
Hyperalphalipoproteinemia and Beyond: The Role of HDL in Cardiovascular Diseases


 ,
,  , and
, and 
Abstract
1. Introduction
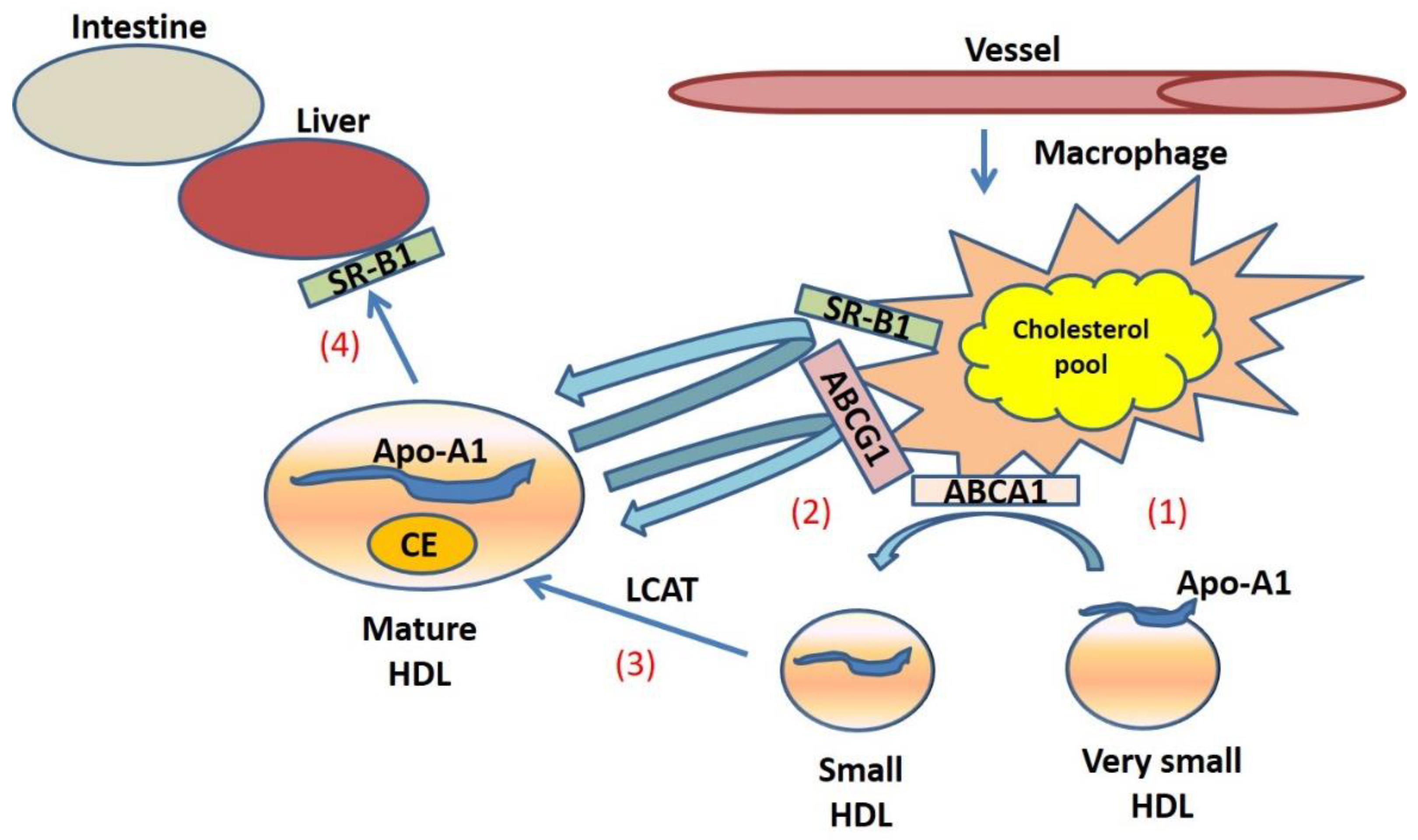
2. HDLs Physiology
3. Primary Causes of HALP
3.1. CETP Deficiency
3.2. Hepatic Lipase and APO-CIII Deficiency
3.3. Scavenger Receptor Class B Type I (SR-BI)
3.4. Endothelial Lipase (EL)
3.5. Polygenic Causes of Hyperalphalipoproteinemia
3.6. HALP and Cardiovascular Risk
3.7. Pharmacological Targets to Increase HDL-C
4. CETP Inhibitors
5. Future Directions
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, H.; Gong, Y.; Wu, Q.; Ye, X.; Yu, B.; Lu, C.; Jiang, W.; Ye, J.; Fu, Z. Rare Diseases Related with Lipoprotein Metabolism. Adv. Exp. Med. Biol. 2020, 1276, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Maruyama, T.; Hirano, K.-I.; Sakai, N.; Nakajima, N.; Matsuzawa, Y. Molecular mechanisms, lipoprotein abnormalities and atherogenicity of hyperalphalipoproteinemia. Atherosclerosis 2000, 152, 271–285. [Google Scholar] [CrossRef]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 1989, 79, 8–15. [Google Scholar] [CrossRef]
- Franceschini, G. Epidemiologic evidence for high-density lipoprotein cholesterol as a risk factor for coronary artery disease. Am. J. Cardiol. 2001, 88, 9–13. [Google Scholar] [CrossRef]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Haase, C.L.; Tybjærg-Hansen, A.; Qayyum, A.A.; Schou, J.; Nordestgaard, B.G.; Frikke-Schmidt, R. LCAT, HDL Cholesterol and Ischemic Cardiovascular Disease: A Mendelian Randomization Study of HDL Cholesterol in 54,500 Individuals. J. Clin. Endocrinol. Metab. 2012, 97, E248–E256. [Google Scholar] [CrossRef]
- Zhong, S.; Sharp, D.S.; Grove, J.S.; Bruce, C.; Yano, K.; Curb, J.D.; Tall, A.R. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J. Clin. Investig. 1996, 97, 2917–2923. [Google Scholar] [CrossRef] [PubMed]
- Curb, J.D.; Abbott, R.D.; Rodriguez, B.L.; Masaki, K.; Chen, R.; Sharp, D.S.; Tall, A.R. A prospective study of HDL-C and cholesteryl ester transfer protein gene mutations and the risk of coronary heart disease in the elderly. J. Lipid Res. 2004, 45, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Rohatgi, A.; Sacks, F.M.; Guyton, J.R. JCL roundtable: High-density lipoprotein function and reverse cholesterol transport. J. Clin. Lipidol. 2018, 12, 1086–1094. [Google Scholar] [CrossRef]
- Calabresi, L.; Gomaraschi, M.; Franceschini, G. Endothelial Protection by High-Density Lipoproteins. Arter. Thromb. Vasc. Biol. 2003, 23, 1724–1731. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in Atherosclerosis—No Longer a Theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef]
- Kajani, S.; Curley, S.; McGillicuddy, F.C. Unravelling HDL—Looking beyond the Cholesterol Surface to the Quality Within. Int. J. Mol. Sci. 2018, 19, 1971. [Google Scholar] [CrossRef] [PubMed]
- Brewer, H.; Fairwell, T.; LaRue, A.; Ronan, R.; Houser, A.; Bronzert, T. The amino acid sequence of human Apoa-I, an apolipoprotein isolated from high density lipoproteins. Biochem. Biophys. Res. Commun. 1978, 80, 623–630. [Google Scholar] [CrossRef]
- Okada, T.; Ohama, T.; Takafuji, K.; Kanno, K.; Matsuda, H.; Sairyo, M.; Zhu, Y.; Saga, A.; Kobayashi, T.; Masuda, D.; et al. Shotgun proteomic analysis reveals proteome alterations in HDL of patients with cholesteryl ester transfer protein deficiency. J. Clin. Lipidol. 2019, 13, 317–325. [Google Scholar] [CrossRef]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C.; Gillard, B.K.; Xu, B.; Gotto, A.M.; Pownall, H.J. Revisiting Reverse Cholesterol Transport in the Context of High-Density Lipoprotein Free Cholesterol Bioavailability. Methodist DeBakey Cardiovasc. J. 2019, 15, 47–54. [Google Scholar] [CrossRef]
- Linton, M.F.; Tao, H.; Linton, E.F.; Yancey, P.G. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Trends Endocrinol. Metab. 2017, 28, 461–472. [Google Scholar] [CrossRef]
- Pownall, H.J.; Rosales, C.; Gillard, B.K.; Gotto, A.M. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat. Rev. Cardiol. 2021, 1–12. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; De Lemos, J.A.; et al. HDL Cholesterol Efflux Capacity and Incident Cardiovascular Events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.-C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol Efflux and Atheroprotection. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef]
- Beazer, J.D.; Patanapirunhakit, P.; Gill, J.M.; Graham, D.; Karlsson, H.; Ljunggren, S.; Mulder, M.T.; Freeman, D.J. High-density lipoprotein’s vascular protective functions in metabolic and cardiovascular disease—Could extracellular vesicles be at play? Clin. Sci. 2020, 134, 2977–2986. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Vega, M.; Massó, F.; Páez, A.; Vargas-Alarcón, G.; Coral-Vázquez, R.; Mas-Oliva, J.; Carreón-Torres, E.; Pérez-Méndez, Ó. HDL-Mediated Lipid Influx to Endothelial Cells Contributes to Regulating Intercellular Adhesion Molecule (ICAM)-1 Expression and eNOS Phosphorylation. Int. J. Mol. Sci. 2018, 19, 3394. [Google Scholar] [CrossRef] [PubMed]
- Kudinov, V.A.; Alekseeva, O.Y.; Torkhovskaya, T.I.; Baskaev, K.K.; Artyushev, R.I.; Saburina, I.N.; Markin, S.S. High-Density Lipoproteins as Homeostatic Nanoparticles of Blood Plasma. Int. J. Mol. Sci. 2020, 21, 8737. [Google Scholar] [CrossRef] [PubMed]
- Radulović, S.; Gottschalk, B.; Hörl, G.; Zardoya-Laguardia, P.; Schilcher, I.; Hallström, S.; Vujić, N.; Schmidt, K.; Trieb, M.; Graier, W.; et al. Endothelial lipase increases eNOS activating capacity of high-density lipoprotein. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158612. [Google Scholar] [CrossRef]
