
The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment



Abstract
1. Introduction
2. Materials and Methods
2.1. Environmental Sampling
2.2. Nucleic Acid Extraction and 16S rRNA Amplicon Sequencing
2.3. Sequence Read Analysis
2.4. Enrichment Cultivation Strategy
2.5. Whole-Genome Sequencing
2.6. 16S rRNA Amplification
2.7. Metagenomic Binning Analysis
2.8. Whole-Genome Comparison
3. Results and Discussion
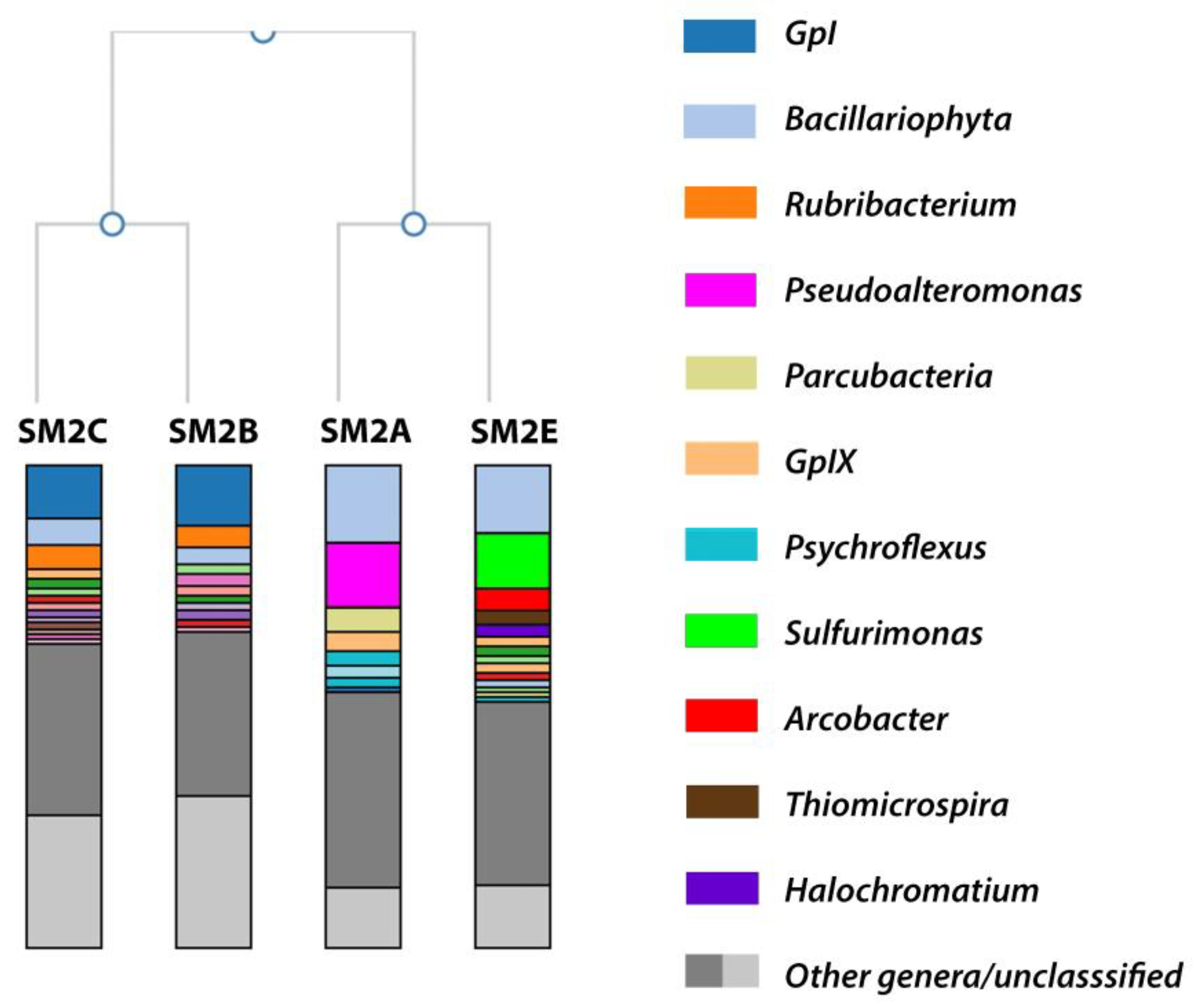
3.1. Metagenomic Analysis: Taxonomic Overview
