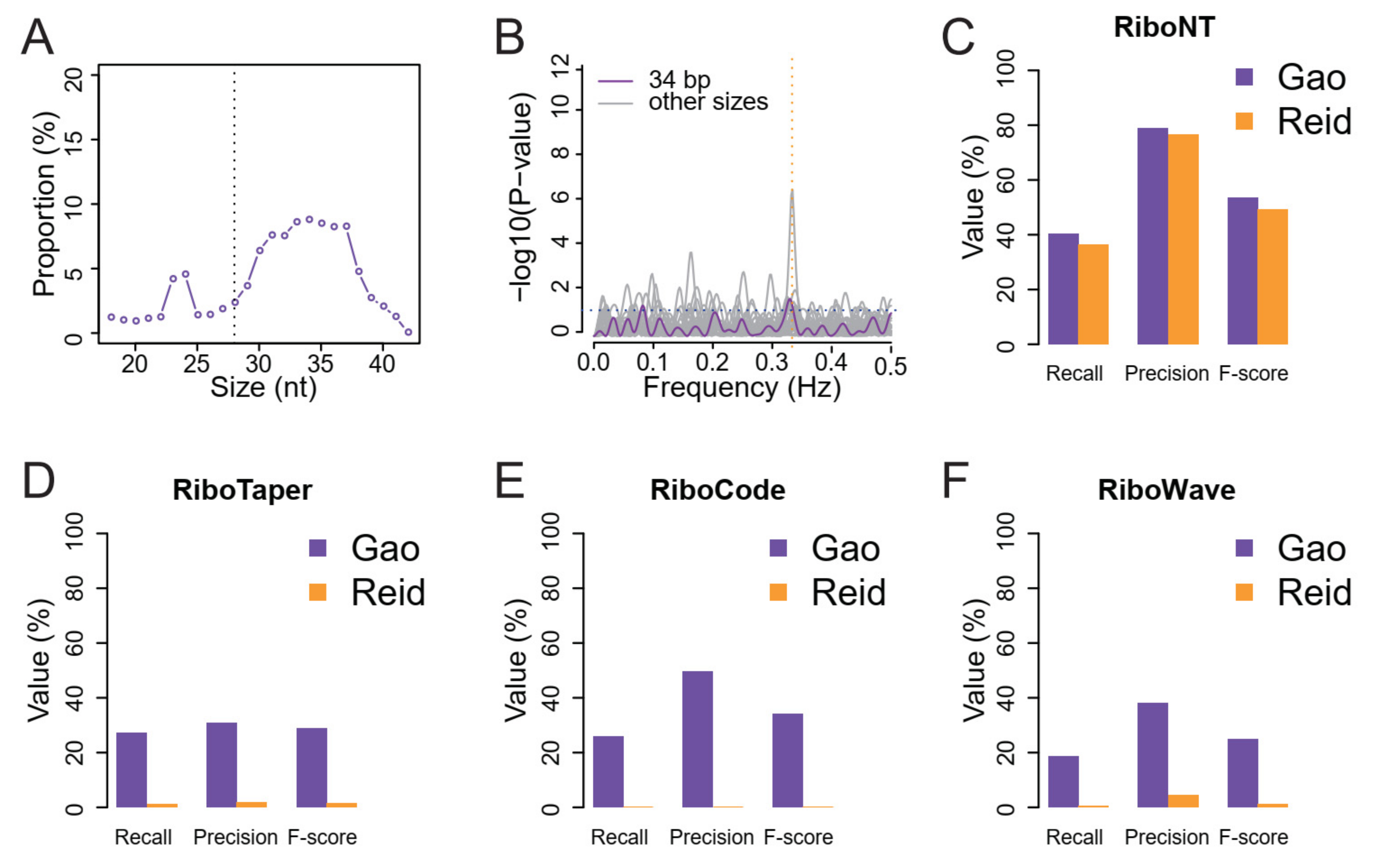
2.3. Validation of Predicted ORFs Using MS Datasets
2.4. Identification and Analysis of ORFs from Human and A. thaliana Low-Quality RPFs
3. Results
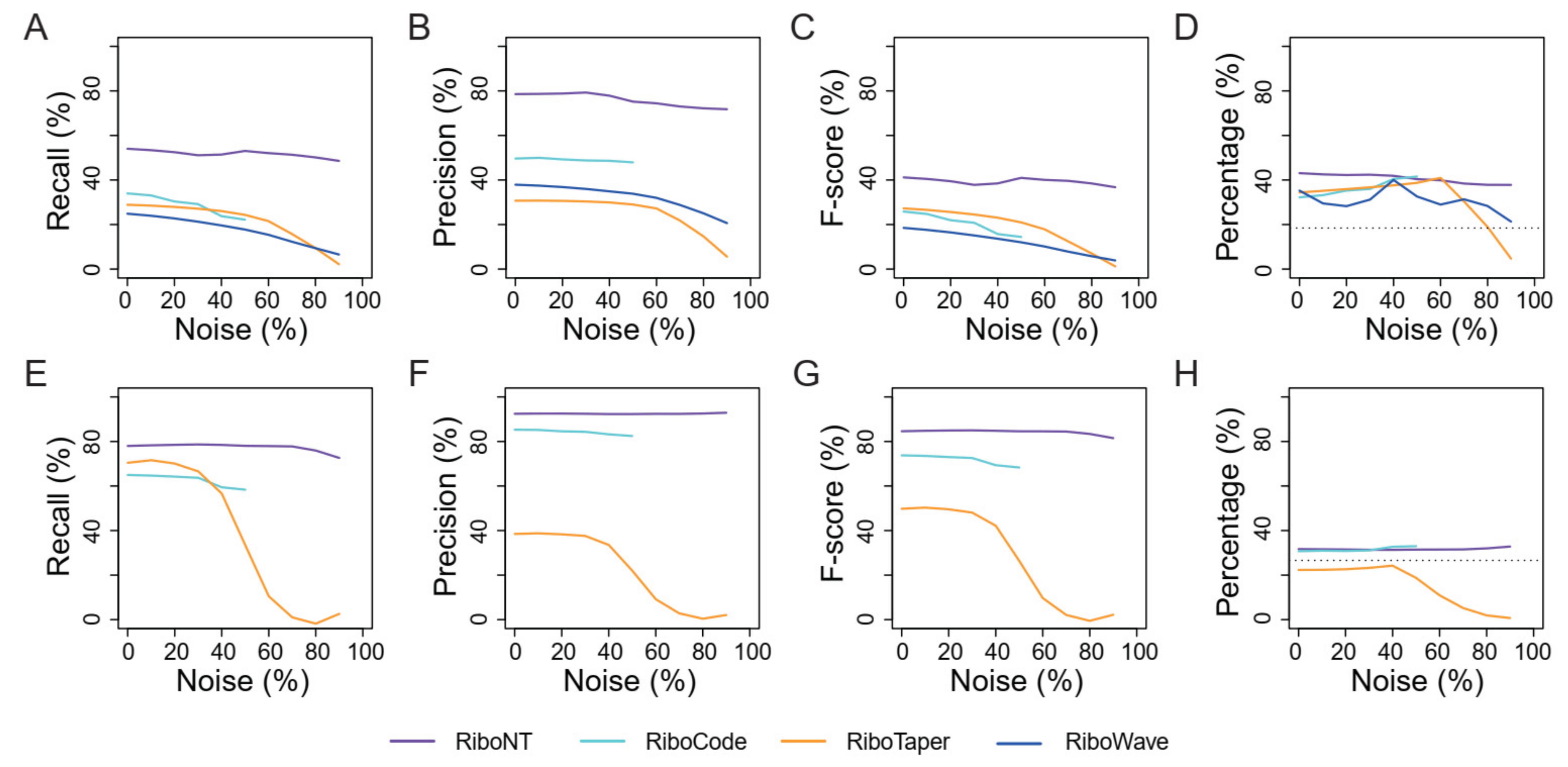
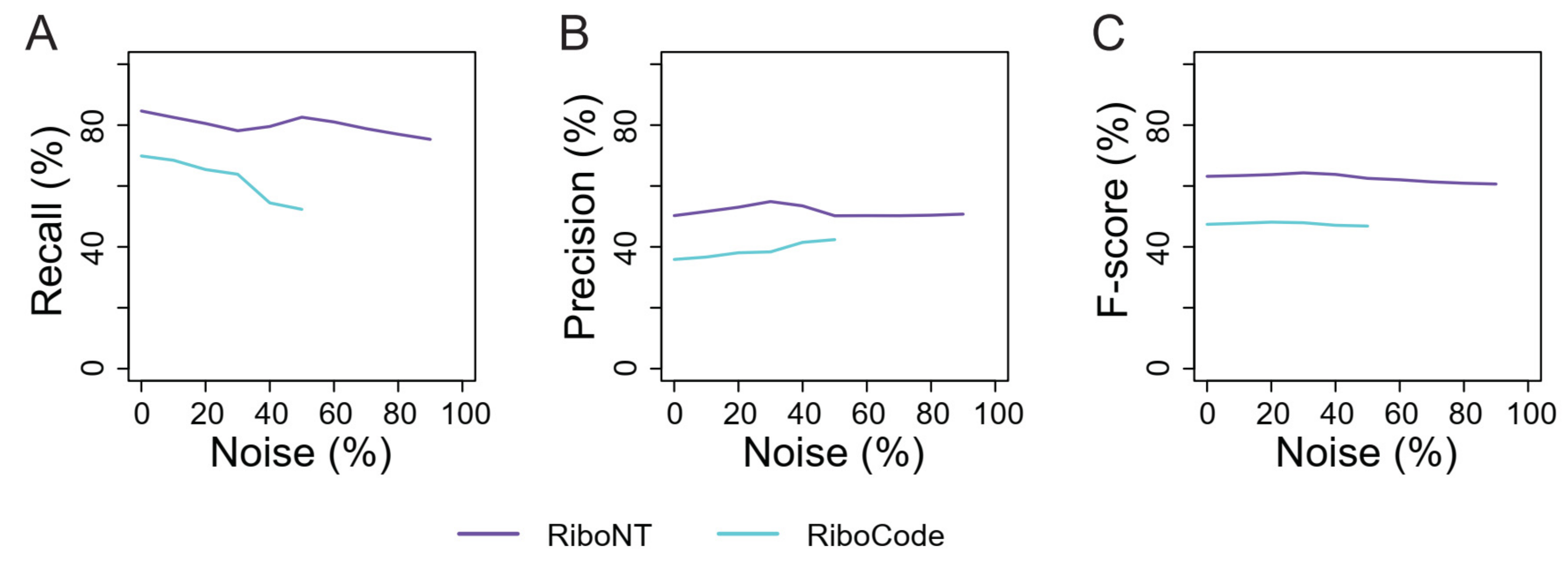
3.1. Identification of Annotated ORFs
3.2. Identification of Translation Initiation Sites
3.3. Identification of Small ORFs
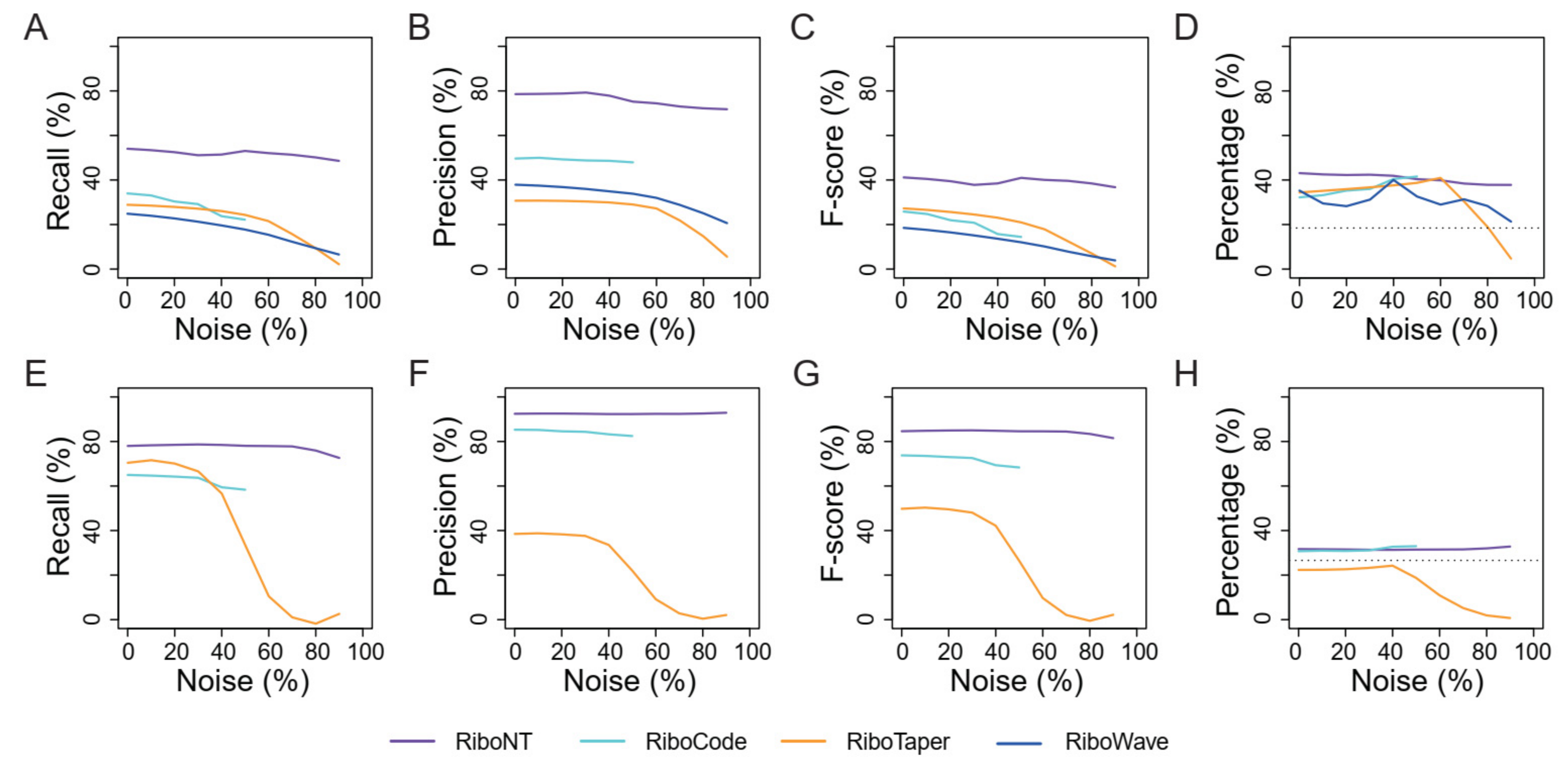
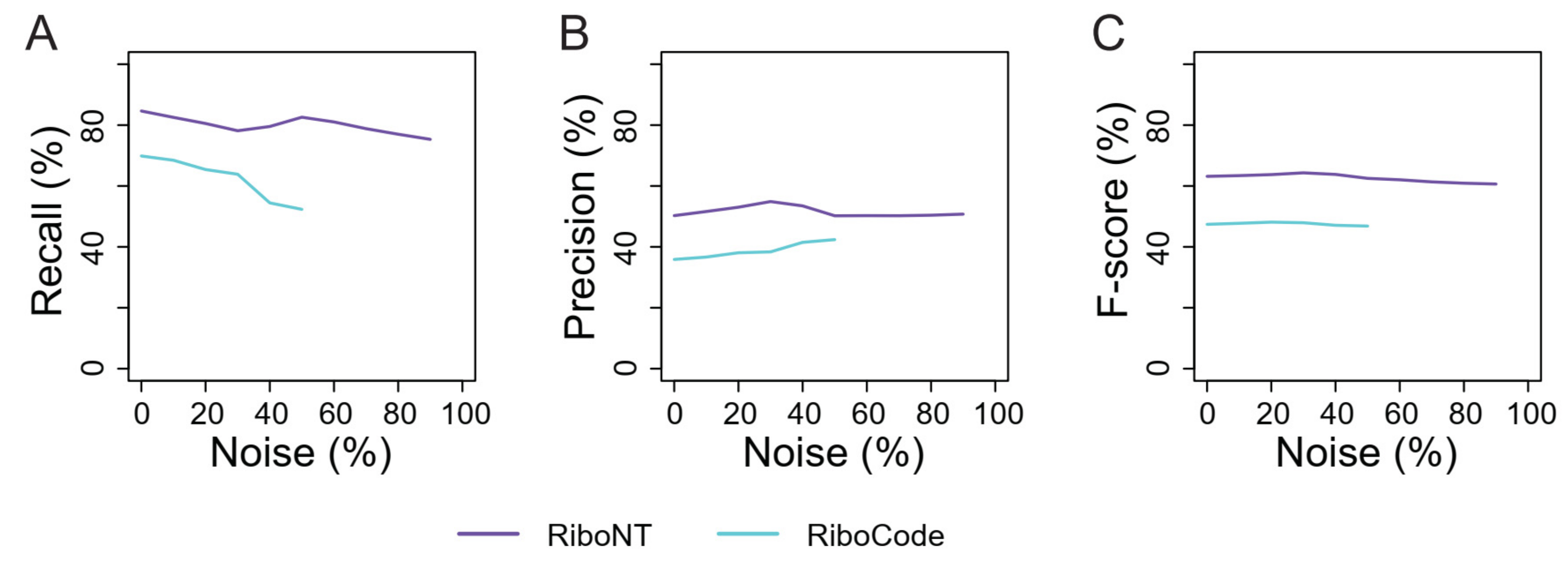
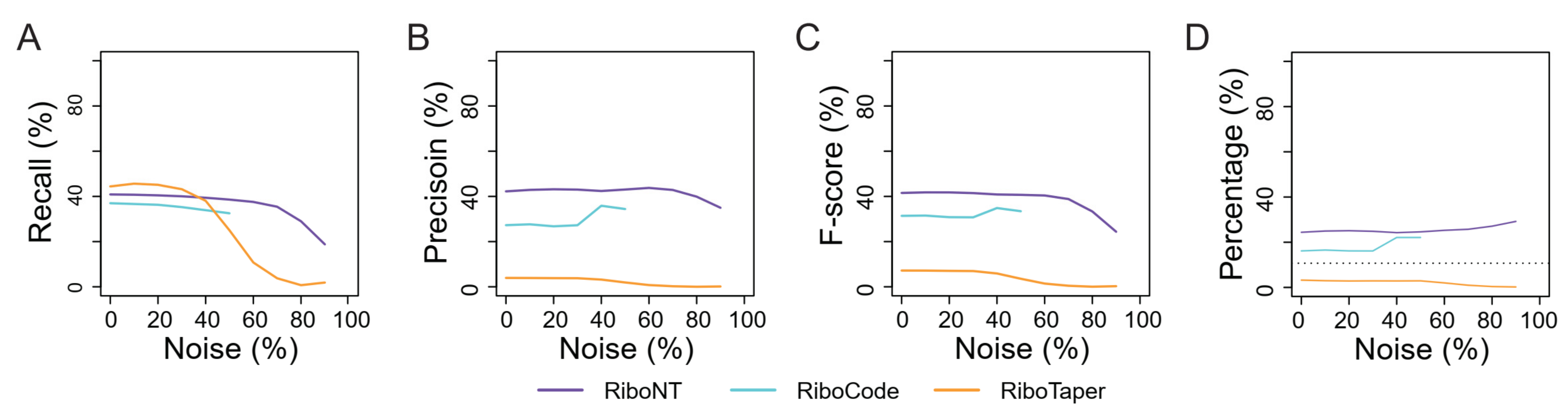
3.4. Identification of Translated ORFs from Human RPFs with Poor Periodicity
3.5. Application of RiboNT to a Dataset of Arabidopsis RPFs with Poor Periodicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kastenmayer, J.P.; Ni, L.; Chu, A.; Kitchen, L.E.; Au, W.-C.; Yang, H.; Carter, C.D.; Wheeler, D.; Davis, R.W.; Boeke, J.D.; et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Res. 2006, 16, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Wan, J.; Liu, B.; Ma, M.; Shen, B.; Qian, S.-B. Quantitative profiling of initiating ribosomes in vivo. Nat. Methods 2015, 12, 147–153. [Google Scholar] [CrossRef]
