
Antiplatelet Effects of PCSK9 Inhibitors in Primary Hypercholesterolemia

 , ,
, ,  ,
,  , and
, and 
Abstract
1. Introduction
2. Pathophysiology
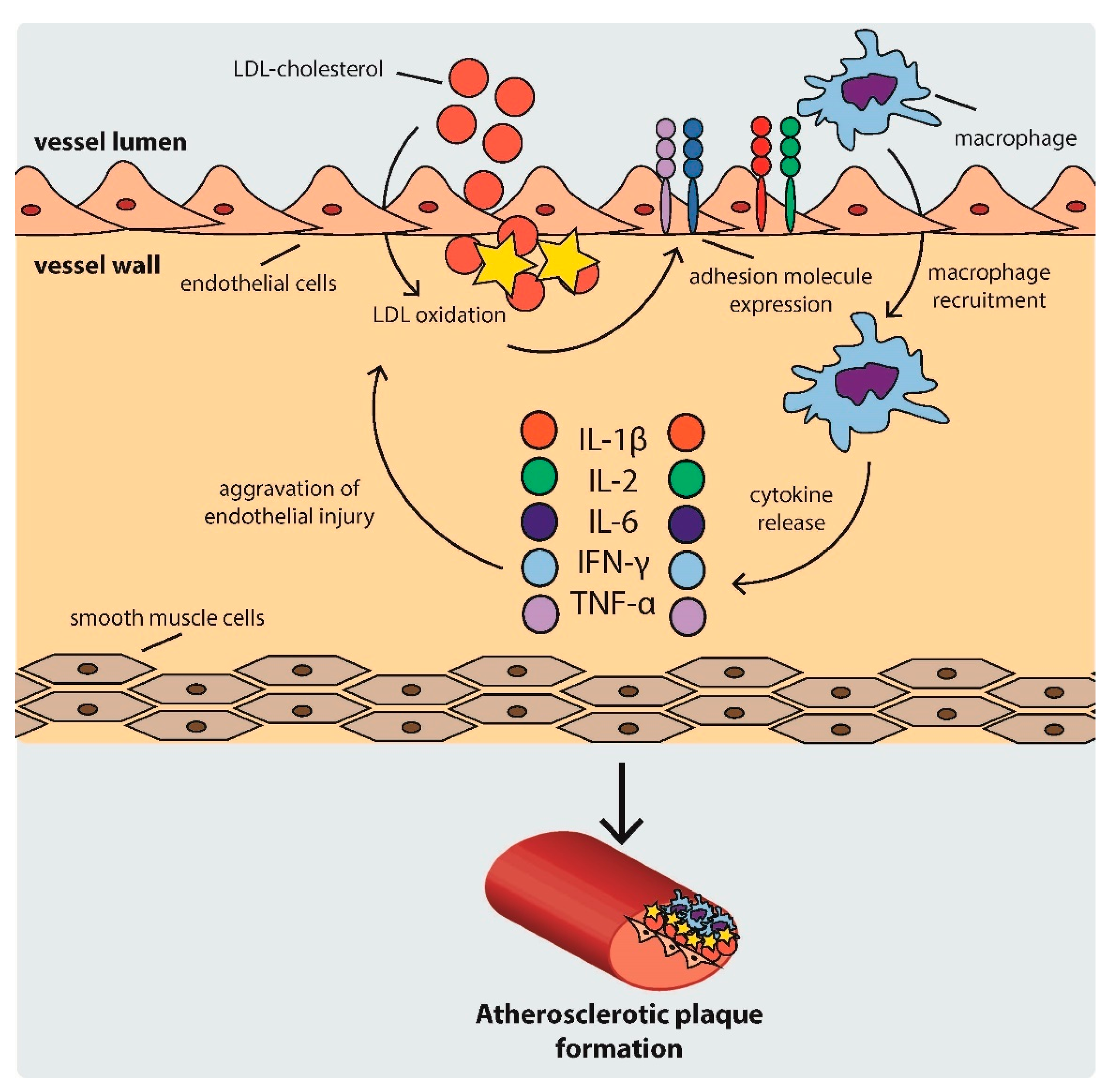
2.1. Development of Atherosclerotic Plaques
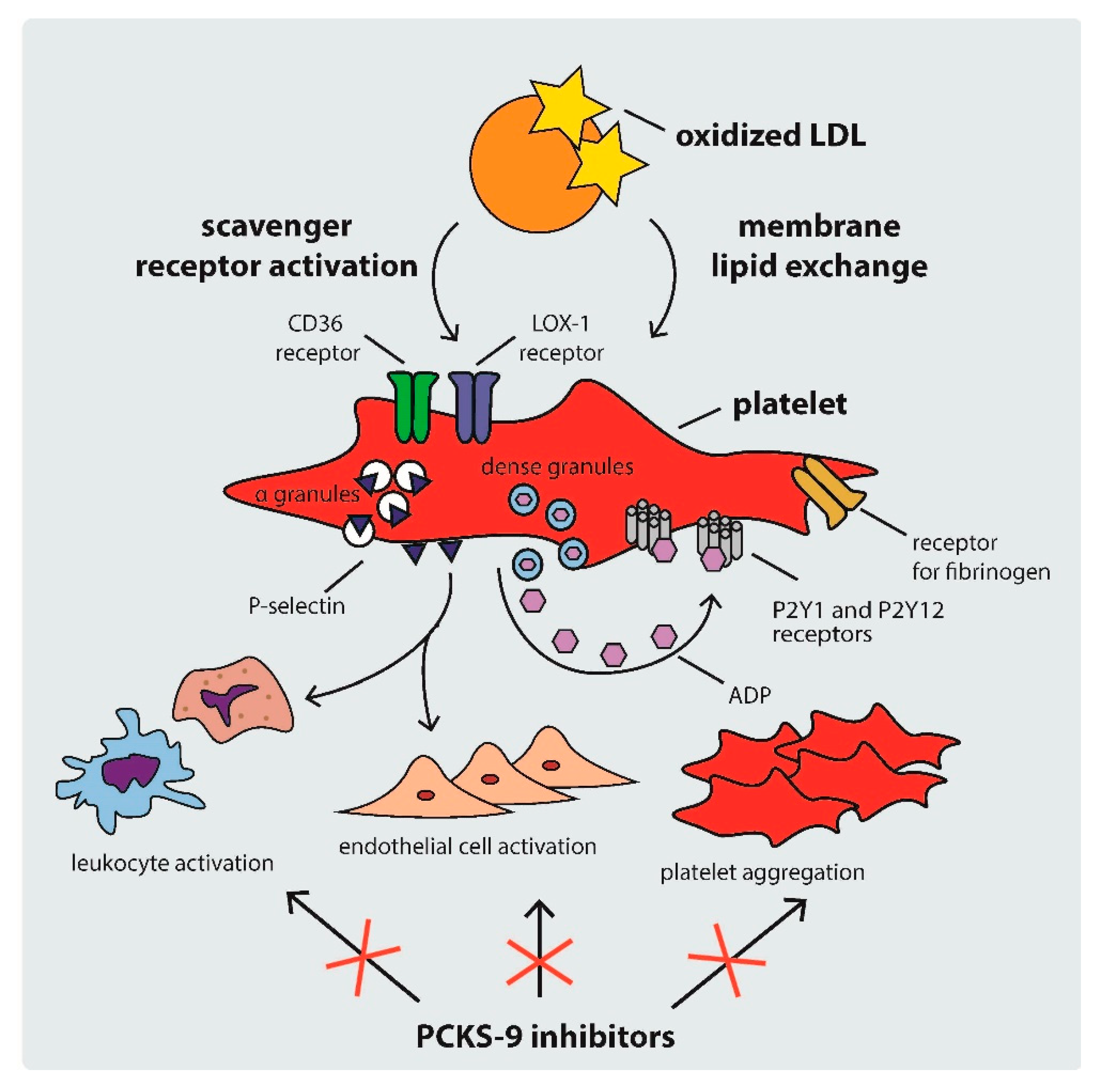
2.2. Mechanisms of Platelet Activation in Hypercholesterolemia
2.3. Platelet Activation, Atherotogenesis, and Atherothrombosis
Platelet-Derived Extracellular Vesicles
3. Markers of Platelet Activation in PH
3.1. Mean Platelet Volume
3.2. Circulating PEV Levels
3.3. Platelet-Derived Inflammatory Biomarkers
3.4. Platelet–Leukocyte Aggregates
3.5. Platelet-Activating Factor Acetylhydrolase
4. PCSK9 and PCSK9 Inhibitors
4.1. The Role of PCSK9
4.2. PCKS9 and Platelets
4.3. PCSK9 Inhibitors
5. Antiplatelet Effects of PCSK-9 Inhibitors
5.1. PCSK9 Inhibitors
5.2. Statins and PCSK9 Inhibitors
5.3. Ezetimibe and PCSK9 Inhibitors
5.4. Antithrombotic Therapy and PCSK9 Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akioyamen, L.E.; Genest, J.; Shan, S.D.; Reel, R.L.; Albaum, J.M.; Chu, A.; Tu, J.V. Estimating the prevalence of heterozygous familial hypercholesterolaemia: A systematic review and meta-analysis. BMJ Open 2017, 7, e016461. [Google Scholar] [CrossRef]
- Langslet, G.; Emery, M.; Wasserman, S.M. Evolocumab (AMG 145) for primary hypercholesterolemia. Expert Rev. Cardiovasc. Ther. 2015, 13, 477–488. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ôunpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors Collaboration; Di Angelantonio, E.; Gao, P.; Pennells, L.; Kaptoge, S.; Caslake, M.; Thompson, A.; Butterworth, A.S.; Sarwar, N.; Wormser, D.; et al. Lipid-related markers and cardiovascular disease prediction. JAMA 2012, 307, 2499–2506. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Choi, S.; Kim, K.; Kim, S.M.; Lee, G.; Park, S.Y.; Kim, Y.; Son, J.S.; Yun, J.; Park, S.M. Effect of Change in Total Cholesterol Levels on Cardiovascular Disease Among Young Adults. J. Am. Hear. Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Cha, K.S.; Lee, H.W.; Oh, J.-H.; Choi, J.H.; Lee, H.C.; Hong, T.J.; Jeong, M.H.; Chae, S.C.; Kim, Y.J. Predictive and protective role of high-density lipoprotein cholesterol in acute myocardial infarction. Cardiol. J. 2013, 26, 176–185. [Google Scholar] [CrossRef]
- Van der Graaf, A.; Hutten, B.A.; Kastelein, J.J.; Vissers, M.N. Premature cardiovascular disease in young women with heterozygous familial hypercholesterolemia. Expert. Rev. Cardiovasc. Ther. 2006, 4, 345–351. [Google Scholar] [CrossRef]
- Navar-Boggan, A.M.; Peterson, E.D.; D’Agostino, S.R.B.; Neely, B.; Sniderman, A.D.; Pencina, M.J. Hyperlipidemia in Early Adulthood Increases Long-Term Risk of Coronary Heart Disease. Circulation 2015, 131, 451–458. [Google Scholar] [CrossRef]
- Vuorio, A.; Watts, G.F.; Schneider, W.J.; Tsimikas, S.; Kovanen, P.T. Familial hypercholesterolemia and elevated lipoprotein(a): Double heritable risk and new therapeutic opportunities. J. Intern. Med. 2019, 287, 2–18. [Google Scholar] [CrossRef]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int. J. Mol. Sci. 2018, 19, 3426. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.; Stein, E.; Shendure, J.; Hobbs, H.H.; Cohen, J.C. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum. Mol. Genet. 2010, 19, 4313–4318. [Google Scholar] [CrossRef] [PubMed]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; A Cooper, J.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 2013, 381, 1293–1301. [Google Scholar] [CrossRef]
- Yu, L.; Gupta, S.; Xu, F.; Liverman, A.D.B.; Moschetta, A.; Mangelsdorf, D.J.; Repa, J.J.; Hobbs, H.H.; Cohen, J.C. Expression of ABCG5 and ABCG8 Is Required for Regulation of Biliary Cholesterol Secretion. J. Biol. Chem. 2005, 280, 8742–8747. [Google Scholar] [CrossRef]