3.1.1. SM2B/C Analysis
3.1.2. SM2A Analysis
3.1.3. SM2E Analysis
3.2. Genomic Analysis of Enrichment Cultures
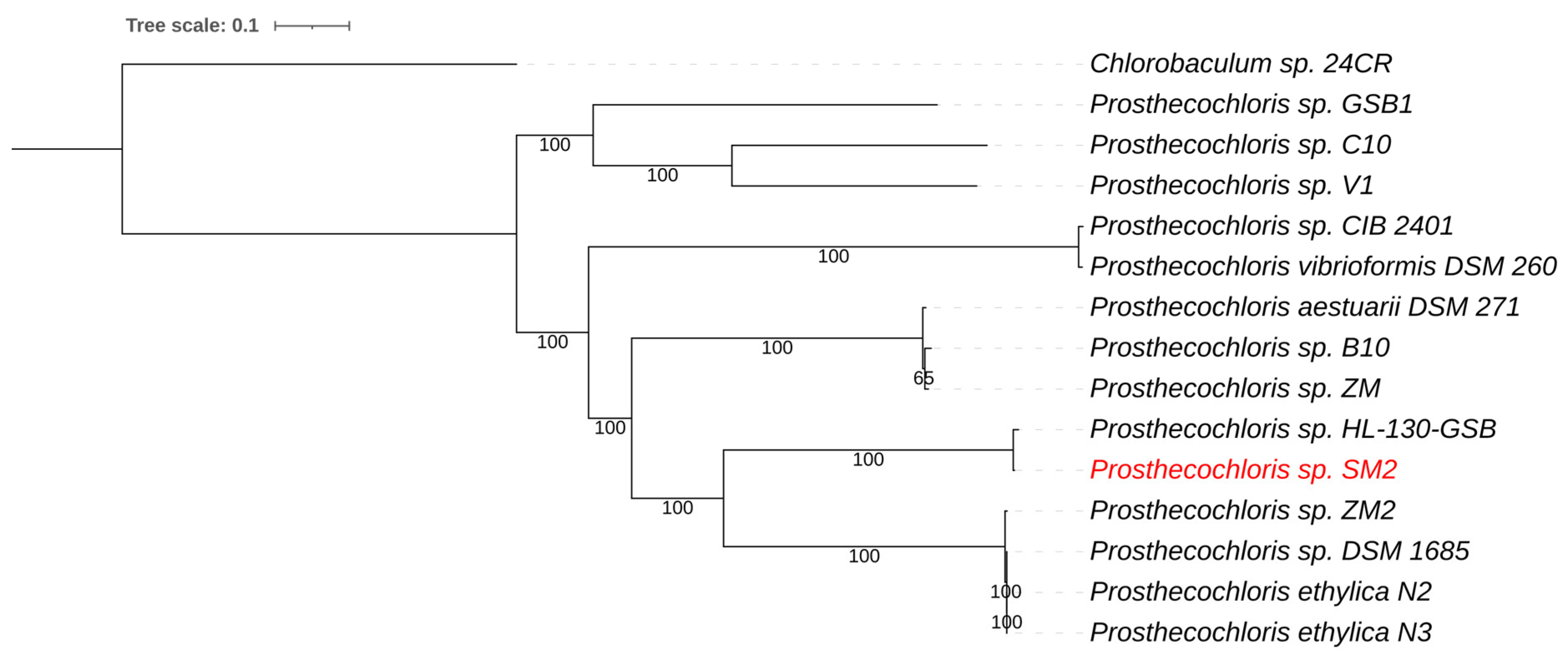
3.2.1. Prosthecochloris sp. SM2
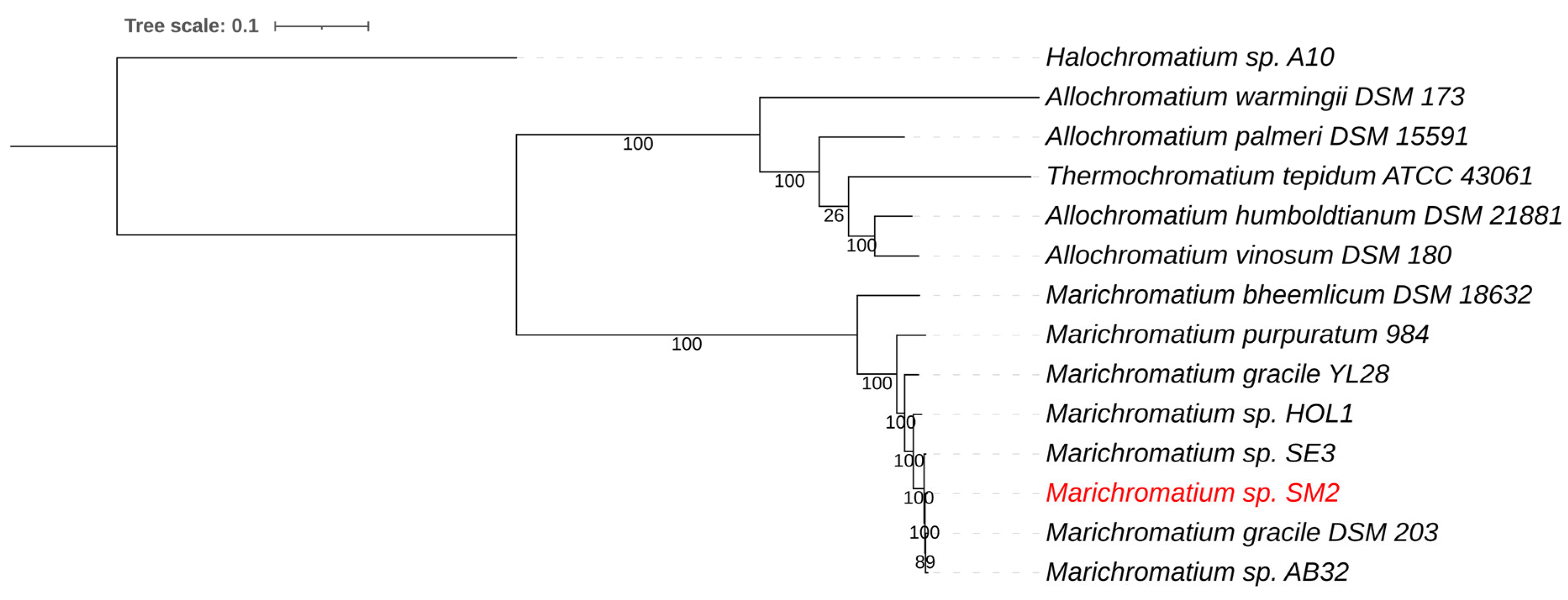
3.2.2. Marichromatium sp. SM2
3.2.3. Sulfurospirillum
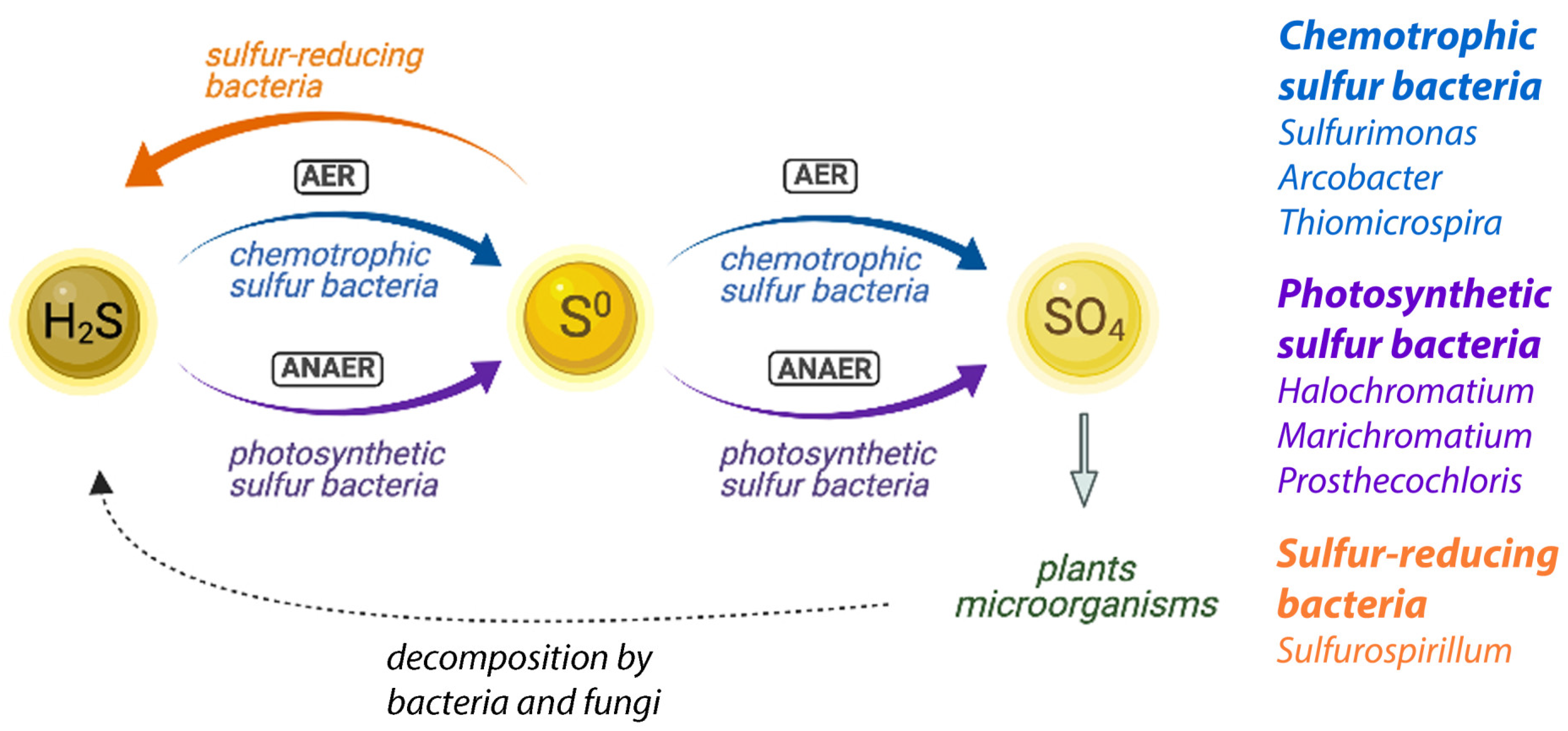
3.3. The Salt Marsh Sulfur Cycle
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gibbens, S. The remnants of a vast prehistoric sea lie hidden in Nebraska’s endangered marshes. In National Geographic; National Geographic Society: Washington, DC, USA, 2020. [Google Scholar]
- Johnsgard, P.A. The Nature of Nebraska: Ecology and Biodiversity; The University of Nebraska Press: Lincoln, NE, USA, 2001. [Google Scholar]
- Spomer, S.; Higley, L. Population status and distribution of the Salt Creek Tiger Beetle, Cicindela nevadica lincolniana Casey (Coleoptera: Cicindelidae). J. Kans. Entomol. Soc. 1993, 66, 392–398. [Google Scholar]
- Brosius, T.R.; Higley, L.G. Behavioral niche partitioning in a sympatric tiger beetle assemblage and implications for the endangered Salt Creek tiger beetle. PeerJ 2013, 1, e169. [Google Scholar] [CrossRef]
- Ungar, I.; Hogan, W.; McClelland, M. Plant communities of saline soils at Lincoln, Nebraska. Am. Midl. Nat. 1969, 82, 564–577. [Google Scholar] [CrossRef]
- Panella, M. Nebraska’s At-Risk Species Wildlife; Nebraska Game & Parks Commission: Lincoln, NE, USA, 2012; pp. 146–147.