- Spealman, P.; Naik, A.W.; May, G.E.; Kuersten, S.; Freeberg, L.; Murphy, R.F.; McManus, J. Conserved non-AUG uORFs revealed by a novel regression analysis of ribosome profiling data. Genome Res. 2018, 28, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Huh, W.-K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nat. Cell Biol. 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Hayden, C.A.; Jorgensen, R.A. Identification of novel conserved peptide uORF homology groups in Arabidopsis and rice reveals ancient eukaryotic origin of select groups and preferential association with transcription factor-encoding genes. BMC Biol. 2007, 5, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreev, D.E.; O’Connor, P.B.F.; Loughran, G.; Dmitriev, S.E.; Baranov, P.V.; Shatsky, I.N. Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Res. 2016, 45, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calviello, L.; Ohler, U. Beyond Read-Counts: Ribo-seq Data Analysis to Understand the Functions of the Transcriptome. Trends Genet. 2017, 33, 728–744. [Google Scholar] [CrossRef]
- Heyer, E.; Moore, M.J. Redefining the Translational Status of 80S Monosomes. Cell 2016, 164, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.; Calviello, L.; Wu, H.-Y.L.; Li, F.-W.; Rothfels, C.J.; Ohler, U.; Benfey, P.N. Super-resolution ribosome profiling reveals unannotated translation events in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, E7126–E7135. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Le, B.; Ma, X.; Li, S.; You, C.; Yu, Y.; Zhang, B.; Liu, L.; Gao, L.; Shi, T.; et al. Biogenesis of phased siRNAs on membrane-bound polysomes in Arabidopsis. eLife 2016, 5, 120. [Google Scholar] [CrossRef] [Green Version]
- Rahim, K.J.; Burr, W.S.; Thomson, D.J. Appendix A: Multitaper R Package in Applications of Multitaper Spectral Analysis to Nonstationary Data. Ph.D. Thesis, Queen’s University, Kingston, ON, Canada, 2014. [Google Scholar]
- Calviello, L.; Mukherjee, N.; Wyler, E.; Zauber, H.; Hirsekorn, A.; Selbach, M.; Landthaler, M.; Obermayer, B.; Ohler, U. Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods 2015, 13, 165–170. [Google Scholar] [CrossRef]
- Makambi, K. Weighted inverse chi-square method for correlated significance tests. J. Appl. Stat. 2003, 30, 225–234. [Google Scholar] [CrossRef]
