Abstract
Collodion baby is a congenital, transient phenotype encountered in approximately 70–90% of autosomal recessive congenital ichthyosis and is an important entity of neonatal erythroderma. The clinical outcome after this severe condition is variable. Genetic mutations of components of the epidermal lipoxygenase pathway have been implicated in the majority of self-improving collodion ichthyosis (SICI). In SICI, the shedding of the collodion membrane reveals clear skin or only mild residual manifestation of ichthyosis. Here we report the case of a girl born with a severe form of collodion baby phenotype, whose skin almost completely cleared within the first month of life. At the age of 3 years, only mild symptoms of a keratinization disorder remained. However, the severity of erythema and scaling showed mild fluctuations over time. To objectively evaluate the skin changes of the patient, we assessed the ichthyosis severity index. Upon sequencing of the ALOX12B gene, we identified a previously unreported heterozygous nonsense mutation, c.1607G>A (p.Trp536Ter) with the recurrent, heterozygous mutation c.1562A>G (p.Tyr521Cys). Thereby, our findings expand the genotypic spectrum of SICI. In addition, we summarize the spectrum of further genetic diseases that can present at birth as collodion baby, in particular the SICI.
1. Introduction
Autosomal recessive congenital ichthyosis (ARCI) is a major subgroup of the non-syndromic forms of congenital ichthyosis characterized by abnormal skin cornification with hyperkeratosis, diffuse scaling and variable degree of erythroderma. ARCI is a rare condition, the reported prevalence varies between 1:33,000 and 1:300,000 [1,2,3]. ARCI has diverse clinical manifestations. To date, the mutational spectrum encompasses ten genes that encode proteins (enzymes, transport proteins) responsible for the formation of the stratum corneum [4,5]. Subtypes of ARCI traditionally include lamellar ichthyosis (LI) with large, dark, plate-like scales without erythroderma, and congenital ichthyosiform erythroderma (CIE) with generalized fine scaling and erythroderma. Although LI and CIE are considered two distinct clinical entities, patients often show overlapping features [6]. The most severe subtype of ARCI is Harlequin ichthyosis (HI), which is triggered by inactivating mutations of the ABCA12 gene [7,8]. About 70–90% of ARCI newborns are born encased in a parchment-like membrane, referred to as the collodion membrane [9,10]. It is noteworthy that the prevalence might be even higher, since due to the occasionally mild clinical presentation, most of the cases may go unreported [11]. The term collodion baby (CB) refers to an early, transient phenotype, most commonly encountered in the case of ARCI, rather than a distinct disease entity of ichthyoses. In the majority of cases, the shedding of the collodion membrane is followed by the development of LI or CIE. About 10–20% of CBs show clear skin or only mild symptoms of ichthyosis later on, which was previously referred to as self-healing collodion baby (SHCB). However, the term self-improving collodion ichthyosis (SICI) is preferable instead of SHCB as this minor group of ARCI patients usually still show residue symptoms of LI or CIE. Vahlquist et al. proposed the umbrella term pleomorphic ichthyosis for cases characterized by marked skin changes at birth and subsequently mild symptoms of ichthyosis encompassing SICI, ichthyosis prematurity syndrome, bathing-suit ichthyosis and congenital ichthyosis with mild scaling [4,12]. SICI is predominantly associated with mutations in the ALOX12B, ALOXE3 and less often TGM1 genes [1,13,14]. Lately, the mutation spectrum was expanded with the CYP4F22 gene [9,14]. The severity of ichthyosis in later life in the case of CB is diverse, ranging from HI to variable degree of LI/CIE and SICI. While genetic testing is an important tool for the diagnosis of ichthyosis, the outcome of CB usually cannot be accurately predicted at birth [15]. Here, in addition to a brief review of the literature regarding the diversity of genotype-phenotype correlation of CB, we report a case of a SICI with a novel mutation of the ALOX12B gene that further expands the genotypic spectrum of SICI.
2. Materials and Methods
2.1. Disease Severity Assessment
We applied the disease severity score for newborns with collodion membrane of Rubio-Gomez et al. to describe the status of the newborn patient [15]. In addition, to objectively assessing the skin changes of the patient, we used the ichthyosis severity index of Marukian et al. [16]. Erythema and scaling were evaluated on the upper back region of the patient.
2.2. Mutation Analysis
Genomic DNA was isolated from peripheral blood leukocytes of the patient and her parents with a Roche MagNA Pure Compact system (Roche Diagnostics, Mannheim, Germany) or with a BioRobot EZ1 DSP Workstation (QIAGEN; Hilden, Germany), for TGM1 or ALOX12B and ALOXE3, respectively. After the amplification of the coding regions and flanking introns of the TGM1 (primer pairs for PCR were as described previously [17,18]), ALOX12B and ALOXE3 genes (using primer sequences displayed on the UCSC Genome Browser, http://www.genome.ucsc.edu, accessed on 24 April 2019), DNA sequencing was performed on amplification products. Sequencing data were analyzed in order to screen for any genetic variations.
Written informed consent was obtained from the parents of the patient, and the study was conducted according to the Principles of the Declaration of Helsinki.