- Malmstrom, T. Saline Wetlands Conservation Partnership 2018 Progress Report; Lincoln Parks and Recreation Department: Lincoln, NE, USA, 2019. Available online: https://www.lincoln.ne.gov/files/sharedassets/public/parks-amp-rec/saline-wetlands/progressrpt2018.pdf (accessed on 13 May 2021).
- Van Gemerden, H. Microbial mats: A joint venture. Mar. Geol. 1993, 113, 3–25. [Google Scholar] [CrossRef]
- Pfennig, N. The phototrophic bacteria and their role in the sulfur cycle. Plant Soil 1975, 43, 1–16. [Google Scholar] [CrossRef]
- Bowen, J.; Crump, B.; Deegan, L.; Hobbie, J.E. Salt marsh sediment bacteria: Their distribution and response to external nutrient inputs. ISME J. 2009, 3, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl. Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal database project: Data and tools for high throughput rRNA analysis. Nucl. Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Weaver, P.F.; Wall, J.D.; Gest, H. Characterization of Rhodopseudomonas capsulata. Arch. Microbiol. 1975, 105, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucl. Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2014, 25, 1043–1055. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.J.B. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucl. Acids Res 2019, 47, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Spellerberg, I.F.; Peter, J.F. A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon-Wiener’ Index. Glob. Ecol. Biogeogr. 2003, 12, 177–179. [Google Scholar] [CrossRef]
- Tuomisto, H. A diversity of beta diversities: Straightening up a concept gone awry. Part 1. Defining beta diversity as a function of alpha and gamma diversity. Ecography 2010, 33, 2–22. [Google Scholar] [CrossRef]
- Hasle, G.R.; Syvertsen, E.E.; Steidinger, K.A.; Tangen, K. Marine diatoms. In Identifying Marine Diatoms and Dinoflagellates; Tomas, C.R., Ed.; Academic Press: Cambridge, MA, USA, 1996; pp. 5–385. ISBN 978-0-08-053441-1. [Google Scholar]
- Fourtanier, E.; Kociolek, J.P. Catalogue of the diatom genera. Diatom Res. 1999, 14, 1–190. [Google Scholar] [CrossRef]
- Stal, L.J.; Gemerden, H.; Krumbein, W.E. Structure and development of a benthic marine microbial mat. FEMS Microbiol. Lett. 1985, 31, 111–125. [Google Scholar] [CrossRef]
- Bolhuis, H.; Stal, L.J. Analysis of bacterial and archaeal diversity in coastal microbial mats using massive parallel 16S rRNA gene tag sequencing. ISME J. 2011, 5, 1701–1712. [Google Scholar] [CrossRef]
- Boldareva, E.N.; Moskalenko, A.A.; Makhneva, Z.K.; Tourova, T.P.; Kolganova, T.V.; Gorlenko, V.M. Rubribacterium polymorphum gen. nov., sp. nov., a novel alkaliphilic nonsulfur purple bacterium from an Eastern Siberian soda lake. Microbiology 2009, 78, 732–740. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Soliman, G.S.H.; Trüper, H.G. The Wadi Natrun: Chemical composition and microbial mass development in alkaline brines of eutrophic desert lakes. Geomicrobiol. J. 1979, 1, 219–234. [Google Scholar] [CrossRef]
- Kompantseva, E.I.; Bryantseva, I.A.; Komova, A.V.; Namsaraev, B.B. The structure of phototrophic communities of soda lakes of the Southeastern Transbaikal Region. Microbiology 2007, 76, 211–219. [Google Scholar] [CrossRef]
- Kompantseva, E.I.; Komova, A.V.; Krauzova, V.I.; Kolganova, T.V.; Panteleeva, E.E. Purple nonsulfur bacteria in weakly and moderately mineralized soda lakes of the Southern Transbaikal region and Northeastern Mongolia. Microbiology 2009, 78, 246–254. [Google Scholar] [CrossRef]