- Cervellati, C.; Vigna, G.B.; Trentini, A.; Sanz, J.M.; Zimetti, F.; Dalla Nora, E.; Morieri, M.L.; Zuliani, G.; Passaro, A. Paraoxonase-1 activities in individuals with different HDL circulating levels: Implication in reverse cholesterol transport and early vascular damage. Atherosclerosis 2019, 285, 64–70. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B.; Ansell, B.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Translation of High-Density Lipoprotein Function into Clinical Practice. Circulation 2013, 128, 1256–1267. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Yamashita, S.; Kameda, K.; Kubo, M.; Tarui, S.; Hara, I. Marked hyper-HDL2-cholesterolemia associated with premature corneal opacity: A case report. Atherosclerosis 1984, 53, 207–212. [Google Scholar] [CrossRef]
- Deiana, L.; Pes, G.M.; Carru, C.; Campus, G.V.; Tidore, M.G.; Cherchi, G.M. Extremely high HDL levels in a patient with multiple symmetric lipomatosis. Clin. Chim. Acta 1993, 223, 143–147. [Google Scholar] [CrossRef]
- Patsch, W.; Kuisk, I.; Glueck, C.; Schonfeld, G. Lipoproteins in familial hyperalphalipoproteinemia. Arter. Off. J. Am. Hear. Assoc. Inc. 1981, 1, 156–161. [Google Scholar] [CrossRef]
- Ruiz-Ramie, J.J.; Barber, J.L.; Sarzynski, M.A. Effects of exercise on HDL functionality. Curr. Opin. Lipidol. 2019, 30, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Nova, E.; Mauro-Martín, I.S.; Díaz-Prieto, L.E.; Marcos, A. Wine and beer within a moderate alcohol intake is associated with higher levels of HDL-c and adiponectin. Nutr. Res. 2019, 63, 42–50. [Google Scholar] [CrossRef]
- Hannuksela, M.; Marcel, Y.L.; Kesäniemi, Y.A.; Savolainen, M.J. Reduction in the concentration and activity of plasma cholesteryl ester transfer protein by alcohol. J. Lipid Res. 1992, 33, 737–744. [Google Scholar] [CrossRef]
- Ramasamy, I. Update on the molecular biology of dyslipidemias. Clin. Chim. Acta 2016, 454, 143–185. [Google Scholar] [CrossRef]
- Godsland, I.F. Biology: Risk factor modification by OCs and HRT lipids and lipoproteins. Maturitas 2004, 47, 299–303. [Google Scholar] [CrossRef]
- Titov, V.N.; Tvorogova, M.G.; Kantardzhian, I.G.; Alamdarova, I.A.; Negovskaia, A.V. High density lipoprotein cholesterol in sec-ondary hyperlipoproteinemias. Lab Delo. 1983, 12, 11–17. [Google Scholar]
- Guevara, M.; Ng, B. Positive effect of hydroxychloroquine on lipid profiles of patients with rheumatoid arthritis: A Veterans Affair cohort. Eur. J. Rheumatol. 2021, 8, 62–66. [Google Scholar] [CrossRef]
- Hargrove, G.M.; Junco, A.; Wong, N.C. Hormonal regulation of apolipoprotein AI. J. Mol. Endocrinol. 1999, 22, 103–111. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; Soulaidopoulos, S.; Sfikakis, P.P.; Dimitroulas, T.; Kitas, G.D. Effect of Biologics on Cardiovascular Inflammation: Mechanistic Insights and Risk Reduction. J. Inflamm. Res. 2021, 14, 1915–1931. [Google Scholar] [CrossRef] [PubMed]
- Raygor, V.; Khera, A. New Recommendations and Revised Concepts in Recent Guidelines on the Management of Dyslipidemias to Prevent Cardiovascular Disease: The 2018 ACC/AHA and 2019 ESC/EAS Guidelines. Curr. Cardiol. Rep. 2020, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Agellon, L.B.; Quinet, E.M.; Gillette, T.G.; Drayna, D.T.; Brown, M.L.; Tall, A.R. Organization of the human cholesteryl ester transfer protein gene. Biochemistry 1990, 29, 1372–1376. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.C.F.; Raposo, H.F. Cholesteryl Ester Transfer Protein and Lipid Metabolism and Cardiovascular Diseases. Adv. Exp. Med. Biol. 2020, 1276, 15–25. [Google Scholar] [CrossRef]
- Ohnishi, T.; Tan, C.; Yokoyama, S. Selective Transfer of Cholesteryl Ester over Triglyceride by Human Plasma Lipid Transfer Protein between Apolipoprotein-Activated Lipid Microemulsions. Biochemistry 1994, 33, 4533–4542. [Google Scholar] [CrossRef]
- Tall, A. Plasma Lipid Transfer Proteins. Annu. Rev. Biochem. 1995, 64, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, J.; Mabuchi, H.; Yoshimura, A.; Michishita, I.; Takeda, M.; Itoh, H.; Sakai, Y.; Sakai, T.; Ueda, K.; Takeda, R. Deficiency of serum cholesteryl-ester transfer activity in patients with familial hyperalphalipoproteinaemia. Atherosclerosis 1985, 58, 175–186. [Google Scholar] [CrossRef]
- Yamashita, S.; Matsuzawa, Y.; Okazaki, M.; Kako, H.; Yasugi, T.; Akioka, H.; Hirano, K.; Tarui, S. Small polydisperse low density lipoproteins in familial hyperalphalipoproteinemia with complete deficiency of cholesteryl ester transfer activity. Atherosclerosis 1988, 70, 7–12. [Google Scholar] [CrossRef]
- Yokoyama, S.; Ueshima, H.; Miida, T.; Nakamura, M.; Takata, K.; Fukukawa, T.; Goto, T.; Harada-Shiba, M.; Sano, M.; Kato, K.; et al. High-Density Lipoprotein Levels Have Markedly Increased Over the Past Twenty Years in Japan. J. Atheroscler. Thromb. 2014, 21, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Nagano, M.; Yamashita, S.; Hirano, K.-I.; Takano, M.; Maruyama, T.; Ishihara, M.; Sagehashi, Y.; Kujiraoka, T.; Tanaka, K.; Hattori, H.; et al. Molecular mechanisms of cholesteryl ester transfer protein deficiency in Japanese. J. Atheroscler. Thromb. 2004, 11, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Sakai, N.; Ishigami, M.; Hirano, K.-I.; Arai, T.; Okada, S.; Okuda, E.; Ohya, A.; Nakajima, N.; Kadowaki, K.; et al. Prevalence and phenotypic spectrum of cholesteryl ester transfer protein gene mutations in Japanese hyperalphalipoproteinemia. Atherosclerosis 2003, 166, 177–185. [Google Scholar] [CrossRef]
- Inazu, A.; Jiang, X.C.; Haraki, T.; Yagi, K.; Kamon, N.; Koizumi, J.; Mabuchi, H.; Takeda, R.; Takata, K.; Moriyama, Y. Genetic cholesteryl ester transfer protein deficiency caused by two prevalent mutations as a major determinant of increased levels of high density lipoprotein cholesterol. J. Clin. Investig. 1994, 94, 1872–1882. [Google Scholar] [CrossRef]