- Al-Eitan, L.N.; Elsaqa, B.Z.; Almasri, A.Y.; A Aman, H.; Khasawneh, R.H.; A Alghamdi, M. Influence of PSRC1, CELSR2, and SORT1 Gene Polymorphisms on the Variability of Warfarin Dosage and Susceptibility to Cardiovascular Disease. Pharm. Pers. Med. 2020, 13, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Chiva-Blanch, G.; Badimon, L. Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles. J. Clin. Med. 2019, 8, 2059. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, H.; Carbone, F.; Montecucco, F.; Pirro, M.; Sahebkar, A. Oxidative burden in familial hypercholesterolemia. J. Cell. Physiol. 2018, 233, 5716–5725. [Google Scholar] [CrossRef] [PubMed]
- Mickiewicz, A.; Borowiec-Wolna, J.; Bachorski, W.; Gilis-Malinowska, N.; Gałąska, R.; Raczak, G.; Chmara, M.; Wasąg, B.; Jaguszewski, M.J.; Fijałkowski, M.; et al. Long-term lipoprotein apheresis in the treatment of severe familial hypercholesterolemia refractory to high intensity statin therapy: Three year experience at a lipoprotein apheresis centre. Cardiol. J. 2020, 26, 669–679. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Hegele, R.A.; Fazio, S.; Cannon, C.P. The Evolving Future of PCSK9 Inhibitors. J. Am. Coll. Cardiol. 2018, 72, 314–329. [Google Scholar] [CrossRef]
- Haddad, L.; Day, I.N.; Hunt, S.; Williams, R.R.; E Humphries, S.; Hopkins, P.N. Evidence for a third genetic locus causing familial hypercholesterolemia. A non-LDLR, non-APOB kindred. J. Lipid Res. 1999, 40, 1113–1122. [Google Scholar] [CrossRef]
- Varret, M.; Rabès, J.-P.; Saint-Jore, B.; Cenarro, A.; Marinoni, J.-C.; Civeira, F.; Devillers, M.; Krempf, M.; Coulon, M.; Thiart, R.; et al. A Third Major Locus for Autosomal Dominant Hypercholesterolemia Maps to 1p34.1-p. Am. J. Hum. Genet. 1999, 64, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Guedeney, P.; Giustino, G.; Sorrentino, S.; E Claessen, B.; Camaj, A.; Kalkman, D.N.; Vogel, B.; Sartori, S.; De Rosa, S.; Baber, U.; et al. Efficacy and safety of alirocumab and evolocumab: A systematic review and meta-analysis of randomized controlled trials. Eur. Heart J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; Russo, I. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Basiak, M.; Kosowski, M.; Cyrnek, M.; Bułdak, Ł.; Maligłówka, M.; Machnik, G.; Okopień, B. Pleiotropic Effects of PCSK-9 Inhibitors. Int. J. Mol. Sci. 2021, 22, 3144. [Google Scholar] [CrossRef] [PubMed]
- Schwenke, D.C.; E Carew, T. Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arter. Off. J. Am. Hear. Assoc. Inc. 1989, 9, 908–918. [Google Scholar] [CrossRef]
- Heo, K.-S.; Fujiwara, K.; Abe, J.-I. Disturbed-flow-mediated vascular reactive oxygen species induce endothelial dysfunction. Circ. J. 2011, 75, 2722–2730. [Google Scholar] [CrossRef]
- Van Haelst, P.L.; van Doormaal, J.J.; Asselbergs, F.W.; van Roon, A.M.; Veeger, N.J.; Henneman, M.M.; Smit, A.J.; Tervaert, J.W.; May, J.F.; Gans, R.O. Correlates of endothelial function and their relationship with inflammation in patients with familial hypercholesterolaemia. Clin. Sci. 2003, 104, 627–632. [Google Scholar] [CrossRef]
- Rahman, T.; Hamzan, N.S.; Mokhsin, A.; Rahmat, R.; Ibrahim, Z.O.; Razali, R.; Thevarajah, M.; Nawawi, H. Enhanced status of inflammation and endothelial activation in subjects with familial hypercholesterolaemia and their related unaffected family members: A case control study. Lipids Heal. Dis. 2017, 16, 1–12. [Google Scholar] [CrossRef]
- Badrnya, S.; Schrottmaier, W.; Kral, J.B.; Yaiw, K.-C.; Volf, I.; Schabbauer, G.; Söderberg-Nauclér, C.; Assinger, A. Platelets Mediate Oxidized Low-Density Lipoprotein–Induced Monocyte Extravasation and Foam Cell Formation. Arter. Thromb. Vasc. Biol. 2014, 34, 571–580. [Google Scholar] [CrossRef]