- Milford, A.D.; Achenbach, L.A.; Jung, D.O.; Madigan, M.T. Rhodobaca bogoriensis gen. nov. and sp. nov. alcaliphilic purple nonsulfur bacterium from African Rift valley soda lakes. Arch. Microbiol. 2000, 174, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Boldareva, E.N.; Akimov, V.N.; Boychenko, V.A.; Stadnichuk, I.N.; Moskalenko, A.A.; Makhneva, Z.K.; Gorlenko, V.M. Rhodobaca barguzinensis sp. nov., a new alkaliphilic purple sulfur bacterium isolated from a soda lake of the Barguzin valley (Buryat Republic, Eastern Siberia). Microbiology 2008, 77, 206–218. [Google Scholar] [CrossRef]
- Holmström, C.; Kjelleberg, S. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol. Ecol. 1999, 30, 285–293. [Google Scholar] [CrossRef]
- Harris, J.K.; Kelley, S.T.; Pace, N.R. New perspective on uncultured bacterial phylogenetic division OP11. Appl. Environ. Microbiol. 2004, 70, 845–849. [Google Scholar] [CrossRef]
- Nelson, W.; Stegen, J. The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front. Microbiol. 2015, 6, 713. [Google Scholar] [CrossRef]
- Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Miller, C.S.; Castelle, C.J.; VerBerkmoes, N.C.; Wilkins, M.J.; Hettich, R.L.; Lipton, M.S.; Williams, K.H.; et al. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 2012, 337, 1661–1665. [Google Scholar] [CrossRef]
- Bowman, J.P.; McCammon, S.A.; Lewis, T.; Skerratt, J.H.; Brown, J.L.; Nichols, D.S.; McMeekin, T.A. Psychroflexus torquis gen. nov., sp. nov., a psychrophilic species from Antarctic sea ice, and reclassification of Flavobacterium gondwanense (Dobson et al. 1993) as Psychroflexus gondwanense gen. nov., comb. nov. Microbiology 1998, 144, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Kang, J.Y.; Jahng, K.Y. Psychroflexus salarius sp. nov., isolated from Gomso salt pan. Int. J. Syst. Evol. Microbiol. 2014, 64, 3467–3472. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Kang, S.J.; Jung, Y.T.; Oh, T.K. Psychroflexus salinarum sp. nov., isolated from a marine solar saltern. Int. J. Syst. Evol. Microbiol. 2009, 59, 2404–2407. [Google Scholar] [CrossRef][Green Version]
- Chen, Y.G.; Cui, X.L.; Wang, Y.X.; Tang, S.K.; Zhang, Y.Q.; Li, W.J.; Liu, J.H.; Peng, Q.; Xu, L.H. Psychroflexus sediminis sp. nov., a mesophilic bacterium isolated from salt lake sediment in China. Int. J. Syst. Evol. Microbiol. 2009, 59, 569–573. [Google Scholar] [CrossRef]
- Seiler, H.; Bleicher, A.; Busse, H.J.; Hüfner, J.; Scherer, S. Psychroflexus halocasei sp. nov., isolated from a microbial consortium on a cheese. Int. J. Syst. Evol. Microbiol. 2012, 62, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Ventosa, A. Unusual micro-organisms from unusual habitats: Hypersaline environments. In Prokaryotic Diversity: Mechanisms and Significance; Cambridge University Press: Cambridge, UK, 2006; pp. 223–254. [Google Scholar]
- Galisteo, C.; Sánchez-Porro, C.; de la Haba, R.R.; López-Hermoso, C.; Fernández, A.B.; Farias, M.E.; Ventosa, A. Characterization of Salinivibrio socompensis sp. nov., a new halophilic bacterium isolated from the high-altitude hypersaline lake Socompa, Argentina. Microorganisms 2019, 7, 241. [Google Scholar] [CrossRef]
- Han, Y.; Perner, M. The globally widespread genus Sulfurimonas: Versatile energy metabolisms and adaptations to redox clines. Front. Microbiol. 2015, 6, 989. [Google Scholar] [CrossRef]