- Nagano, M.; Yamashita, S.; Hirano, K.-I.; Ito, M.; Maruyama, T.; Ishihara, M.; Sagehashi, Y.; Oka, T.; Kujiraoka, T.; Hattori, H.; et al. Two novel missense mutations in the CETP gene in Japanese hyperalphalipoproteinemic subjects: High-throughput assay by Invader® assay. J. Lipid Res. 2002, 43, 1011–1018. [Google Scholar] [CrossRef]
- Gotoda, T.; Kinoshita, M.; Shimano, H.; Harada, K.; Shimada, M.; Ohsuga, J.; Teramoto, T.; Yazaki, Y.; Yamada, N. Cholesteryl Ester Transfer Protein Deficiency Caused by a Nonsense Mutation Detected in the Patient′s Macrophage mRNA. Biochem. Biophys. Res. Commun. 1993, 194, 519–524. [Google Scholar] [CrossRef]
- Thompson, J.F.; Reynolds, J.M.; Williams, S.P.; Wood, L.S.; Paciga, S.A.; Lloyd, D.B. Frequency and function of CETP variants among individuals of Asian ancestry. Atherosclerosis 2009, 202, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Teh, E.M.; Dolphin, P.J.; Breckenridge, W.C.; Tan, M.-H. Human plasma CETP deficiency: Identification of a novel mutation in exon 9 of the CETP gene in a Caucasian subject from North America. J. Lipid Res. 1998, 39, 442–456. [Google Scholar] [CrossRef]
- Rhyne, J.; Ryan, M.J.; White, C.; Chimonas, T.; Miller, M. The two novel CETP mutations Gln87X and Gln165X in a compound heterozygous state are associated with marked hyperalphalipoproteinemia and absence of significant coronary artery disease. J. Mol. Med. 2006, 84, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Van der Steeg, W.A.; Hovingh, G.K.; Klerkx, A.H.E.M.; Hutten, B.A.; Nootenboom, I.C.; Levels, J.H.M.; van Tol, A.; Dallinga-Thie, G.M.; Zwinderman, A.H.; Kastelein, J.J.P.; et al. Cholesteryl ester transfer protein and hyperalphalipoproteinemia in Caucasians. J. Lipid Res. 2007, 48, 674–682. [Google Scholar] [CrossRef]
- Cefalu’, A.B.; Noto, D.; Magnolo, L.; Pinotti, E.; Gomaraschi, M.; Martini, S.; Vigna, G.B.; Calabresi, L.; Tarugi, P.; Averna, M.R. Novel mutations of CETP gene in Italian subjects with hyeralphalipoproteinemia. Atherosclerosis 2009, 204, 202–207. [Google Scholar] [CrossRef]
- Calabresi, L.; Nilsson, P.; Pinotti, E.; Gomaraschi, M.; Favari, E.; Adorni, M.P.; Bernini, F.; Sirtori, C.R.; Calandra, S.; Franceschini, G.; et al. A novel homozygous mutation in CETP gene as a cause of CETP deficiency in a caucasian kindred. Atherosclerosis 2009, 205, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Pirim, D.; Wang, X.; Niemsiri, V.; Radwan, Z.H.; Bunker, C.H.; Hokanson, J.E.; Hamman, R.F.; Barmada, M.M.; Demirci, F.Y.; Kamboh, M.I. Resequencing of the CETP gene in American whites and African blacks: Association of rare and common variants with HDL-cholesterol levels. Metabolism 2016, 65, 36–47. [Google Scholar] [CrossRef]
- Okada, T.; Ohama, T.; Okazaki, M.; Kanno, K.; Matsuda, H.; Sairyo, M.; Zhu, Y.; Saga, A.; Kobayashi, T.; Masuda, D.; et al. Particle number analysis of lipoprotein subclasses by gel permeation HPLC in patients with cholesteryl ester transfer protein deficiency. PLoS ONE 2018, 13, e0190875. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Matsuzawa, Y.; Hirano, K.; Yamashita, S.; Nozaki, S.; Ueyama, Y.; Kubo, M.; Tarui, S. Detection of two species of low density lipoprotein particles in cholesteryl ester transfer protein deficiency. Arter. Thromb. A J. Vasc. Biol. 1991, 11, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Tsukada, T.; Murase, T.; Matsumoto, K. Particle size analysis of high density lipoproteins in patients with genetic cholesteryl ester transfer protein deficiency. Clin. Chim. Acta 2000, 301, 103–117. [Google Scholar] [CrossRef]
- Ishigami, M.; Yamashita, S.; Sakai, N.; Aral, T.; Hirano, K.-I.; Hiraoka, H.; Kameda-Takemura, K.; Matsuzawa, Y. Large and Cholesteryl Ester-Rich High-Density Lipoproteins in Cholesteryl Ester Transfer Protein (CETP) Deficiency Can Not Protect Macrophages from Cholesterol Accumulation Induced by Acetylated Low-Density Lipoproteins. J. Biochem. 1994, 116, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Nakamura, R.; Takata, R.; Saito, Y.; Yamashita, S.; Horiuchi, S.; Matsuda, I. Structural and functional differences of sub-species of apoA-I-containing lipoprotein in patients with plasma cholesteryl ester transfer protein deficiency. J. Lipid Res. 1995, 36, 696–704. [Google Scholar] [CrossRef]
- Inazu, A.; Brown, M.L.; Hesler, C.B.; Agellon, L.B.; Koizumi, J.; Takata, K.; Maruhama, Y.; Mabuchi, H.; Tall, A.R. Increased High-Density Lipoprotein Levels Caused by a Common Cholesteryl-Ester Transfer Protein Gene Mutation. N. Engl. J. Med. 1990, 323, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y.; Okamura, T.; Inazu, A.; Doi, M.; Iso, H.; Mouri, Y.; Ishikawa, Y.; Suzuki, H.; Iida, M.; Koizumi, J.; et al. A Low Prevalence of Coronary Heart Disease among Subjects with Increased High-Density Lipoprotein Cholesterol Levels, Including Those with Plasma Cholesteryl Ester Transfer Protein Deficiency. Prev. Med. 1998, 27, 659–667. [Google Scholar] [CrossRef]
- Borggreve, S.E.; Hillege, H.L.; Wolffenbuttel, B.H.R.; De Jong, P.E.; Zuurman, M.W.; Van Der Steege, G.; Van Tol, A.; Dullaart, R.P.F. An Increased Coronary Risk Is Paradoxically Associated with Common Cholesteryl Ester Transfer Protein Gene Variations That Relate to Higher High-Density Lipoprotein Cholesterol: A Population-Based Study. J. Clin. Endocrinol. Metab. 2006, 91, 3382–3388. [Google Scholar] [CrossRef]
- Kolovou, V.; Diakoumakou, O.; Papazafiropoulou, A.K.; Katsiki, N.; Fragopoulou, E.; Vasiliadis, I.; Degiannis, D.; Duntas, L.; Antonopoulou, S.; Kolovou, G. Biomarkers and Gene Polymorphisms in Members of Long- and Short-lived Families: A Longevity Study. Open Cardiovasc. Med. J. 2018, 12, 59–70. [Google Scholar] [CrossRef]
- Pirim, D.; Bunker, C.H.; Hokanson, J.E.; Hamman, R.F.; Demirci, F.Y.; Kamboh, M.I. Hepatic lipase (LIPC) sequencing in individuals with extremely high and low high-density lipoprotein cholesterol levels. PLoS ONE 2020, 15, e0243919. [Google Scholar] [CrossRef]