- Moore, K.J.; Freeman, M.W. Scavenger receptors in atherosclerosis: Beyond lipid uptake. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef]
- Jukema, R.A.; Ahmed, T.A.N.; Tardif, J.-C. Does low-density lipoprotein cholesterol induce inflammation? If so, does it matter? Current insights and future perspectives for novel therapies. BMC Med. 2019, 17, 1–9. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 1–32. [Google Scholar] [CrossRef]
- Gąsecka, A.; Rogula, S.; Szarpak, Ł.; Filipiak, K.J. LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis. Life 2021, 11, 39. [Google Scholar] [CrossRef]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Valiyaveettil, M.; Podrez, E.A. Platelet hyperreactivity, scavenger receptors and atherothrombosis. J. Thromb. Haemost. 2009, 7, 218–221. [Google Scholar] [CrossRef]
- Hoebe, K.; Georgel, P.; Rutschmann, S.; Du, X.; Mudd, S.; Crozat, K.; Sovath, S.; Shamel, L.; Hartung, T.; Zähringer, U.; et al. CD36 is a sensor of diacylglycerides. Nat. Cell Biol. 2005, 433, 523–527. [Google Scholar] [CrossRef]
- Bodart, V.; Febbraio, M.; Demers, A.; McNicoll, N.; Pohankova, P.; Perreault, A.; Sejlitz, T.; Escher, E.; Silverstein, R.; Lamontagne, D.; et al. CD36 Mediates the Cardiovascular Action of Growth Hormone-Releasing Peptides in the Heart. Circ. Res. 2002, 90, 844–849. [Google Scholar] [CrossRef]
- A Podrez, E.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Silverstein, R.L. CD36 and ERK5 link dyslipidemia to apoptotic-like platelet procoagulant function. Curr. Opin. Hematol. 2019, 26, 357–365. [Google Scholar] [CrossRef]
- Marcus, A.J.; Silk, S.T.; Safier, L.B.; Ullman, H.L. Superoxide production and reducing activity in human platelets. J. Clin. Investig. 1977, 59, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.K.; Jeong, M.H.; Bae, H.R.; Han, J.Y.; Jeong, S.J.; Jin, H.J.; Lim, Y.J.; Kim, S.H.; Kim, J.W. Activated platelets induce secretion of interleukin-1beta, monocyte chemotactic protein-1, and macrophage inflammatory protein-1alpha and surface expression of intercellular adhesion molecule-1 on cultured endothelial cells. J. Korean Med. Sci. 2000, 15, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Siegel-Axel, D.; Daub, K.; Seizer, P.; Lindemann, S.; Gawaz, M. Platelet lipoprotein interplay: Trigger of foam cell formation and driver of atherosclerosis. Cardiovasc. Res. 2008, 78, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ogura, S.; Chen, J.; Little, P.J.; Moss, J.; Liu, P. LOX-1 in atherosclerosis: Biological functions and pharmacological modifiers. Cell. Mol. Life Sci. 2013, 70, 2859–2872. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, T.; Kakino, A.; Fujita, Y. LOX-1: A multiligand receptor at the crossroads of response to danger signals. Curr. Opin. Lipidol. 2012, 23, 439–445. [Google Scholar] [CrossRef]
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Moriwaki, H.; Murase, T.; Sawamura, T.; Masaki, T.; Hashimoto, N.; Kita, T. Expression of Lectinlike Oxidized Low-Density Lipoprotein Receptor-1 in Human Atherosclerotic Lesions. Circulation 1999, 99, 3110–3117. [Google Scholar] [CrossRef]
- Price, J.; Lord, J.M.; Harrison, P. Inflammaging and platelet hyperreactivity: A new therapeutic target? J. Thromb. Haemost. 2020, 18, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Kögl, C.; Kulschar, R.; Schaipp, B. Transfer of phosphatidylcholine, phosphatidylethanolamine and sphingomyelin from low- and high-density lipoprotein to human platelets. Biochem. J. 1996, 315, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, L.; Lam, J.Y.; Hung, J.; Letchacovski, G.; Solymoss, C.B.; Waters, D. Hyperlipidemia and coronary disease. Correction of the increased thrombogenic potential with cholesterol reduction. Circulation 1995, 92, 3172–3177. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Lievens, D.; von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost 2011, 106, 827–838. [Google Scholar] [CrossRef]