3. Results
3.1. Case Report
Here we report the case of a three-year-old girl, who was born to non-consanguineous parents at 37 gestational weeks following an uncomplicated pregnancy, through a normal vaginal delivery as a CB. The parents had no relevant history of skin diseases. The newborn was covered in an opaque membrane with underlying erythroderma (Figure 1A–C). The intertriginous regions and the trunk presented with several fissures. Marked ectropion and eclabium could be noted as well. Routine neonatal assessment was otherwise normal with an Apgar score of 9/9 at 1 and 5 min, respectively. Due to the marked skin changes, the newborn was transferred to a perinatal intensive care unit. The skin status dynamically changed during the first few days postpartum. Due to the compression of the shiny, tight collodion membrane, the extremities appeared edematous and fingers were fixed in a contracture (Figure 1D). Elevated inflammatory markers and positive skin and blood culture showed signs of a multimicrobial infection, which was treated successfully with combined intravenous antibiotic therapy. The newborn had severe anemia secondary to the infection, for which she received blood transfusion. Her skin improved following treatment with topical emollients; the ectropion and eclabium healed and she was emitted from the intensive care unit at the age of 18 days. By the age of one month, the shedding of the collodion membrane revealed erythroderma with fine white scaling. Over time, the severity of the erythema and scaling fluctuated (Figure 2). Currently, at age of three, the patient has mild residual manifestation of ichthyosis, namely xerosis and mild erythematous macules.
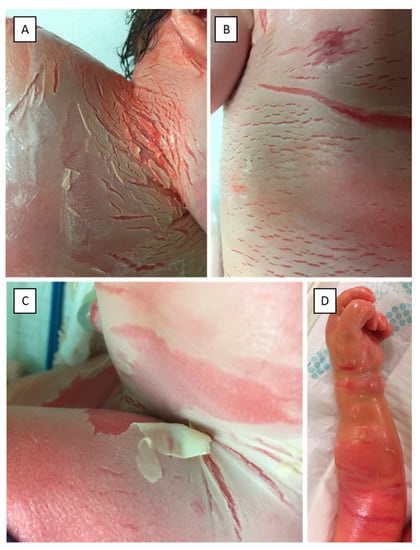
Figure 1.
Clinical pictures of the neck (A), chest (B) and inguinal region (C) of the newborn patient. The newborn was covered in an opaque membrane with underlying erythroderma. The neck, trunk and inguinal region presented with several fissures. Picture (D) taken at 2 days of age shows compression by the tight collodion, which covers the edematous hand and the arm of the patient.
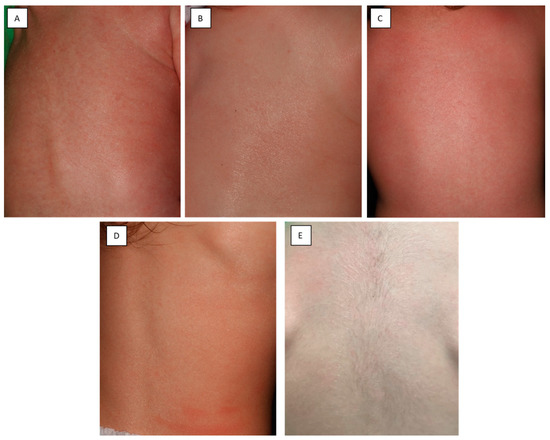
Figure 2.
Clinical photographs of the back of the patient taken at 1 month (A), 3 months (B), 6 months (C), 1.5 years (D) and 3 years (E) of age. The severity of erythroderma fluctuated over time.
3.2. Disease Severity
Collodion membrane severity score was 12 points out of 15, which accounts for high severity. Changes of the ichthyosis severity index after the neonatal period, including the erythema and scaling score, are seen in Figure 3. Scaling was mild after 1 month of age, while the erythema score fluctuated during the observed time period.
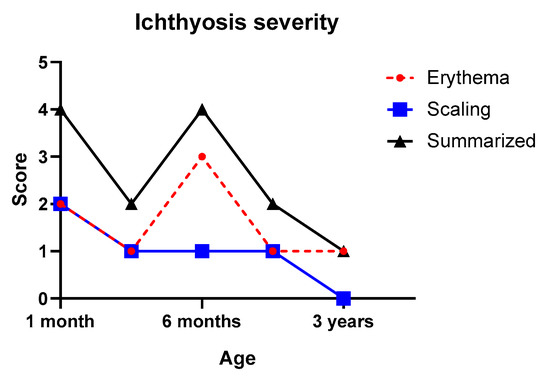
Figure 3.
Changes of ichthyosis severity index of the patient. Erythema and scaling were evaluated on the upper back region of the patient.
3.3. Mutation Analysis
Sequencing of ALOX12B revealed a previously described pathogenic, missense mutation, c.1562A>G (p.Tyr521Cys) in one allele (Figure 4) and a previously not reported, nonsense mutation c.1607G>A (p.Trp536Ter) in the other allele (Figure 5). Thus, the patient had a compound heterozygote mutation in the ALOX12B gene.