- Chatterjee, C.; Sparks, D.L. Hepatic Lipase, High Density Lipoproteins, and Hypertriglyceridemia. Am. J. Pathol. 2011, 178, 1429–1433. [Google Scholar] [CrossRef]
- Tani, M.; Horvath, K.V.; Lamarche, B.; Couture, P.; Burnett, J.R.; Schaefer, E.J.; Asztalos, B.F. High-density lipoprotein subpopulation profiles in lipoprotein lipase and hepatic lipase deficiency. Atherosclerosis 2016, 253, 7–14. [Google Scholar] [CrossRef]
- Rouhani, N.; Young, E.; Chatterjee, C.; Sparks, D.L. HDL Composition Regulates Displacement of Cell Surface-Bound Hepatic Lipase. Lipids 2008, 43, 793–804. [Google Scholar] [CrossRef]
- Kobayashi, J. Which is the Best Predictor for the Development of Atherosclerosis Among Circulating Lipoprotein Lipase, Hepatic Lipase, and Endothelial Lipase? J. Atheroscler. Thromb. 2019, 26, 758–759. [Google Scholar] [CrossRef]
- Carlson, L.A.; Holmquist, L.; Nilsson-Ehle, P. Deficiency of Hepatic Lipase Activity in Post-heparin Plasma in Familial Hyper-α-Triglyceridemia. Acta Med. Scand. 2009, 219, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, W.; Little, J.; Alaupovic, P.; Wang, C.; Kuksis, A.; Kakis, G.; Lindgren, F.; Gardiner, G. Lipoprotein abnormalities associated with a familial deficiency of hepatic lipase. Atherosclerosis 1982, 45, 161–179. [Google Scholar] [CrossRef]
- Zambon, A.; Deeb, S.S.; Pauletto, P.; Crepaldi, G.; Brunzell, J.D. Hepatic lipase: A marker for cardiovascular disease risk and response to therapy. Curr. Opin. Lipidol. 2003, 14, 179–189. [Google Scholar] [CrossRef]
- McCaskie, P.; Cadby, G.; Hung, J.; McQuillan, B.; Chapman, C.; Carter, K.; Thompson, P.; Palmer, L.; Beilby, J.; McQuillan, B.; et al. The C-480T hepatic lipase polymorphism is associated with HDL-C but not with risk of coronary heart disease. Clin. Genet. 2006, 70, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.T. Quantile-Dependent Expressivity and Gene-Lifestyle Interactions Involving High-Density Lipoprotein Cholesterol. Lifestyle Genom. 2021, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Little, J.; Connelly, P.W. Compound heterozygosity for mutant hepatic lipase in familial hepatic lipase deficiency. Biochem. Biophys. Res. Commun. 1991, 179, 78–84. [Google Scholar] [CrossRef]
- Knudsen, P.; Antikainen, M.; Ehnholm, S.; Uusi-Oukari, M.; Tenkanen, H.; Lahdenpera, S.; Kahri, J.; Tilly-Kiesi, M.; Bensadoun, A.; Taskinen, M.R.; et al. A compound heterozygote for hepatic lipase gene mutations Leu334>Phe and Thr383>Met: Cor-relation between hepatic lipase activity and phenotypic expression. J. Lipid Res. 1996, 37, 825–834. [Google Scholar] [CrossRef]
- Brand, K.; Dugi, K.A.; Brunzell, J.D.; Nevin, D.N.; Santamarina-Fojo, S. A novel A-->G mutation in intron I of the hepatic lipase gene leads to alternative splicing resulting in enzyme deficiency. J. Lipid Res. 1996, 37, 213–223. [Google Scholar] [CrossRef]
- Hegele, R.; Lupien, P.; Gagne, C.; Brun, L.; Little, J.; Connelly, P.; Vezina, C.; Moorjani, S. A Hepatic Lipase Gene Mutation Associated with Heritable Lipolytic Deficiency. J. Clin. Endocrinol. Metab. 1991, 72, 730–732. [Google Scholar] [CrossRef]
- Ruel, I.L.; Couture, P.; Gagné, C.; Deshaies, Y.; Simard, J.; Hegele, R.A.; Lamarche, B. Characterization of a novel mutation causing hepatic lipase deficiency among French Canadians. J. Lipid Res. 2003, 44, 1508–1514. [Google Scholar] [CrossRef]
- Gangabadage, C.S.; Zdunek, J.; Tessari, M.; Nilsson, S.; Olivecrona, G.; Wijmenga, S.S. Structure and Dynamics of Human Apolipoprotein CIII. J. Biol. Chem. 2008, 283, 17416–17427. [Google Scholar] [CrossRef]
- Pollin, T.I.; Damcott, C.M.; Shen, H.; Ott, S.H.; Shelton, J.; Horenstein, R.B.; Post, W.; McLenithan, J.C.; Bielak, L.F.; Peyser, P.A.; et al. A Null Mutation in Human APOC3 Confers a Favorable Plasma Lipid Profile and Apparent Cardioprotection. Science 2008, 322, 1702–1705. [Google Scholar] [CrossRef]
- Crosby, J.H.; Peloso, G.M.; Auer, P.L.; Crosslin, D.R.; Stitziel, N.; Lange, L.A.; Lu, Y.; Tang, Z.Z.; Zhang, H.; Hindy, G.; et al. Loss-of-Function Mutations in APOC3, Triglycerides, and Coronary Disease. N. Engl. J. Med. 2014, 371, 22–31. [Google Scholar] [CrossRef]
- Bochem, A.; Van Capelleveen, J.; Dallinga-Thie, G.; Schimmel, A.; Motazacker, M.; Tietjen, I.; Singaraja, R.; Hayden, M.; Kastelein, J.; Stroes, E.; et al. Two novel mutations in apolipoprotein C3 underlie atheroprotective lipid profiles in families. Clin. Genet. 2013, 85, 433–440. [Google Scholar] [CrossRef]
- Von Eckardstein, A.; Holz, H.; Sandkamp, M.; Weng, W.; Funke, H.; Assmann, G. Apolipoprotein C-III(Lys58----Glu). Identification of an apolipoprotein C-III variant in a family with hyperalphalipoproteinemia. J. Clin. Investig. 1991, 87, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Dai, Y.; Mineo, C. Novel Functions of Endothelial Scavenger Receptor Class B Type I. Curr. Atheroscler. Rep. 2021, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, R.B.; Lammers, B.; Meurs, I.; Korporaal, S.J.; De Haan, W.; Zhao, Y.; Kruijt, J.K.; Praticò, D.; Schimmel, A.W.; Holleboom, A.G.; et al. Restoration of High-Density Lipoprotein Levels by Cholesteryl Ester Transfer Protein Expression in Scavenger Receptor Class B Type I (SR-BI) Knockout Mice Does Not Normalize Pathologies Associated with SR-BI Deficiency. Arter. Thromb. Vasc. Biol. 2010, 30, 1439–1445. [Google Scholar] [CrossRef][Green Version]
- Hoekstra, M.; Meurs, I.; Koenders, M.; Out, R.; Hildebrand, R.B.; Kruijt, J.K.; Van Eck, M.; Van Berkel, T.J.C. Absence of HDL cholesteryl ester uptake in mice via SR-BI impairs an adequate adrenal glucocorticoid-mediated stress response to fasting. J. Lipid Res. 2008, 49, 738–745. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Sahoo, D. Functional Characterization of Newly-Discovered Mutations in Human SR-BI. PLoS ONE 2012, 7, e45660. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brunham, L.R.; Tietjen, I.; Bochem, A.E.; Singaraja, R.R.; Franchini, P.L.; Radomski, C.; Mattice, M.; Legendre, A.; Hovingh, G.K.; Kastelein, J.J.P.; et al. Novel mutations in scavenger receptor BI associated with high HDL cholesterol in humans. Clin. Genet. 2011, 79, 575–581. [Google Scholar] [CrossRef]