- Daub, K.; Seizer, P.; Stellos, K.; Krämer, B.F.; Bigalke, B.; Schaller, M.; Fateh-Moghadam, S.; Gawaz, M.; Lindemann, S. Oxidized LDL-Activated Platelets Induce Vascular Inflammation. Semin. Thromb. Hemost. 2010, 36, 146–156. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Schmitt, M.M. Platelets and their chemokines in atherosclerosis-clinical applications. Front. Physiol. 2014, 5, 294. [Google Scholar] [CrossRef] [PubMed]
- Daub, K.; Langer, H.; Seizer, P.; Stellos, K.; May, A.E.; Goyal, P.; Bigalke, B.; Schönberger, T.; Geisler, T.; Siegel-Axel, D.; et al. Platelets induce differentiation of human CD34 + progenitor cells into foam cells and endothelial cells. FASEB J. 2006, 20, 2559–2561. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Huczek, Z.; Kapłon-Cieślicka, A.; Lacroix, R.; Opolski, G.; et al. Randomized controlled trial protocol to investigate the antiplatelet therapy effect on extracellular vesicles (AFFECT EV) in acute myocardial infarction. Platelets 2018, 31, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Böing, A.N.; Filipiak, K.J.; Nieuwland, R. Platelet extracellular vesicles as biomarkers for arterial thrombosis. Platelets 2016, 28, 228–234. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; Siljander, P.R.-M. Platelet-Derived Extracellular Vesicles. In Platelets; Elsevier: Amsterdam, The Netherlands, 2019; pp. 401–416. [Google Scholar]
- Boilard, E. Thematic Review Series: Exosomes and Microvesicles: Lipids as Key Components of their Biogenesis and Functions Extracellular vesicles and their content in bioactive lipid mediators: More than a sack of microRNA. J. Lipid Res. 2018, 59, 2037–2046. [Google Scholar] [CrossRef]
- Suades, R.; Padró, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219. [Google Scholar] [CrossRef]
- Feng, C.; Chen, Q.; Fan, M.; Guo, J.; Liu, Y.; Ji, T.; Zhu, J.; Zhao, X. Platelet-derived microparticles promote phagocytosis of oxidized low-density lipoprotein by macrophages, potentially enhancing foam cell formation. Ann. Transl. Med. 2019, 7, 477. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Lovren, F.; Verma, S. Evolving Role of Microparticles in the Pathophysiology of Endothelial Dysfunction. Clin. Chem. 2013, 59, 1166–1174. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper-Lenkiewicz, O.M.; Kamińska, J.; Kemona, H.; Dymicka-Piekarska, V. Mean Platelet Volume (MPV): New Perspectives for an Old Marker in the Course and Prognosis of Inflammatory Conditions. Mediat. Inflamm. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Icli, A.; Aksoy, F.; Nar, G.; Kaymaz, H.; Alpay, M.F.; Nar, R.; Guclu, A.; Arslan, A.; Dogan, A. Increased Mean Platelet Volume in Familial Hypercholesterolemia. Angiol. 2015, 67, 146–150. [Google Scholar] [CrossRef]
- Suades, R.; Padró, T.; Alonso, R.; Mata, P.; Badimon, L. High levels of TSP1+/CD142+ platelet-derived microparticles characterise young patients with high cardiovascular risk and subclinical atherosclerosis. Thromb. Haemost. 2015, 114, 1310–1321. [Google Scholar] [CrossRef]
- Escate, R.; Padró, T.; Suades, R.; Camino, S.; Muñiz, O.; Diaz-Diaz, J.L.; Sionis, A.; Mata, P.; Badimon, L. High miR-133a levels in the circulation anticipates presentation of clinical events in familial hypercholesterolaemia patients. Cardiovasc. Res. 2021, 117, 109–122. [Google Scholar] [CrossRef]
- Hovland, A.; Narverud, I.; Øyri, L.K.L.; Bogsrud, M.P.; Aagnes, I.; Ueland, T.; Mulder, M.; Leijten, F.; Langslet, G.; Wium, C.; et al. Subjects with familial hypercholesterolemia have lower aortic valve area and higher levels of inflammatory biomarkers. J. Clin. Lipidol. 2021, 15, 134–141. [Google Scholar] [CrossRef]