- Xiao, Z.; Huang, R.; Xing, X.; Chen, Y.; Deng, H.; Yang, X. De novo annotation and characterization of the translatome with ribosome profiling data. Nucleic Acids Res. 2018, 46, e61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, Y.; Xie, Z. Computational resources for ribosome profiling: From database to Web server and software. Briefings Bioinform. 2017, 20, 144–155. [Google Scholar] [CrossRef]
- Xu, Z.; Hu, L.; Shi, B.; Geng, S.; Xu, L.; Wang, D.; Lu, Z.J. Ribosome elongating footprints denoised by wavelet transform comprehensively characterize dynamic cellular translation events. Nucleic Acids Res. 2018, 46, e109. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Bielow, C.; Mastrobuoni, G.; Kempa, S. Proteomics Quality Control: Quality Control Software for MaxQuant Results. J. Proteome Res. 2015, 15, 777–787. [Google Scholar] [CrossRef]
- De Witt, R.N.; Kroukamp, H.; Volschenk, H. Proteome response of two natural strains ofSaccharomyces cerevisiaewith divergent lignocellulosic inhibitor stress tolerance. FEMS Yeast Res. 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Kretzschmar, F.K.; Mengel, L.A.; Müller, A.O.; Schmitt, K.; Blersch, K.F.; Valerius, O.; Braus, G.H.; Ischebeck, T. PUX10 Is a Lipid Droplet-Localized Scaffold Protein That Interacts with CELL DIVISION CYCLE48 and Is Involved in the Degradation of Lipid Droplet Proteins. Plant Cell 2018, 30, 2137–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rytz, T.C.; Miller, M.J.; McLoughlin, F.; Augustine, R.; Marshall, R.S.; Juan, Y.-T.; Charng, Y.-Y.; Scalf, M.; Smith, L.M.; Vierstra, R.D. SUMOylome Profiling Reveals a Diverse Array of Nuclear Targets Modified by the SUMO Ligase SIZ1 during Heat Stress. Plant Cell 2018, 30, 1077–1099. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Reid, D.W.; Shenolikar, S.; Nicchitta, C.V. Simple and inexpensive ribosome profiling analysis of mRNA translation. Methods 2015, 91, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazin, J.; Baerenfaller, K.; Gosai, S.J.; Gregory, B.D.; Crespi, M.; Bailey-Serres, J. Global analysis of ribosome-associated noncoding RNAs unveils new modes of translational regulation. Proc. Natl. Acad. Sci. USA 2017, 114, E10018–E10027. [Google Scholar] [CrossRef] [Green Version]