- Vergeer, M.; Korporaal, S.J.; Franssen, R.; Meurs, I.; Out, R.; Hovingh, G.K.; Hoekstra, M.; Sierts, J.A.; Dallinga-Thie, G.M.; Motazacker, M.M.; et al. Genetic Variant of the Scavenger Receptor BI in Humans. N. Engl. J. Med. 2011, 364, 136–145. [Google Scholar] [CrossRef]
- Helgadottir, A.; Sulem, P.; Thorgeirsson, G.; Gretarsdottir, S.; Thorleifsson, G.; Jensson, B.Ö.; Arnadottir, G.A.; Olafsson, I.; Eyjolfsson, G.I.; Sigurdardottir, O.; et al. Rare SCARB1 mutations associate with high-density lipoprotein cholesterol but not with coronary artery disease. Eur. Heart J. 2018, 39, 2172–2178. [Google Scholar] [CrossRef]
- Davidson, J.; Rotondo, D. Scavenger receptor B1 mutation, elevated HDL cholesterol and a paradoxical increase in atherosclerosis. Curr. Opin. Lipidol. 2016, 27, 541–542. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sethi, A.; Yanek, L.R.; Knapper, C.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; Becker, D.M.; Mathias, R.A.; Remaley, A.T.; Becker, L.C. SCARB1Gene Variants Are Associated with the Phenotype of Combined High High-Density Lipoprotein Cholesterol and High Lipoprotein (a). Circ. Cardiovasc. Genet. 2016, 9, 408–418. [Google Scholar] [CrossRef]
- Schilcher, I.; Ledinski, G.; Radulović, S.; Hallström, S.; Eichmann, T.; Madl, T.; Zhang, F.; Leitinger, G.; Kolb-Lenz, D.; Darnhofer, B.; et al. Endothelial lipase increases antioxidative capacity of high-density lipoprotein. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 1363–1374. [Google Scholar] [CrossRef]
- Jahangiri, A.; Rader, D.J.; Marchadier, D.; Curtiss, L.K.; Bonnet, D.J.; Rye, K.-A. Evidence that endothelial lipase remodels high density lipoproteins without mediating the dissociation of apolipoprotein A-I. J. Lipid Res. 2005, 46, 896–903. [Google Scholar] [CrossRef]
- Schilcher, I.; Kern, S.; Hrzenjak, A.; Eichmann, T.O.; Stojakovic, T.; Scharnagl, H.; Duta-Mare, M.; Kratky, D.; Marsche, G.; Frank, S. Impact of Endothelial Lipase on Cholesterol Efflux Capacity of Serum and High-density Lipoprotein. Sci. Rep. 2017, 7, 12485. [Google Scholar] [CrossRef]
- Escolà-Gil, J.C.; Chen, X.; Julve, J.; Quesada, H.; Santos, D.; Metso, J.; Tous, M.; Jauhiainen, M.; Blanco-Vaca, F. Hepatic lipase- and endothelial lipase-deficiency in mice promotes macrophage-to-feces RCT and HDL antioxidant properties. Biochim. et Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 691–697. [Google Scholar] [CrossRef]
- Edmondson, A.C.; Brown, R.J.; Kathiresan, S.; Cupples, L.A.; Demissie, S.; Manning, A.K.; Jensen, M.K.; Rimm, E.B.; Wang, J.; Rodrigues, A.; et al. Loss-of-function variants in endothelial lipase are a cause of elevated HDL cholesterol in humans. J. Clin. Investig. 2009, 119, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Khetarpal, S.A.; Edmondson, A.C.; Raghavan, A.; Neeli, H.; Jin, W.; Badellino, K.O.; Demissie, S.; Manning, A.K.; DerOhannessian, S.L.; Wolfe, M.L.; et al. Mining the LIPG Allelic Spectrum Reveals the Contribution of Rare and Common Regulatory Variants to HDL Cholesterol. PLoS Genet. 2011, 7, e1002393. [Google Scholar] [CrossRef]
- Singaraja, R.R.; Sivapalaratnam, S.; Hovingh, K.; Dubé, M.-P.; Castro-Perez, J.; Collins, H.L.; Adelman, S.J.; Riwanto, M.; Manz, J.; Hubbard, B.; et al. The Impact of Partial and Complete Loss-of-Function Mutations in Endothelial Lipase on High-Density Lipoprotein Levels and Functionality in Humans. Circ. Cardiovasc. Genet. 2013, 6, 54–62. [Google Scholar] [CrossRef]
- Boekholdt, S.M.; Thompson, J.F. Natural genetic variation as a tool in understanding the role of CETP in lipid levels and disease. J. Lipid Res. 2003, 44, 1080–1093. [Google Scholar] [CrossRef]
- Wu, Z.; Lou, Y.; Qiu, X.; Liu, Y.; Lu, L.; Chen, Q.; Jin, W. Association of cholesteryl ester transfer protein (CETP) gene polymorphism, high density lipoprotein cholesterol and risk of coronary artery disease: A meta-analysis using a Mendelian randomization approach. BMC Med. Genet. 2014, 15, 1–17. [Google Scholar] [CrossRef]
- Thompson, A.; Di Angelantonio, E.; Sarwar, N.; Erqou, S.; Saleheen, D.; Dullaart, R.P.F.; Keavney, B.; Ye, Z.; Danesh, J. Association of Cholesteryl Ester Transfer Protein Genotypes with CETP Mass and Activity, Lipid Levels, and Coronary Risk. JAMA 2008, 299, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Klerkx, A.H.; Tanck, M.W.; Kastelein, J.J.; Molhuizen, H.O.; Jukema, J.W.; Zwinderman, A.H.; Kuivenhoven, J.A. Haplotype analysis of the CETP gene: Not TaqIB, but the closely linked -629C->A polymorphism and a novel promoter variant are independently associated with CETP concentration. Hum. Mol. Genet. 2003, 12, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ordovas, J.M.; Cupples, L.A.; Corella, D.; Otvos, J.D.; Osgood, D.; Martinez, A.; Lahoz, C.; Coltell, O.; Wilson, P.W.F.; Schaefer, E.J. Association of Cholesteryl Ester Transfer Protein–TaqIB Polymorphism with Variations in Lipoprotein Subclasses and Coronary Heart Disease Risk. Arter. Thromb. Vasc. Biol. 2000, 20, 1323–1329. [Google Scholar] [CrossRef]