- Collado, A.; Marques, P.; Domingo, E.; Perello, E.; González-Navarro, H.; Martinez-Hervás, S.; Real, J.T.; Piqueras, L.; Ascaso, J.F.; Sanz, M.-J. Novel Immune Features of the Systemic Inflammation Associated with Primary Hypercholesterolemia: Changes in Cytokine/Chemokine Profile, Increased Platelet and Leukocyte Activation. J. Clin. Med. 2018, 8, 18. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef]
- Tsimihodimos, V.; Karabina, S.-A.P.; Tambaki, A.P.; Bairaktari, E.; Miltiadous, G.; Goudevenos, J.A.; Cariolou, M.A.; Chapman, M.J.; Tselepis, A.D.; Elisaf, M. Altered distribution of platelet-activating factor-acetylhydrolase activity between LDL and HDL as a function of the severity of hypercholesterolemia. J. Lipid Res. 2002, 43, 256–263. [Google Scholar] [CrossRef]
- Melendez, Q.M.; Krishnaji, S.T.; Wooten, C.J.; Lopez, D. Hypercholesterolemia: The role of PCSK. Arch. Biochem. Biophys. 2017, 625-626, 39–53. [Google Scholar] [CrossRef]
- Li, S.; Li, J.-J. PCSK9: A key factor modulating atherosclerosis. J. Atheroscler. Thromb. 2015, 22, 221–230. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S.; Anderson, R.G.W.; Russell, D.; Schneider, W.J. Receptor-Mediated Endocytosis: Concepts Emerging from the LDL Receptor System. Annu. Rev. Cell Biol. 1985, 1, 1–39. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D.; Spady, D.K. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 1993, 34, 1637–1659. [Google Scholar] [CrossRef]
- Lakoski, S.G.; Lagace, T.A.; Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Genetic and Metabolic Determinants of Plasma PCSK9 Levels. J. Clin. Endocrinol. Metab. 2009, 94, 2537–2543. [Google Scholar] [CrossRef]
- Feingold, K.R.; Moser, A.H.; Shigenaga, J.K.; Patzek, S.M.; Grunfeld, C. Inflammation stimulates the expression of PCSK. Biochem. Biophys. Res. Commun. 2008, 374, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Jiang, L.; Peng, J.; Ren, Z.; Wei, D.; Wu, C.; Pan, L.; Jiang, Z.; Liu, L. PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF-kappaB activation in THP-1-derived macrophages. Int. J. Mol. Med. 2012, 30, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Enhances Platelet Activation, Thrombosis, and Myocardial Infarct Expansion by Binding to Platelet CD. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef]
- McDonagh, M.; Peterson, K.; Holzhammer, B.; Fazio, S. A Systematic Review of PCSK9 Inhibitors Alirocumab and Evolocumab. J. Manag. Care Spéc. Pharm. 2016, 22, 641–653. [Google Scholar] [CrossRef]
- Kastelein, J.J.; Ginsberg, H.N.; Langslet, G.; Hovingh, G.K.; Ceska, R.; Dufour, R.; Blom, D.; Civeira, F.; Krempf, M.; Lorenzato, C.; et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur. Heart J. 2015, 36, 2996–3003. [Google Scholar] [CrossRef]
- Ogura, M. PCSK9 inhibition in the management of familial hypercholesterolemia. J. Cardiol. 2018, 71, 1–7. [Google Scholar] [CrossRef]
- Tomlinson, B.; Hu, M.; Zhang, Y.; Chan, P.; Liu, Z.M. Alirocumab for the treatment of hypercholesterolemia. Expert Opin. Biol. Ther. 2017, 17, 633–643. [Google Scholar] [CrossRef]
- Robinson, J.G.; Nedergaard, B.S.; Rogers, W.J.; Fialkow, J.; Neutel, J.M.; Ramstad, D.; Somaratne, R.; Legg, J.C.; Nelson, P.; Scott, R.; et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: The LAPLACE-2 randomized clinical trial. JAMA 2014, 311, 1870–1882. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- E Kosmas, C.; Estrella, A.M.; Skavdis, A.; Genao, E.P.; Martinez, I.; Guzman, E. Inclisiran for the Treatment of Cardiovascular Disease: A Short Review on the Emerging Data and Therapeutic Potential. Ther. Clin. Risk Manag. 2020, 16, 1031–1037. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Bittner, V. Pleiotropic Effects of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) Inhibitors? Circulation 2016, 134, 1695–1696. [Google Scholar] [CrossRef]