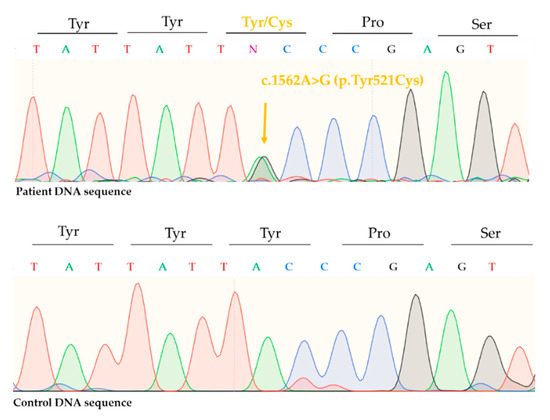
Figure 4.
Direct sequencing revealed a heterozygous, recurrent, pathogenic, missense mutation on the ALOX12B gene c.1562A>G (p.Tyr521Cys).
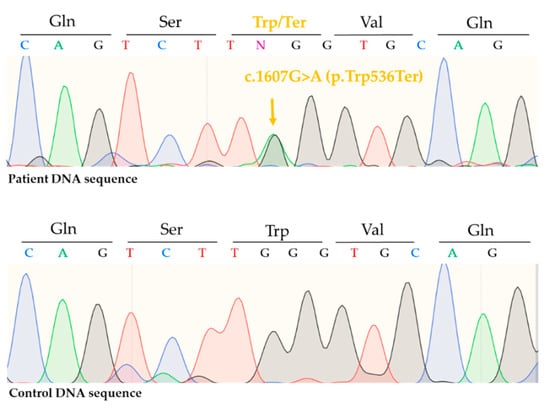
Figure 5.
Direct sequencing revealed a novel, nonsense mutation on the ALOX12B gene in heterozygous form c.1607G>A (p.Trp536Ter).
Upon sequencing of TGM1 and ALOXE3, wild type alleles were detected. Parental testing revealed c.1607G>A (p.Trp536Ter) heterozygous mutation of ALOX12B in the mother and c.1562A>G (p.Tyr521Cys) heterozygous mutation of ALOX12B in the paternal sample.
4. Discussion
ARCI is a genetically heterogeneous group of cornification disorders with diverse and often dynamically changing or overlapping phenotypes. The presence of collodion membrane at birth is often the initial presentation of ARCI. Later, CB develops into CIE/LI or, on rare occasions, heals spontaneously. This latter phenotype is known as SICI, where patients show clear or almost clear skin with mild signs of ichthyosis.
Here we reported the case of a CB, whose symptoms improved significantly after birth. Sequencing of the ALOX12B gene revealed c.1526A>G (p.Tyr521Cys), a frequently reported, well-documented mutation and a novel, pathogenic nonsense mutation, c.1607G>A (p.Trp536Ter). Vahlquist et al. reported five SICI patients with the c.1526A>G (p.Tyr521Cys) mutation [19]. Before that, this particular mutation was only documented in case of other forms of ARCI (LI/CIE) [19]. In a new meta-analysis exploring the genotypic spectrum of ALOX12B and ALOXE3 mutations, c.1526A>G (p.Tyr521Cys) was the most frequent mutation, with an allelic frequency of 22% (61 out of 282 alleles) [5]. Although c.1526A>G (p.Tyr521Cys) frequently occurs in SICI cases, it was detected in numerous LI and CIE patients as well, both in homozygote and compound heterozygote form [5]. Based on this meta-analysis, CB phenotype was documented in 25 out of 44 cases with either heterozygous or homozygous form of c.1526A>G (p.Tyr521Cys) mutation, including five SICI cases. On the other hand, to the best of our knowledge, the other mutation of the patient, c.1607G>A (p.Trp536Ter), has not yet been reported in the literature. The novel c.1607G>A (p.Trp536Ter) mutation affects exon 12 of ALOXB12 gene and causes a premature stop codon.
Of note, in case of mutations of genes encoding the lipoxygenase enzymes, the outcome of CB cannot be accurately predicted neither based on genetic analysis nor the initial clinical presentation [5]. In addition, in our experience, the severity of the disease, e.g., the degree of erythema, can show relapses and remissions that probably are influenced by variable exogenous and endogenous factors. The background of the dramatic improvement of the skin status in SICI after birth is unknown. In the case of TGM1 mutations, two mutated alleles were found to be sensitive to hydrostatic pressure that results in inactive transglutaminase-1 enzyme in utero, which explains the severe phenotype in the newborn that later resolves in normal environmental conditions [20]. Such an explanation does not exist in the case of the lipoxygenase pathway genes. However, the fact that patients with identical mutations can demonstrate different outcomes suggests the role of yet undiscovered factors. A multicentric study confirmed a similar trend as Rubio-Gomez et al.—toward a higher collodion membrane severity score in the case of non-syndromic forms of ichthyosis compared to syndromic forms [15,21]. However, this study could not establish a strong link between the collodion membrane score and the clinical outcome of the disease. The authors hypothesized that it was due to the fact that genes that cause non-syndromic forms of ichthyosis are related to a higher differentiation state of keratinocytes. Our patient had a high ichthyosis severity score, which fits in with the trend that these two studies established. However, the outcome of the SICI phenotype could not be predicted based on this scoring system. While collodion membrane at birth occurs most commonly in ARCI, it must be noted that other diseases can present at birth as CB. These include other forms of syndromic and non-syndromic forms of ichthyosis, hypohidrotic ectodermal dysplasia, palmoplantar keratosis with leukokeratosis anogenitalis [15], congenital hypothyroidism [22,23], alpha-ketoadipic aciduria [24,25] and koraxitrachitic syndrome [26] (Table 1 and Table 2).