- Brousseau, M.E.; O’Connor, J.J.; Ordovas, J.M.; Collins, D.; Otvos, J.D.; Massov, T.; McNamara, J.R.; Rubins, H.B.; Robins, S.J.; Schaefer, E.J. Cholesteryl Ester Transfer ProteinTaqI B2B2 Genotype Is Associated with Higher HDL Cholesterol Levels and Lower Risk of Coronary Heart Disease End Points in Men With HDL Deficiency. Arter. Thromb. Vasc. Biol. 2002, 22, 1148–1154. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Freeman, D.J.; Samani, N.J.; Wilson, V.; McMahon, A.D.; Braund, P.S.; Cheng, S.; Caslake, M.J.; Packard, C.J.; Gaffney, D. A polymorphism of the cholesteryl ester transfer protein gene predicts cardiovascular events in non-smokers in the West of Scotland Coronary Prevention Study. Eur. Hear. J. 2003, 24, 1833–1842. [Google Scholar] [CrossRef]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Jiang, X.-C.; Poirier, O.; Lackner, K.J.; Meyer, J.; Cambien, F.; Tiret, L. Common genetic variation of the cholesteryl ester transfer protein gene strongly predicts future cardiovascular death in patients with coronary artery disease. J. Am. Coll. Cardiol. 2003, 41, 1983–1989. [Google Scholar] [CrossRef]
- Lu, H.; Inazu, A.; Moriyama, Y.; Higashikata, T.; Kawashiri, M.-A.; Yu, W.; Huang, Z.; Okamura, T.; Mabuchi, H. Haplotype analyses of cholesteryl ester transfer protein gene promoter: A clue to an unsolved mystery of TaqIB polymorphism. J. Mol. Med. 2003, 81, 246–255. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Yang, Z.-X.; Zhou, D.-F.; Su, Y.; Tang, L.; Qiu, Y.-M.; Cai, W.-W. CETP polymorphisms confer genetic contribution to centenarians of Hainan, south of China. Asian Pac. J. Trop. Med. 2016, 9, 872–876. [Google Scholar] [CrossRef]
- Motazacker, M.M.; Peter, J.; Treskes, M.; Shoulders, C.C.; Kuivenhoven, J.A.; Hovingh, G.K. Evidence of a Polygenic Origin of Extreme High-Density Lipoprotein Cholesterol Levels. Arter. Thromb. Vasc. Biol. 2013, 33, 1521–1528. [Google Scholar] [CrossRef]
- Boes, E.; Coassin, S.; Kollerits, B.; Heid, I.M.; Kronenberg, F. Genetic-epidemiological evidence on genes associated with HDL cholesterol levels: A systematic in-depth review. Exp. Gerontol. 2009, 44, 136–160. [Google Scholar] [CrossRef] [PubMed]
- Weissglas-Volkov, D.; Pajukanta, P. Genetic causes of high and low serum HDL-cholesterol. J. Lipid Res. 2010, 51, 2032–2057. [Google Scholar] [CrossRef]
- Kuwano, T.; Bi, X.; Cipollari, E.; Yasuda, T.; Lagor, W.R.; Szapary, H.J.; Tohyama, J.; Millar, J.S.; Billheimer, J.T.; Lyssenko, N.N.; et al. Overexpression and deletion of phospholipid transfer protein reduce HDL mass and cholesterol efflux capacity but not macrophage reverse cholesterol transport. J. Lipid Res. 2017, 58, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Oates, C.P.; Koenig, D.; Rhyne, J.; Bogush, N.; O’Connell, J.; Mitchell, B.D.; Miller, M. Novel polymorphisms associated with hyperalphalipoproteinemia and apparent cardioprotection. J. Clin. Lipidol. 2018, 12, 110–115. [Google Scholar] [CrossRef]
- Huggins, G.S.; Papandonatos, G.; Erar, B.; Belalcazar, L.M.; Brautbar, A.; Ballantyne, C.; Kitabchi, A.E.; Wagenknecht, L.E.; Knowler, W.C.; Pownall, H.; et al. Do Genetic Modifiers of High-Density Lipoprotein Cholesterol and Triglyceride Levels also Modify Their Response to a Lifestyle Intervention in the Setting of Obesity and Type-2 Diabetes Mellitus? Circ. Cardiovasc. Genet. 2013, 6, 391–399. [Google Scholar] [CrossRef]
- Kathiresan, S.; Willer, C.J.; Peloso, G.M.; Demissie, S.; Musunuru, K.; Schadt, E.E.; Kaplan, L.; Bennett, D.; Li, Y.; Tanaka, T.; et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2008, 41, 56–65. [Google Scholar] [CrossRef]
- Gofman, J.W.; Young, W.; Tandy, R. Ischemic Heart Disease, Atherosclerosis, and Longevity. Circulation 1966, 34, 679–697. [Google Scholar] [CrossRef]
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High density lipoprotein as a protective factor against coronary heart disease: The Framingham Study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef]
- Rye, K.-A.; Barter, P.J. Cardioprotective functions of HDLs. J. Lipid Res. 2014, 55, 168–179. [Google Scholar] [CrossRef]
- Feghaly, J.; Mooradian, A.D. The Rise and Fall “ing” of the HDL Hypothesis. Drugs 2020, 80, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Hamer, M.; O’Donovan, G.; Stamatakis, E. High-Density Lipoprotein Cholesterol and Mortality. Arter. Thromb. Vasc. Biol. 2018, 38, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Rahilly-Tierney, C.R.; Spiro, A.; Vokonas, P.; Gaziano, J.M. Relation Between High-Density Lipoprotein Cholesterol and Survival to Age 85 Years in Men (from the VA Normative Aging Study). Am. J. Cardiol. 2011, 107, 1173–1177. [Google Scholar] [CrossRef]
- Feitosa, M.F.; Lunetta, K.L.; Wang, L.; Wojczynski, M.K.; Kammerer, C.M.; Perls, T.; Schupf, N.; Christensen, K.; Murabito, J.M.; Province, M.A. Gene discovery for high-density lipoprotein cholesterol level change over time in prospective family studies. Atherosclerosis 2020, 297, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.; Genest, J. HDL cholesterol and ASCVD risk stratification: A debate. Atherosclerosis 2019, 283, 7–12. [Google Scholar] [CrossRef]
- Boekholdt, S.M.; Arsenault, B.J.; Hovingh, G.K.; Mora, S.; Pedersen, T.R.; LaRosa, J.C.; Welch, K.; Amarenco, P.; DeMicco, D.A.; Tonkin, A.M.; et al. Levels and Changes of HDL Cholesterol and Apolipoprotein A-I in Relation to Risk of Cardiovascular Events Among Statin-Treated Patients. Circulation 2013, 128, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Demler, O.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events. Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Rader, D.J. Macrophage Reverse Cholesterol Transport. Circulation 2006, 113, 2548–2555. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Hear. J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.M.; Nordestgaard, B.G. Is It Time for New Thinking About High-Density Lipoprotein? Arter. Thromb. Vasc. Biol. 2018, 38, 484–486. [Google Scholar] [CrossRef]
- Agerholm-Larsen, B.; Tybjærg-Hansen, A.; Schnohr, P.; Steffensen, R.; Nordestgaard, B.G. Common Cholesteryl Ester Transfer Protein Mutations, Decreased HDL Cholesterol, and Possible Decreased Risk of Ischemic Heart Disease. Circulation 2000, 102, 2197–2203. [Google Scholar] [CrossRef]