- Paciullo, F.; Momi, S.; Gresele, P. PCSK9 in Haemostasis and Thrombosis: Possible Pleiotropic Effects of PCSK9 Inhibitors in Cardiovascular Prevention. Thromb. Haemost. 2019, 119, 359–367. [Google Scholar] [CrossRef]
- Barale, C.; Frascaroli, C.; Senkeev, R.; Cavalot, F.; Russo, I. Simvastatin Effects on Inflammation and Platelet Activation Markers in Hypercholesterolemia. BioMed Res. Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; A Stein, E.; Dufour, R.; Turner, T.; Civeira, F.; Burgess, L.; Langslet, G.; Scott, R.; Olsson, A.G.; Sullivan, D.; et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 331–340. [Google Scholar] [CrossRef]
- El-Seweidy, M.M.; Amin, R.S.; Atteia, H.H.; El-Zeiky, R.R.; Al-Gabri, N.A. Dyslipidemia induced inflammatory status, platelet activation and endothelial dysfunction in rabbits: Protective role of 10-Dehydrogingerdione. Biomed. Pharmacother. 2019, 110, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Q.; Wang, J.; Guo, C.; Kleiman, K.; Meng, H.; Knight, J.S.; Eitzman, D.T. Proprotein convertase subtilisin/kexin type 9 (PCSK9) Deficiency is Protective Against Venous Thrombosis in Mice. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Barale, C.; Bonomo, K.; Noto, F.; Traversa, M.; Cavalot, F.; Iozzia, M.; Frascaroli, C.; Guerrasio, A.; Russo, I. Effects of PCSK9 inhibitors on platelet function in adults with hypercholesterolemia. Atherosclerosis 2017, 263, e30–e31. [Google Scholar] [CrossRef]
- Landmesser, U.; Haghikia, A.; A Leiter, L.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Stoekenbroek, R.; Kastelein, J.J.P.; Ray, K.K. Effect of inclisiran, the small-interfering RNA against proprotein convertase subtilisin/kexin type 9, on platelets, immune cells, and immunological biomarkers: A pre-specified analysis from ORION. Cardiovasc. Res. 2021, 117, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.M.; Oemrawsingh, R.M.; Garcia-Garcia, H.M.; Boersma, E.; van Geuns, R.-J.; Serruys, P.W.; Kardys, I.; Akkerhuis, K.M. PCSK9 in relation to coronary plaque inflammation: Results of the ATHEROREMO-IVUS study. Atherosclerosis 2016, 248, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Tokgözoğlu, L.; Corsini, A.; Sirtori, C.R. PCSK9 inhibition and inflammation: A narrative review. Atherosclerosis 2019, 288, 146–155. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Sabouri-Rad, S.; Gotto, A.M.; Pirro, M.; Banach, M.; Awan, Z.; Barreto, G.E.; Sahebkar, A. PCSK9 and inflammation: A review of experimental and clinical evidence. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.L.; Liao, F.J.; Fang, L.; Zhong, F.; Liu, W.; Li, J.Q. Effect and mechanism of PCSK9 on lectin-like oxidized low-density lipoprotein receptor-1 mediated oxidized low-density lipoprotein uptake by THP-1 derived macrophages. Zhonghua Xin Xue Guan Bing Za Zhi 2019, 47, 367–373. [Google Scholar]
- Maulucci, G.; Cipriani, F.; Russo, D.; Casavecchia, G.; Di Staso, C.; Di Martino, L.; Ruggiero, A.; Di Biase, M.; Brunetti, N.D. Improved endothelial function after short-term therapy with evolocumab. J. Clin. Lipidol. 2018, 12, 669–673. [Google Scholar] [CrossRef]
- Ridker, P.M.; Mora, S.; Rose, L. Percent reduction in LDL cholesterol following high-intensity statin therapy: Potential implications for guidelines and for the prescription of emerging lipid-lowering agents. Eur. Heart J. 2016, 37, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Wei, W.