Table 1.
Variable forms of ARCI as the most common cause of the collodion baby phenotype with the reported frequency in the literature.
Table 2.
Rare causes of the collodion baby phenotype.
It is important to note that while these diseases can present as CB at birth, most of them are rare exceptions in the literature. So far, two cases with congenital hypothyroidism and one with alpha-ketoadipic aciduria were reported to be born as CB [22,23,24]. Although koraxitrachitic syndrome is frequently cited in association to rare causes of the CB phenotype, there are very few publications about this rare entity [26]. A recent article reported the case of a Harlequin fetus-like newborn, who improved in the first weeks and presented with palmoplantar keratoderma and leukokeratosis anogenitalis caused by KDSR mutation. Autosomal recessive mutations of KDSR are the cause of erythrokeratodermia variabilis et progressiva 4 as well where a vernix-like thickened skin was documented at birth, in some cases besides a true collodion membrane [55]. Another interesting point is the difference of CB and Harlequin ichthyosis at birth. By definition, Harlequin fetus is a neonatal phenotype that later develops to the most severe form of ichthyosis. According to the First Ichthyosis Consensus Conference in Sorèze, the Harlequin fetus is a severe form of CB with thick plate-like, cornified skin [59]. However, due to the marked skin changes and different clinical consequences, it is often referred to as a distinct disease entity in the literature [10,60]. Additionally, it is important to note that there are neonatal conditions that can be confused with the CB phenotype, including ichthyosis prematurity syndrome (OMIM# 604194) and keratitis-ichthyosis-deafness syndrome (OMIM# 600157). The marked skin changes at birth resemble excessive amounts of vernix caseosa in these cases [61]. However, due to the lack of a unified nomenclature, some of these cases are reported as a collodion membrane in the literature [9].
5. Conclusions
In conclusion, we described the case of a SICI, where the patient had a compound heterozygous mutation in the ALOX12B gene. Here, we identified a previously unreported, pathogenic, nonsense mutation c.1607G>A (p.Trp536Ter) along with a recurrent, pathogenic, missense mutation, c.1562A>G, (p.Tyr521Cys). Our case supports the notion that the clinical interpretation and the determination of the prognosis based on genotype–phenotype correlation regarding SICI is challenging. The clinical outcome of a CB cannot always be accurately predicted based on the severity of the clinical presentation at birth; thus, genetic testing is essential. However, in the case of mutations of the lipoxygenase enzyme genes, an association between specific mutations and the mature phenotype could not yet be confirmed. Thus, counseling and education of the parents are of great importance in these cases.
Author Contributions
Conceptualization, M.M. and P.A.; investigation, M.M., P.A., I.K., É.C., K.B., B.M., N.N., M.S., Z.S., D.P., S.Z. and K.F.; resources, M.M., N.Á., M.S., N.N. and B.M.; data curation, P.A.; writing—original draft preparation, P.A.; writing—review and editing, N.K., B.M., K.B., S.Z., D.P. and K.F.; visualization, P.A., S.Z., D.P. and K.F.; supervision, M.M.; project administration, M.M. and B.M.; funding acquisition, M.M., P.A., N.K., S.Z., K.F., D.P. and S.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Research, Development and Innovation Office of Hungary—NKFIH (FK_131916, 2019 (Semmelweis University, M.M.)), EFOP-3.6.3-VEKOP-16-2017-00009 (P.A., S.Z., D.P., K.F.), the ÚNKP-20-4-II-SE-7 (N.K.) and ÚNKP-20-3-I-SE-24 (S.Z.) New National Excellence Program of the Ministry For Innovation and Technology from the source of the National Research, Development and Innovation Fund of Hungary.
Institutional Review Board Statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Scientific and Research Committee of Medical Research Council, Hungary (4937-8/2020/EÜIG) and the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed Consent Statement
Written informed consent was obtained from the parents of the patient to participate. Patients signed informed consent regarding publishing their data.