- Juntawong, P.; Girke, T.; Bazin, J.; Bailey-Serres, J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, E203–E212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Chavez, A.; Tung, A.; Chan, Y.; Kaas, C.; Yin, Y.; Cecchi, R.; Garnier, S.L.; Kelsic, E.D.; Schubert, M.; et al. High-throughput creation and functional profiling of DNA sequence variant libraries using CRISPR–Cas9 in yeast. Nat. Biotechnol. 2018, 36, 540–546. [Google Scholar] [CrossRef]
- Raj, A.; Wang, S.H.; Shim, H.; Harpak, A.; Li, Y.I.; Engelmann, B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Thousands of novel translated open reading frames in humans inferred by ribosome footprint profiling. eLife 2016, 5, e13328. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Song, R.; Regev, A.; Struhl, K. Many lncRNAs, 5′UTRs, and pseudogenes are translated and some are likely to express functional proteins. eLife 2015, 4, e08890. [Google Scholar] [CrossRef]
- Bartholomäus, A.; Kolte, B.; Mustafayeva, A.; Goebel, I.; Fuchs, S.; Benndorf, B.; Engelmann, S.; Ignatova, Z. smORFer: A modular algorithm to detect small ORFs in prokaryotes. Nucleic. Acids. Res. 2021, gkab477. [Google Scholar] [CrossRef]