-Q.; Chung, C.P.; Levinson, R.T.; Bastarache, L.; Denny, J.C.; Stein, C.M. The effect of genetic variation in PCSK9 on the LDL-cholesterol response to statin therapy. Pharm. J. 2017, 17, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.A.; Panza, G.; Pescatello, L.S.; Chipkin, S.; Gipe, D.; Shao, W.; White, C.M.; Thompson, P.D. Serum PCSK9 Levels Distinguish Individuals Who Do Not Respond to High-Dose Statin Therapy with the Expected Reduction in LDL-C. J. Lipids 2014, 2014, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendía, L.E.; Guerrero-Romero, F.; Golledge, J.; Watts, G.F. Effect of statin therapy on plasma proprotein convertase subtilisin kexin 9 (PCSK9) concentrations: A systematic review and meta-analysis of clinical trials. Diabetes Obes. Metab. 2015, 17, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.A.; Thompson, P.D. Statins and Their Effect on PCSK9—Impact and Clinical Relevance. Curr. Atheroscler. Rep. 2016, 18, 1–6. [Google Scholar] [CrossRef]
- Kim, C.J.; Han, E.J.; Chu, E.-H.; Hwang, B.-H.; Kim, J.-J.; Seung, K.-B.; Kim, S.H.; O, J.H.; Chang, K. Effect of moderate-intensity statin therapy on plaque inflammation in patients with acute coronary syndrome: A prospective interventional study evaluated by 18F-FDG PET/CT of the carotid artery. Cardiol. J. 2020, 27, 762–771. [Google Scholar] [CrossRef]
- Yano, H.; Horinaka, S.; Ishimitsu, T. Effect of evolocumab therapy on coronary fibrous cap thickness assessed by optical coherence tomography in patients with acute coronary syndrome. J. Cardiol. 2020, 75, 289–295. [Google Scholar] [CrossRef]
- Steffens, D.; Bramlage, P.; Scheeff, C.; Kasner, M.; Hassanein, A.; Friebel, J.; Rauch-Kröhnert, U. PCSK9 inhibitors and cardiovascular outcomes. Expert Opin. Biol. Ther. 2019, 20, 35–47. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Kim, Y.U.; Jukema, J.W.; Pordy, R.; Roe, M.T.; et al. Peripheral Artery Disease and Venous Thromboembolic Events After Acute Coronary Syndrome: Role of Lipoprotein(a) and Modification by Alirocumab: Prespecified Analysis of the ODYSSEY OUTCOMES Randomized Clinical Trial. Circulation 2020, 141, 1608–1617. [Google Scholar] [CrossRef]
- Marston, N.A.; Gurmu, Y.; Melloni, G.E.; Bonaca, M.; Gencer, B.; Sever, P.S.; Pedersen, T.R.; Keech, A.C.; Roselli, C.; Lubitz, S.A.; et al. The Effect of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) Inhibition on the Risk of Venous Thromboembolism. Circulation 2020, 141, 1600–1607. [Google Scholar] [CrossRef]
- Camargo, L.; França, C.; Izar, M.; Bianco, H.; Lins, L.; Barbosa, S.; Pinheiro, L.; Fonseca, F. Effects of simvastatin/ezetimibe on microparticles, endothelial progenitor cells and platelet aggregation in subjects with coronary heart disease under antiplatelet therapy. Braz. J. Med. Biol. Res. 2014, 47, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Roth, E.M.; Taskinen, M.-R.; Ginsberg, H.N.; Kastelein, J.J.; Colhoun, H.M.; Robinson, J.G.; Merlet, L.; Pordy, R.; Baccara-Dinet, M.T. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: Results of a 24week, double-blind, randomized Phase 3 trial. Int. J. Cardiol. 2014, 176, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Becher, T.; Schulze, T.J.; Schmitt, M.; Trinkmann, F.; El-Battrawy, I.; Akin, I.; Kälsch, T.; Borggrefe, M.; Stach, K. Ezetimibe inhibits platelet activation and uPAR expression on endothelial cells. Int. J. Cardiol. 2017, 227, 858–862. [Google Scholar] [CrossRef]
- Fazio, S. The role of PCSK9 in intestinal lipoprotein metabolism: Synergism of statin and ezetimibe. Atheroscler. Suppl. 2015, 17, 23–26. [Google Scholar] [CrossRef]
- Cui, C.-J.; Li, S.; Li, J.-J. PCSK9 and its modulation. Clin. Chim. Acta 2015, 440, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Olie, R.H.; Van Der Meijden, P.E.J.; Spronk, H.M.H.; Cate, H.T. Antithrombotic Therapy: Prevention and Treatment of Atherosclerosis and Atherothrombosis. Organotypic Models Drug Dev. 2020, 1–28. [Google Scholar] [CrossRef]