- Ko, D.T.; Alter, D.A.; Guo, H.; Koh, M.; Lau, G.; Austin, P.C.; Booth, G.L.; Hogg, W.; Jackevicius, C.A.; Lee, D.; et al. High-Density Lipoprotein Cholesterol and Cause-Specific Mortality in Individuals without Previous Cardiovascular Conditions. J. Am. Coll. Cardiol. 2016, 68, 2073–2083. [Google Scholar] [CrossRef]
- Bowe, B.; Xie, Y.; Xian, H.; Balasubramanian, S.; Zayed, M.A.; Al-Aly, Z. High Density Lipoprotein Cholesterol and the Risk of All-Cause Mortality among U.S. Veterans. Clin. J. Am. Soc. Nephrol. 2016, 11, 1784–1793. [Google Scholar] [CrossRef]
- Siddiqi, H.K.; Kiss, D.; Rader, D. HDL-cholesterol and cardiovascular disease. Curr. Opin. Cardiol. 2015, 30, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Sharma, A.; Krishnamoorthy, P.; Garg, J.; Virmani, D.; Sharma, T.; Stefanini, G.; Kostis, J.B.; Mukherjee, D.; Sikorskaya, E. Role of Niacin in Current Clinical Practice: A Systematic Review. Am. J. Med. 2017, 130, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Schandelmaier, S.; Briel, M.; Saccilotto, R.; Olu, K.K.; Arpagaus, A.; Hemkens, L.; Nordmann, A.J. Niacin for primary and secondary prevention of cardiovascular events. Cochrane Database Syst. Rev. 2017, 6, CD009744. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, G.; Favari, E.; Calabresi, L.; Simonelli, S.; Bondioli, A.; Adorni, M.P.; Zimetti, F.; Gomaraschi, M.; Coutant, K.; Rossomanno, S.; et al. Differential effects of fenofibrate and extended-release niacin on high-density lipoprotein particle size distribution and cholesterol efflux capacity in dyslipidemic patients. J. Clin. Lipidol. 2013, 7, 414–422. [Google Scholar] [CrossRef]
- D’Andrea, E.; Hey, S.P.; Ramirez, C.L.; Kesselheim, A.S. Assessment of the Role of Niacin in Managing Cardiovascular Disease Outcomes: A Systematic Review and Meta-analysis. JAMA Netw. Open 2019, 2, e192224. [Google Scholar] [CrossRef] [PubMed]
- The AIM-HIGH Investigators. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef]
- The HPS2-THRIVE Collaborative Group. Effects of Extended-Release Niacin with Laropiprant in High-Risk Patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Chapman, M.J.; Robillard, P.; Krempf, M.; Laville, M.; Borén, J. Niacin action in the atherogenic mixed dyslipidemia of metabolic syndrome: Insights from metabolic biomarker profiling and network analysis. J. Clin. Lipidol. 2018, 12, 810–821. [Google Scholar] [CrossRef]
- Canner, P.L.; Berge, K.G.; Wenger, N.K.; Stamler, J.; Friedman, L.; Prineas, R.J.; Friedewald, W. Fifteen year mortality in Coronary Drug Project patients: Long-term benefit with niacin. J. Am. Coll. Cardiol. 1986, 8, 1245–1255. [Google Scholar] [CrossRef]
- Taylor, A.J.; Sullenberger, L.E.; Lee, H.J.; Lee, J.K.; Grace, K.A. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2. Circulation 2004, 110, 3512–3517. [Google Scholar] [CrossRef]
- Villines, T.C.; Stanek, E.J.; Devine, P.J.; Turco, M.; Miller, M.; Weissman, N.J.; Griffen, L.; Taylor, A.J. The ARBITER 6-HALTS Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6–HDL and LDL Treatment Strategies in Atherosclerosis): Final Results and the Impact of Medication Adherence, Dose, and Treatment Duration. J. Am. Coll. Cardiol. 2010, 55, 2721–2726. [Google Scholar] [CrossRef]
- Whitney, E.J.; Krasuski, R.A.; Personius, B.E.; Michalek, J.E.; Maranian, A.M.; Kolasa, M.W.; Monick, E.; Brown, B.G.; Gotto, A.M. A Randomized Trial of a Strategy for Increasing High-Density Lipoprotein Cholesterol Levels: Effects on Progression of Coronary Heart Disease and Clinical Events. Ann. Intern. Med. 2005, 142, 95–104. [Google Scholar] [CrossRef]
- Blankenhorn, D.H.; Nessim, S.A.; Johnson, R.L.; Sanmarco, M.E.; Azen, S.P.; Cashin-Hemphill, L. Beneficial effects of combined coles-tipol-niacin therapy on coronary atherosclerosis and coronary venous bypass grafts. JAMA 1987, 257, 3233–3240. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.; Danielson, M.; Ekberg, I.; Klintemar, B.; Rosenhamer, G. Reduction of myocardial reinfarction by the combined treatment with clofibrate and nicotinic acid. Atherosclerosis 1977, 28, 81–86. [Google Scholar] [CrossRef]
- Williams, P.T.; Zhao, X.-Q.; Marcovina, S.M.; Brown, B.G.; Krauss, R.M. Levels of Cholesterol in Small LDL Particles Predict Atherosclerosis Progression and Incident CHD in the HDL-Atherosclerosis Treatment Study (HATS). PLoS ONE 2013, 8, e56782. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Brandrup-Wognsen, G.; Palmer, M.; Barter, P.J. Meta-analysis of Comparative Efficacy of Increasing Dose of Atorvastatin Versus Rosuvastatin Versus Simvastatin on Lowering Levels of Atherogenic Lipids (from VOYAGER). Am. J. Cardiol. 2010, 105, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Luna, D.; Ortiz-Rodriguez, M.A.; Medina-Briseño, L.; Carreón-Torres, E.; Izquierdo-Vega, J.A.; Sharma, A.; Cancino-Díaz, J.C.; Pérez-Méndez, O.; Belefant-Miller, H.; Betanzos-Cabrera, G. Current Therapies Focused on High-Density Lipoproteins Associated with Cardiovascular Disease. Molecules 2018, 23, 2730. [Google Scholar] [CrossRef]
- Tall, A.R.; Rader, D.J. Trials and Tribulations of CETP Inhibitors. Circ. Res. 2018, 122, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Bots, M.L.; Visseren, F.; Evans, G.W.; Riley, W.A.; Revkin, J.H.; Tegeler, C.H.; Shear, C.L.; Duggan, W.T.; Vicari, R.M.; Grobbee, D.E.; et al. Torcetrapib and carotid intima-media thickness in mixed dyslipidaemia (RADIANCE 2 study): A randomised, double-blind trial. Lancet 2007, 370, 153–160. [Google Scholar] [CrossRef]
- Barter, P.J.; Rye, K.-A.; Tardif, J.-C.; Waters, D.D.; Boekholdt, S.M.; Breazna, A.; Kastelein, J.J. Effect of Torcetrapib on Glucose, Insulin, and Hemoglobin A 1c in Subjects in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) Trial. Circulation 2011, 124, 555–562. [Google Scholar] [CrossRef]