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The Department of Dermatology, Venereology, and Dermatooncology, Semmelweis University is a Reference Centre of the ERN-Skin: European Reference Network on Rare and Undiagnosed Skin Disorders. We thank Rita Mátrahegyi for her assistance in clinical photography, and Mercédesz Mazán and Adrien Suba Flóriánné for DNA isolation and Sanger sequencing (TGM1).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pigg, M.H.; Bygum, A.; Gånemo, A.; Virtanen, M.; Brandrup, F.; Zimmer, A.D.; Hotz, A.; Vahlquist, A.; Fischer, J. Spectrum of Autosomal Recessive Congenital Ichthyosis in Scandinavia: Clinical Characteristics and Novel and Recurrent Mutations in 132 Patients. Acta Derm. Venereol. 2016, 96, 932–937. [Google Scholar] [CrossRef]
- Rodríguez-Pazos, L.; Ginarte, M.; Fachal, L.; Toribio, J.; Carracedo, A.; Vega, A. Analysis of TGM1, ALOX12B, ALOXE3, NIPAL4 and CYP4F22 in autosomal recessive congenital ichthyosis from Galicia (NW Spain): Evidence of founder effects. Br. J. Derm. 2011, 165, 906–911. [Google Scholar] [CrossRef] [PubMed]
- González-Del Carmen, M.; Montaño, S.; Reyes-Hernández, O.D.; Vizcaíno-Dorado, P.A.; Leyva-García, N.; Morales-Morfín, J.C.; Diaz-Beltran, W.; Quinto-Santiago, E.; Cariño-Calvo, L.; Magaña, J.J.; et al. High prevalence of autosomal recessive congenital ichthyosis in a Mexican population caused by a new mutation in the TGM1 gene: Epidemiological evidence of a founder effect. Int. J. Dermatol. 2020, 59, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Vahlquist, A.; Fischer, J.; Törmä, H. Inherited Nonsyndromic Ichthyoses: An Update on Pathophysiology, Diagnosis and Treatment. Am. J. Clin. Dermatol. 2018, 19, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Hotz, A.; Kopp, J.; Bourrat, E.; Oji, V.; Komlosi, K.; Giehl, K.; Bouadjar, B.; Bygum, A.; Tantcheva-Poor, I.; Hellström Pigg, M.; et al. Meta-Analysis of Mutations in ALOX12B or ALOXE3 Identified in a Large Cohort of 224 Patients. Genes 2021, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Harting, M.; Brunetti-Pierri, N.; Chan, C.S.; Kirby, J.; Dishop, M.K.; Richard, G.; Scaglia, F.; Yan, A.C.; Levy, M.L. Self-healing collodion membrane and mild nonbullous congenital ichthyosiform erythroderma due to 2 novel mutations in the ALOX12B gene. Arch. Derm. 2008, 144, 351–356. [Google Scholar] [CrossRef]
- Rodríguez-Pazos, L.; Ginarte, M.; Vega, A.; Toribio, J. Autosomal recessive congenital ichthyosis. Actas Dermosifiliogr. 2013, 104, 270–284. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, L.; Wang, L.; Zhang, C. Prenatal diagnosis of a rare variant of harlequin ichthyosis with literature review. BMC Med. Imaging 2021, 21, 56. [Google Scholar] [CrossRef]
- Simpson, J.K.; Martinez-Queipo, M.; Onoufriadis, A.; Tso, S.; Glass, E.; Liu, L.; Higashino, T.; Scott, W.; Tierney, C.; Simpson, M.A.; et al. Genotype-phenotype correlation in a large English cohort of patients with autosomal recessive ichthyosis. Br. J. Derm. 2020, 182, 729–737. [Google Scholar] [CrossRef]
- Pinkova, B.; Buckova, H.; Borska, R.; Fajkusova, L. Types of congenital nonsyndromic ichthyoses. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech. Repub. 2020, 164, 357–365. [Google Scholar] [CrossRef]
- Simalti, A.K.; Sethi, H. Collodion Baby. Med. J. Armed Forces India 2017, 73, 197–199. [Google Scholar] [CrossRef][Green Version]
- Vahlquist, A. Pleomorphic ichthyosis: Proposed name for a heterogeneous group of congenital ichthyoses with phenotypic shifting and mild residual scaling. Acta Derm. Venereol. 2010, 90, 454–460. [Google Scholar] [CrossRef]
- Aradhya, S.S.; Srinivas, S.M.; Hiremagalore, R.; Shanmukappa, A.G. Clinical outcome of collodion baby: A retrospective review. Indian J. Derm. Venereol. Leprol. 2013, 79, 553. [Google Scholar] [CrossRef]
- Noguera-Morel, L.; Feito-Rodríguez, M.; Maldonado-Cid, P.; García-Miñáur, S.; Kamsteeg, E.J.; González-Sarmiento, R.; De Lucas-Laguna, R.; Hernández-Martín, A.; Torrelo, A. Two Cases of Autosomal Recessive Congenital Ichthyosis due to CYP4F22 Mutations: Expanding the Genotype of Self-Healing Collodion Baby. Pediatr. Derm. 2016, 33, e48–e51. [Google Scholar] [CrossRef]