- SamKamaleson, A.; Gonsalves, M.-J. Role of sulfur-oxidizing bacteria on the ecology in tropical mangrove sediments. Reg. Stud. Mar. Sci. 2019, 28, 100574. [Google Scholar]
- Aviles, A.F.; Kyndt, J.A. Sequencing of coastal lagoon samples from the Piñones Lagoon, Puerto Rico, reveals important role of bacterial sulfur metabolism in the lagoon ecosystem. Microbiol. Res. Announc. 2021, 10, e00172-21. [Google Scholar]
- Donachie, S.P.; Bowman, J.P.; On, S.L.W.; Alam, M. Arcobacter halophilus sp. nov., the first obligate halophile in the genus Arcobacter. Int. J. Syst. Evol. Microbiol. 2005, 55, 1271–1277. [Google Scholar] [CrossRef]
- McClung, C.R.; Patriquin, D.G.; Davis, R.E. Campylobacter nitrofigilis sp. nov., a nitrogen-fixing bacterium associated with roots of Spartina alterniflora Loisel. Int. J. Syst. Bacteriol. 1983, 33, 605–612. [Google Scholar] [CrossRef]
- Fera, M.T.; Maugeri, T.L.; Gugliandolo, C.; Beninati, C.; Giannone, M.; La Camera, E.; Carbone, M. Detection of Arcobacter spp. in the coastal environment of the Mediterranean Sea. Appl. Environ. Microbiol. 2004, 70, 1271–1276. [Google Scholar] [CrossRef]
- Wirsen, C.O.; Sievert, S.M.; Cavanaugh, C.M.; Molyneaux, S.J.; Ahmad, A.; Taylor, L.T.; DeLong, E.F.; Taylor, C.D. Characterization of an autotrophic sulfide-oxidizing marine Arcobacter sp. that produces filamentous sulfur. Appl. Environ. Microbiol. 2002, 68, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Wirsen, C. Is Life Thriving Deep Beneath the Seafloor? Woods Hole Oceanographic Institution: Falmouth, MA, USA, 2004; Available online: https://www.whoi.edu/oceanus/feature/is-life-thriving-deep-beneath-the-seafloor/ (accessed on 1 March 2021).
- Boden, R.; Scott, K.M.; Williams, J.; Russel, S.; Antonenen, K.; Rae, A.W.; Hutt, L.P. An evaluation of Thiomicrospira, Hydrogenovibrio and Thioalkalimicrobium: Reclassification of four species of Thiomicrospira to each Thiomicrorhabdus gen. nov. and Hydrogenovibrio, and reclassification of all four species of Thioalkalimicrobium to Thiomicrospira. Int. J. Syst. Evol. Microbiol. 2017, 67, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, D.Y.; Lysenko, A.M.; Mityushina, L.L.; Tourova, T.P.; Jones, B.E.; Rainey, F.A.; Robertson, L.A.; Kuenen, G.J. Thioalkalimicrobium aerophilum gen. nov., sp. nov. and Thioalkalimicrobium sibericum sp. nov., and Thioalkalivibrio versutus gen. nov., sp. nov., Thioalkalivibrio nitratis sp.nov., novel and Thioalkalivibrio denitrificancs sp. nov., novel obligately alkaliphilic and obligately chemolithoautotrophic sulfur-oxidizing bacteria from soda lakes. Int. J. Syst. Evol. Microbiol. 2001, 51, 565–580. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Gorlenko, V.M.; Tourova, T.P.; Tsapin, A.I.; Nealson, K.H.; Kuenen, G.J. Thioalkalimicrobium cyclicum sp. nov. and Thioalkalivibrio janaschiijannaschii sp. nov., novel species of haloalkaliphilic, obligately chemolithoautotrophic sulfur-oxidizing bacteria from hypersaline alkaline mono lake (California). Int. J. Syst. Evol. Microbiol. 2002, 52, 913–920. [Google Scholar] [PubMed]
- Sorokin, D.Y.; Foti, M.; Pinkart, H.C.; Muyzer, G. Sulfur-Oxidizing bacteria in soap lake (Washington state), a meromictic, haloalkaline lake with an unprecedented high sulfide content. Appl. Environ. Microbiol. 2007, 73, 451–455. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Süling, J.; Petri, R. Phylogenetic relationships among the Chromatiaceae, their taxonomic reclassification and description of the new genera Allochromatium, Halochromatium, Isochromatium, Marichromatium, Thiococcus, Thiohalocapsa and Thermochromatium. Int. J. Syst. Bacteriol. 1998, 48, 1129–1143. [Google Scholar] [CrossRef]