- Korish, A.A. Clopidogrel Prophylaxis Abates Myocardial Ischemic Injury and Inhibits the Hyperlipidemia-Inflammation Loop in Hypercholestrolemic Mice. Arch. Med. Res. 2020, 51, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Li, J.; Liang, X.; Zhang, S.; Liu, T.; Liu, J.; Arif, M.; Li, G. Ticagrelor suppresses oxidized low-density lipoprotein-induced endothelial cell apoptosis and alleviates atherosclerosis in ApoE-/- mice via downregulation of PCSK. Mol. Med. Rep. 2018, 19, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Luzak, B.; Boncler, M.; Rywaniak, J.; Wilk, R.; Stanczyk, L.; Czyz, M.; Rysz, J.; Watala, C. The effect of a platelet cholesterol modulation on the acetylsalicylic acid-mediated blood platelet inhibition in hypercholesterolemic patients. Eur. J. Pharmacol. 2011, 658, 91–97. [Google Scholar] [CrossRef]
- Grdinic, A.; Vojvodic, D.; Djukanovic, N.; Colic, M.; Grdinic, A.G.; Ignjatovic, V.; Majstorovic, I.; Ilic, V.; Magic, Z.; Obradovic, S.; et al. PCI and clopidogrel: Antiplatelet responsiveness and patient characteristics. Acta Cardiol. 2011, 66, 333–340. [Google Scholar] [CrossRef]
- Navarese, E.P.; Kołodziejczak, M.; Winter, M.-P.; Alimohammadi, A.; Lang, I.M.; Buffon, A.; Lip, G.Y.; Siller-Matula, J.M. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: The PCSK9-REACT study. Int. J. Cardiol. 2017, 227, 644–649. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| PCSK-9 Inhibitor | Population | Effect | Ref. |
|---|---|---|---|
| Monotherapy | |||
| Alirocumab/evolocumab | Patients with PH (n = 7) | Decrease in P-selectin exposure, with and without agonists | [24] |
| Alirocumab/evolocumab | Patients with hypercholesterolemia (n = 21) | Reduced platelet reactivity to agonists | [95] |
| Alirocumab | Patients with FH (n = 736) | LDL-C lowering | [81] |
| Evolocumab | Patients with FH (n = 331) | LDL-C lowering | [92] |
| 10-Dehydrogingerdione | Rabbits (n = 30) | Decrease in sP-selectin and sCD40L | [93] |
| PCSK9 deficiency | Mice (n = 20) | Lowered risk of venous thrombosis | [94] |
| Polytherapy | |||
| Alirocumab + statin (unspecified) | Patients with hypercholesterolemia (n = 18,924) | Decreased risk of thrombotic events | [110] |
| Evolocumab + statin (unspecified) | Patients after acute coronary syndrome (n = 18,924) | Decreased risk of venous thromboembolism | [111] |
| Evolocumab + rosuvastatin | Patients with de novo acute coronary artery disease (n = 64) | Stabilization of atherosclerotic plaque | [108] |
| Loss-of-funcion mutation in PCSK9 gene + statin (unspecified) | Patients with hypercholesterolemia (n = 2388) | Improved response to statin therapy | [103] |
| Alirocumab/evolocumab + aspirin | Patients with PH (n = 14) | Decrease in P-selectin exposure, with and without stimuli | [24] |
| Alirocumab + aspirin | In vitro study (n = 10) | Decrease in platelet aggregation | [79] |
| Lower levels of PCSK9 + ticagrelor | Patients with acute coronary syndrome (n = 333) | Decrease in platelet aggregation | [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pęczek, P.; Leśniewski, M.; Mazurek, T.; Szarpak, L.; Filipiak, K.J.; Gąsecka, A. Antiplatelet Effects of PCSK9 Inhibitors in Primary Hypercholesterolemia. Life 2021, 11, 466. https://doi.org/10.3390/life11060466
Pęczek P, Leśniewski M, Mazurek T, Szarpak L, Filipiak KJ, Gąsecka A. Antiplatelet Effects of PCSK9 Inhibitors in Primary Hypercholesterolemia. Life. 2021; 11(6):466. https://doi.org/10.3390/life11060466
Chicago/Turabian StylePęczek, Piotr, Mateusz Leśniewski, Tomasz Mazurek, Lukasz Szarpak, Krzysztof J. Filipiak, and Aleksandra Gąsecka. 2021. "Antiplatelet Effects of PCSK9 Inhibitors in Primary Hypercholesterolemia" Life 11, no. 6: 466. https://doi.org/10.3390/life11060466
APA StylePęczek, P., Leśniewski, M., Mazurek, T., Szarpak, L., Filipiak, K. J., & Gąsecka, A. (2021). Antiplatelet Effects of PCSK9 Inhibitors in Primary Hypercholesterolemia. Life, 11(6), 466. https://doi.org/10.3390/life11060466