- Rubio-Gomez, G.A.; Weinstein, M.; Pope, E. Development of a disease severity score for newborns with collodion membrane. J. Am. Acad. Derm. 2014, 70, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Marukian, N.V.; Deng, Y.; Gan, G.; Ren, I.; Thermidor, W.; Craiglow, B.G.; Milstone, L.M.; Choate, K.A. Establishing and Validating an Ichthyosis Severity Index. J. Investig. Dermatol. 2017, 137, 1834–1841. [Google Scholar] [CrossRef]
- Laiho, E.; Ignatius, J.; Mikkola, H.; Yee, V.C.; Teller, D.C.; Niemi, K.M.; Saarialho-Kere, U.; Kere, J.; Palotie, A. Transglutaminase 1 mutations in autosomal recessive congenital ichthyosis: Private and recurrent mutations in an isolated population. Am. J. Hum. Genet. 1997, 61, 529–538. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Becker, K.; Csikós, M.; Sárdy, M.; Szalai, Z.S.; Horváth, A.; Kárpáti, S. Identification of two novel nonsense mutations in the transglutaminase 1 gene in a Hungarian patient with congenital ichthyosiform erythroderma. Exp. Derm. 2003, 12, 324–329. [Google Scholar] [CrossRef]
- Vahlquist, A.; Bygum, A.; Gånemo, A.; Virtanen, M.; Hellström-Pigg, M.; Strauss, G.; Brandrup, F.; Fischer, J. Genotypic and Clinical Spectrum of Self-Improving Collodion Ichthyosis: ALOX12B, ALOXE3, and TGM1 Mutations in Scandinavian Patients. J. Investig. Dermatol. 2010, 130, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, M.; Hennies, H.C.; Ahvazi, B.; Vogel, M.; Reis, A.; Steinert, P.M.; Traupe, H. Self-healing collodion baby: A dynamic phenotype explained by a particular transglutaminase-1 mutation. J. Investig. Derm. 2003, 120, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Cuperus, E.; Bolling, M.C.; de Graaf, M.; van den Akker, P.C.; van Gijn, M.E.; Simon, M.E.H.; Sigurdsson, V.; Pasmans, S. Collodion babies: A 15-year retrospective multicenter study in The Netherlands-Evaluation of severity scores to predict the underlying disease. J. Am. Acad. Derm. 2021, 84, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Dogan, D.G.; Aslan, M.; Karabiber, H. A collodion baby with hypothyroidism. Genet. Couns. 2010, 21, 343–346. [Google Scholar] [PubMed]
- Kurtoğlu, S.; Caksen, H.; Erdoğan, R.; Kisaarslan, A.F. Collodion baby concomitant with congenital hypothyroidism: A patient report and review of the literature. J. Pediatr. Endocrinol. Metab. 1998, 11, 569–573. [Google Scholar] [CrossRef]
- Przyrembel, H.; Bachmann, D.; Lombeck, I.; Becker, K.; Wendel, U.; Wadman, S.K.; Bremer, H.J. Alpha-ketoadipic aciduria, a new inborn error of lysine metabolism; biochemical studies. Clin. Chim. Acta 1975, 58, 257–269. [Google Scholar] [CrossRef][Green Version]
- Hagen, J.; te Brinke, H.; Wanders, R.J.; Knegt, A.C.; Oussoren, E.; Hoogeboom, A.J.; Ruijter, G.J.; Becker, D.; Schwab, K.O.; Franke, I.; et al. Genetic basis of alpha-aminoadipic and alpha-ketoadipic aciduria. J. Inherit. Metab. Dis. 2015, 38, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Verloes, A.; Hermanns-Lê, T.; Lesenfants, S.; Lombet, J.; Lamotte, P.J.; Crèvecoeur-Liégeois, C.; Duchesne, B.; Piérard, G.E. Koraxitrachitic syndrome: A syndromic form of self-healing collodion baby with residual dappled atrophy of the derma. Am. J. Med. Genet. 1999, 86, 454–458. [Google Scholar] [CrossRef]
- Farasat, S.; Wei, M.H.; Herman, M.; Liewehr, D.J.; Steinberg, S.M.; Bale, S.J.; Fleckman, P.; Toro, J.R. Novel transglutaminase-1 mutations and genotype-phenotype investigations of 104 patients with autosomal recessive congenital ichthyosis in the USA. J. Med. Genet. 2009, 46, 103–111. [Google Scholar] [CrossRef]
- Ballin, N.; Hotz, A.; Bourrat, E.; Küsel, J.; Oji, V.; Bouadjar, B.; Brognoli, D.; Hickman, G.; Heinz, L.; Vabres, P.; et al. Genetical, clinical, and functional analysis of a large international cohort of patients with autosomal recessive congenital ichthyosis due to mutations in NIPAL4. Hum. Mutat. 2019, 40, 2318–2333. [Google Scholar] [CrossRef] [PubMed]