- Clerc, R.G.; Stauffer, A.; Weibel, F.; Hainaut, E.; Perez, A.; Hoflack, J.-C.; Bénardeau, A.; Pflieger, P.; Garriz, J.M.; Funder, J.W.; et al. Mechanisms underlying off-target effects of the cholesteryl ester transfer protein inhibitor torcetrapib involve L-type calcium channels. J. Hypertens. 2010, 28, 1676–1686. [Google Scholar] [CrossRef]
- Salahuddin, T.; Kittelson, J.; Tardif, J.-C.; Shah, P.K.; Olsson, A.G.; Nicholls, S.J.; Leitersdorf, E.; Leiter, L.A.; Kallend, D.; Black, D.M.; et al. Association of high-density lipoprotein particle concentration with cardiovascular risk following acute coronary syndrome: A case-cohort analysis of the dal-Outcomes trial. Am. Hear. J. 2020, 221, 60–66. [Google Scholar] [CrossRef]
- Bagdade, J.; Barter, P.; Quiroga, C.; Alaupovic, P. Effects of Torcetrapib and Statin Treatment on ApoC-III and Apoprotein-Defined Lipoprotein Subclasses (from the ILLUMINATE Trial). Am. J. Cardiol. 2017, 119, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Nicholls, S.J.; Kastelein, J.J.P.; Rye, K.-A. Is Cholesteryl Ester Transfer Protein Inhibition an Effective Strategy to Reduce Cardiovascular Risk? Circulation 2015, 132, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Noto, D.; Cefalu’, A.B.; Averna, M.R. Beyond Statins: New Lipid Lowering Strategies to Reduce Cardiovascular Risk. Curr. Atheroscler. Rep. 2014, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Woudberg, N.; Pedretti, S.; Lecour, S.; Schulz, R.; Vuilleumier, N.; James, R.W.; Frias, M.A. Pharmacological Intervention to Modulate HDL: What Do We Target? Front. Pharmacol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Clinical Trial | Description | Effect on HDL-C and CV Outcomes | Reference |
|---|---|---|---|
| AIM-HIGH | 3414 subjects were treated with niacin or placebo on top of high-intensity statin treatment | The niacin-treated arm showed a modest but significant HDL-C increase (25% vs. 12%) but no benefits in terms of cardiovascular outcomes. The trial was stopped earlier due to futility of results. | [144] |
| HPS2-THRIVE | 25673 subjects affected by vascular diseases were randomized to receive extended-release niacin + laropiprant (to reduce the flushing side-effect of niacin) or placebo both on top of statin therapy | After a follow up mean of 3.9 year, the niacin-treated group showed a modest but significant HDL-C increase (6 mg/dl), but no difference in the incidence of CV events. | [145] |
| Niacin Study Group | Males with metabolic syndrome (obese, hypertriglyceridemic, non-diabetic) and low HDL-C levels received niacin for 8 weeks | A decrease in LDL-C and total cholesterol associated to a reduction in inflammation, cell-adhesion and proliferation biomarkers. No significant change of CV events. | [146] |
| Coronary Drug Project (CDP) | 8 341 Males after myocardial infarction treated with niacin or clofibrate vs. placebo | HDL-C increase, LDL-C and TG decrease. No significant change of CV events. | [147] |
| ARBITER-2 | 167 patients with Coronary Artery Disease were treated with ER-niacin 1 g/day vs. placebo on top of stable statin therapy | HDL-C increase by 21%. Progression of cIMT in the niacin group | [148] |
| ARBITER-6 | 208 patients (≥30 years) with CAD or equivalent of CAD risk were treated with ER-niacin vs. ezetimibe on top of statin therapy | HDL-C increase in ER-Niacin group; reduced incidence of cardiovascular events by 5% in ER-Niacin group vs. 1% in EZE-group | [149] |
| AFREGS | 143 patients (<76 years) with low HDL-C and coronary disease were treated with Niacin 0.25–3 g gemfibrozil 1.2 g cholestyramine 2 g vs. placebo | HDL-C increase by 36%; 13.7% decrease of combined cardiovascular events. No significant data. | [150] |
| CLAS | 162 Males after CABG treated with Niacin 3–12 g/day + colestipol 30 g/day vs. placebo | HDL-C increase by 31%; TC decrease by 15–20% and LDL-C decrease by 43%; atherosclerotic regression in 16.2% of patients at 2 years and 17.9% at 4 years, compared with 2.4% and 6.4%, respectively, in the placebo group | [151] |
| Stockholm trial | 558 patients after MI, aged <70 treated with Clofibrate 2×1 g + niacin 3×1 g vs. placebo | TC decrease by 26%; TG decrease by 30%; nonfatal Miocardial Iinfarction decrease by 50% | [152] |
| HATS | 160 patients with CAD and low HDL-C distributed in 4 arms and treated with: Group A: simvastatin 10–20 mg/d plus niacin 2–4 g/d; Group B: antioxidant; Group C: simvastatin + niacin + antioxidant; Group D: placebo | LDL-C decrease by 42%; HDL-C increase by 26%; regression of severe coronary stenosis by 0.4% vs. placebo; 88% decrease of CV events (coronary death, MI or stroke, or revascularization) | [153] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giammanco, A.; Noto, D.; Barbagallo, C.M.; Nardi, E.; Caldarella, R.; Ciaccio, M.; Averna, M.R.; Cefalù, A.B. Hyperalphalipoproteinemia and Beyond: The Role of HDL in Cardiovascular Diseases. Life 2021, 11, 581. https://doi.org/10.3390/life11060581
Giammanco A, Noto D, Barbagallo CM, Nardi E, Caldarella R, Ciaccio M, Averna MR, Cefalù AB. Hyperalphalipoproteinemia and Beyond: The Role of HDL in Cardiovascular Diseases. Life. 2021; 11(6):581. https://doi.org/10.3390/life11060581
Chicago/Turabian StyleGiammanco, Antonina, Davide Noto, Carlo Maria Barbagallo, Emilio Nardi, Rosalia Caldarella, Marcello Ciaccio, Maurizio Rocco Averna, and Angelo Baldassare Cefalù. 2021. "Hyperalphalipoproteinemia and Beyond: The Role of HDL in Cardiovascular Diseases" Life 11, no. 6: 581. https://doi.org/10.3390/life11060581
APA StyleGiammanco, A., Noto, D., Barbagallo, C. M., Nardi, E., Caldarella, R., Ciaccio, M., Averna, M. R., & Cefalù, A. B. (2021). Hyperalphalipoproteinemia and Beyond: The Role of HDL in Cardiovascular Diseases. Life, 11(6), 581. https://doi.org/10.3390/life11060581