- Shao, D.; Ahmed, N.; Soni, N.; O’Brien, E.P. RiboA: A web application to identify ribosome A-site locations in ribosome profiling data. BMC Bioinform. 2021, 22, 156. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity and Dynamics of Mammalian Proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Zhu, W.; Chen, S.; Qian, J.; Li, L. Genome-Wide Identification and Characterization of Small Peptides in Maize. Front. Plant Sci. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, B.; Jiang, M.; Gao, L. RiboNT: A Noise-Tolerant Predictor of Open Reading Frames from Ribosome-Protected Footprints. Life 2021, 11, 701. https://doi.org/10.3390/life11070701
Song B, Jiang M, Gao L. RiboNT: A Noise-Tolerant Predictor of Open Reading Frames from Ribosome-Protected Footprints. Life. 2021; 11(7):701. https://doi.org/10.3390/life11070701
Chicago/Turabian StyleSong, Bo, Mengyun Jiang, and Lei Gao. 2021. "RiboNT: A Noise-Tolerant Predictor of Open Reading Frames from Ribosome-Protected Footprints" Life 11, no. 7: 701. https://doi.org/10.3390/life11070701
APA StyleSong, B., Jiang, M., & Gao, L. (2021). RiboNT: A Noise-Tolerant Predictor of Open Reading Frames from Ribosome-Protected Footprints. Life, 11(7), 701. https://doi.org/10.3390/life11070701