- Hotz, A.; Bourrat, E.; Küsel, J.; Oji, V.; Alter, S.; Hake, L.; Korbi, M.; Ott, H.; Hausser, I.; Zimmer, A.D.; et al. Mutation update for CYP4F22 variants associated with autosomal recessive congenital ichthyosis. Hum. Mutat. 2018, 39, 1305–1313. [Google Scholar] [CrossRef]
- Zimmer, A.D.; Kim, G.J.; Hotz, A.; Bourrat, E.; Hausser, I.; Has, C.; Oji, V.; Stieler, K.; Vahlquist, A.; Kunde, V.; et al. Sixteen novel mutations in PNPLA1 in patients with autosomal recessive congenital ichthyosis reveal the importance of an extended patatin domain in PNPLA1 that is essential for proper human skin barrier function. Br. J. Derm. 2017, 177, 445–455. [Google Scholar] [CrossRef]
- Vahidnezhad, H.; Youssefian, L.; Saeidian, A.H.; Zeinali, S.; Mansouri, P.; Sotoudeh, S.; Barzegar, M.; Mohammadi-Asl, J.; Karamzadeh, R.; Abiri, M.; et al. Gene-Targeted Next Generation Sequencing Identifies PNPLA1 Mutations in Patients with a Phenotypic Spectrum of Autosomal Recessive Congenital Ichthyosis: The Impact of Consanguinity. J. Investig. Derm. 2017, 137, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Youssefian, L.; Vahidnezhad, H.; Saeidian, A.H.; Sotoudeh, S.; Mahmoudi, H.; Daneshpazhooh, M.; Aghazadeh, N.; Adams, R.; Ghanadan, A.; Zeinali, S.; et al. Autosomal recessive congenital ichthyosis: CERS3 mutations identified by a next generation sequencing panel targeting ichthyosis genes. Eur. J. Hum. Genet. 2017, 25, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Eckl, K.M.; Tidhar, R.; Thiele, H.; Oji, V.; Hausser, I.; Brodesser, S.; Preil, M.L.; Onal-Akan, A.; Stock, F.; Müller, D.; et al. Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J. Investig. Derm. 2013, 133, 2202–2211. [Google Scholar] [CrossRef]
- Radner, F.P.; Marrakchi, S.; Kirchmeier, P.; Kim, G.J.; Ribierre, F.; Kamoun, B.; Abid, L.; Leipoldt, M.; Turki, H.; Schempp, W.; et al. Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLOS Genet. 2013, 9, e1003536. [Google Scholar] [CrossRef]
- Takeichi, T.; Nomura, T.; Takama, H.; Kono, M.; Sugiura, K.; Watanabe, D.; Shimizu, H.; Simpson, M.A.; McGrath, J.A.; Akiyama, M. Deficient stratum corneum intercellular lipid in a Japanese patient with lamellar ichthyosis with a homozygous deletion mutation in SDR9C7. Br. J. Derm. 2017, 177, e62–e64. [Google Scholar] [CrossRef]
- Shigehara, Y.; Okuda, S.; Nemer, G.; Chedraoui, A.; Hayashi, R.; Bitar, F.; Nakai, H.; Abbas, O.; Daou, L.; Abe, R.; et al. Mutations in SDR9C7 gene encoding an enzyme for vitamin A metabolism underlie autosomal recessive congenital ichthyosis. Hum. Mol. Genet. 2016, 25, 4484–4493. [Google Scholar] [CrossRef]
- Heinz, L.; Kim, G.-J.; Marrakchi, S.; Christiansen, J.; Turki, H.; Rauschendorf, M.-A.; Lathrop, M.; Hausser, I.; Zimmer, A.D.; Fischer, J. Mutations in SULT2B1 Cause Autosomal-Recessive Congenital Ichthyosis in Humans. Am. J. Hum. Genet. 2017, 100, 926–939. [Google Scholar] [CrossRef]
- Fozia, F.; Nazli, R.; Khan, S.A.; Bari, A.; Nasir, A.; Ullah, R.; Mahmood, H.M.; Sohaib, M.; Alobaid, A.; Ansari, S.A.; et al. Novel Homozygous Mutations in the Genes TGM1, SULT2B1, SPINK5 and FLG in Four Families Underlying Congenital Ichthyosis. Genes 2021, 12, 373. [Google Scholar] [CrossRef]
- Mizrachi-Koren, M.; Geiger, D.; Indelman, M.; Bitterman-Deutsch, O.; Bergman, R.; Sprecher, E. Identification of a novel locus associated with congenital recessive ichthyosis on 12p11.2-q13. J. Investig. Derm. 2005, 125, 456–462. [Google Scholar] [CrossRef]
- Marukian, N.V.; Hu, R.-H.; Craiglow, B.G.; Milstone, L.M.; Zhou, J.; Theos, A.; Kaymakcalan, H.; Akkaya, D.A.; Uitto, J.J.; Vahidnezhad, H.; et al. Expanding the Genotypic Spectrum of Bathing Suit Ichthyosis. JAMA Derm. 2017, 153, 537–543. [Google Scholar] [CrossRef]
- Ferrari, B.; Martínez, J.P.; Luna, P.C.; Larralde, M. Acral self-healing collodion baby: A case series. Int. J. Womens Derm. 2016, 2, 140–142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prado, R.; Ellis, L.Z.; Gamble, R.; Funk, T.; Arbuckle, H.A.; Bruckner, A.L. Collodion baby: An update with a focus on practical management. J. Am. Acad. Derm. 2012, 67, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Srivastava, A.; Srivastava, P.; Betigeri, A.V.K.; Verma, M. Congenital Ichthyosis—Collodion Baby Case Report. J. Clin. Diagn. Res. 2016, 10, SJ01–SJ02. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Williams, M.L.; Choi, E.-H.; Feingold, K.R. Role of cholesterol sulfate in epidermal structure and function: Lessons from X-linked ichthyosis. Biochim. Biophys. Acta 2014, 1841, 353–361. [Google Scholar] [CrossRef]
- Muñoz-Aceituno, E.; Nogera-Morel, L.; Torrelo, A.; Hernandez-Martin, A. Mild collodion baby as a presenting sign of loricrin keratoderma: Report of a case and review of the literature. Clin. Exp. Derm. 2020, 45, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Long, M.C. Ichthyosis with confetti: A rare diagnosis and treatment plan. BMJ Case Rep. 2014, 2014, bcr2014204509. [Google Scholar] [CrossRef]
- Guerra, L.; Diociaiuti, A.; El Hachem, M.; Castiglia, D.; Zambruno, G. Ichthyosis with confetti: Clinics, molecular genetics and management. Orphanet J. Rare Dis. 2015, 10, 115. [Google Scholar] [CrossRef]
- Mégarbané, H.; Mégarbané, A. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome. Orphanet J. Rare Dis. 2011, 6, 29. [Google Scholar] [CrossRef]
- de Almeida, H., Jr.; Has, C.; Rampon, G.; Isaacsson, H.; de Castro, L.A.S. Mend Syndrome: A Case Report with Scanning Electron Microscopy Findings of the Collodion. Membrane. Acta Derm. Venereol. 2017, 97, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Scacchi, M.F.; Pagotto, B.; Correa, N.; Castillo, A.; Paula, C.L.; Boggio, P.; Abad, M.E.; Larralde, M. Collodion baby. Report of 14 patients. Derm. Argent. 2011, 17, 128–133. [Google Scholar]
- Carr, P.C.; Casamiquela, K.M.; Jacks, S.K. Gaucher Disease Type 2 Presenting with Collodion Membrane and Blueberry Muffin Lesions. Pediatric Dermatol. 2016, 33, e20–e22. [Google Scholar] [CrossRef]
- Stone, D.L.; Carey, W.F.; Christodoulou, J.; Sillence, D.; Nelson, P.; Callahan, M.; Tayebi, N.; Sidransky, E. Type 2 Gaucher disease: The collodion baby phenotype revisited. Arch. Dis. Child. Fetal Neonatal Ed. 2000, 82, F163–F166. [Google Scholar] [CrossRef]
- García-Martín, P.; Hernández-Martín, A.; Torrelo, A. Ectodermal Dysplasias: A Clinical and Molecular Review. Actas Dermo-Sifiliográficas (Engl. Ed.) 2013, 104, 451–470. [Google Scholar] [CrossRef]
- Thomas, C.; Suranyi, E.; Pride, H.; Tyler, W. A child with hypohidrotic ectodermal dysplasia with features of a collodion membrane. Pediatr. Derm. 2006, 23, 251–254. [Google Scholar] [CrossRef]
- Boyden, L.M.; Vincent, N.G.; Zhou, J.; Hu, R.; Craiglow, B.G.; Bayliss, S.J.; Rosman, I.S.; Lucky, A.W.; Diaz, L.A.; Goldsmith, L.A.; et al. Mutations in KDSR Cause Recessive Progressive Symmetric Erythrokeratoderma. Am. J. Hum. Genet. 2017, 100, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Lautenschlager, S.; Pittelkow, M.R. Palmoplantar keratoderma and leukokeratosis anogenitalis: The second case of a new disease. Dermatology 1998, 197, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Chiticariu, E.; Bachmann, D.; Flatz, L.; Hohl, D. Palmoplantar Keratoderma with Leukokeratosis Anogenitalis Caused by KDSR Mutations. J. Investig. Derm. 2020, 140, 1662–1665.e1661. [Google Scholar] [CrossRef]
- Arbuckle, H.A.; Morelli, J. Holocarboxylase synthetase deficiency presenting as ichthyosis. Pediatr. Derm. 2006, 23, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Oji, V.; Tadini, G.; Akiyama, M.; Blanchet Bardon, C.; Bodemer, C.; Bourrat, E.; Coudiere, P.; DiGiovanna, J.J.; Elias, P.; Fischer, J.; et al. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Derm. 2010, 63, 607–641. [Google Scholar] [CrossRef] [PubMed]
- Mazereeuw-Hautier, J.; Vahlquist, A.; Traupe, H.; Bygum, A.; Amaro, C.; Aldwin, M.; Audouze, A.; Bodemer, C.; Bourrat, E.; Diociaiuti, A.; et al. Management of congenital ichthyoses: European guidelines of care, part one. Br. J. Derm. 2019, 180, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Bygum, A.; Westermark, P.; Brandrup, F. Ichthyosis prematurity syndrome: A well-defined congenital ichthyosis subtype. J. Am. Acad. Derm. 2008, 59, S71–S74. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).