- Hunter, C.N.; Daldal, F.; Thurnauer, M.C.; Beatty, J.T. The Purple Phototropic Bacteria; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Imhoff, J.F. Biology of green sulfur bacteria. In Encyclopedia of Life Sciences; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Thiel, V.; Drautz-Moses, D.I.; Purbojati, R.W.; Schuster, S.C.; Lindemann, S.; Bryant, D.A. Genome sequence of Prosthecochloris sp. strain HL-130-GSB from the phylum Chlorobi. Genome Announc. 2017, 5, e00538-17. [Google Scholar] [CrossRef] [PubMed]
- Kyndt, J.A.; Van Beeumen, J.J.; Meyer, T.E. Simultaneous genome sequencing of Prosthecochloris ethylica and Desulfuromonas acetoxidans within a syntrophic mixture reveals unique pili and protein interactions. Microorganisms 2020, 8, 1939. [Google Scholar] [CrossRef]
- Muller, J.; Overmann, J. Close interspecies interactions between prokaryotes from sulfureous environments. Front. Microbiol. 2011, 2, 146. [Google Scholar] [CrossRef]
- Freed, S.; Robertson, S.; Meyer, T.; Kyndt, J. Draft whole-genome sequence of the green sulfur photosynthetic bacterium Chlorobaculum sp. strain 24CR, isolated from the Carmel River. Microbiol. Resour. Announc. 2019, 8, e00116-19. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F.; Rahn, T.; Künzel, S.; Keller, A.; Neulinger, S.C. Osmotic adaptation and compatible solute biosynthesis of phototrophic bacteria as revealed from genome analyses. Microorganisms 2020, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Goris, T.; Diekert, G. The genus sulfurospirillum. In Organohalide-Respiring Bacteria; Adrian, L., Löffler, F., Eds.; Springer: Heidelberg/Berlin, Germany, 2016. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Zhen, Y.; Mi, T.; He, H.; Yu, Z. Microbial diversity and community structure of sulfate-reducing and sulfur-oxidizing bacteria in sediment cores from the East China Sea. Front. Microbiol. 2017, 8, 2133. [Google Scholar] [CrossRef]
- Kruse, S.; Goris, T.; Westermann, M.; Adrian, L.; Diekert, G. Hydrogen production by Sulfurospirillum species enables syntrophic interactions of Epsilonproteobacteria. Nat. Commun. 2018, 9, 4872. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Buttet, G.F.; Murray, A.M.; Goris, T.; Burion, M.; Jin, B.; Rolle, M.; Holliger, C.; Maillard, J. Coexistence of two distinct Sulfurospirillum populations respiring tetrachloroethene-genomic and kinetic considerations. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Tourova, T.P.; Muyzer, G. Isolation and characterization of two novel alkalitolerant sulfidogens from a Thiopaq bioreactor, Desulfonatronum alkalitolerans sp. nov., and Sulfurospirillum alkalitolerans sp. nov. Extremophiles 2013, 17, 535–543. [Google Scholar] [CrossRef]
- Margulis, L.; Barghoorn, E.S.; Ashendorf, D.; Banjeree, S.; Chase, D.; Francis, S.; Giovannoni, S.; Stolz, J. The microbial community in the layered sediments at Laguna Figueroa, Baja California, Mexico: Does it have Precambrian analogues? Precambr. Res. 1980, 11, 93–123. [Google Scholar] [CrossRef]
- Nicholson, J.A.; Stolz, J.F.; Pierson, B.K. Structure of a microbial mat at Great Sippewissett Marsh, Cape Cod, Massachusetts. FEMS Microbiol. Ecol. 1987, 45, 343–346. [Google Scholar] [CrossRef]
- Pierson, B.K.; Oesterle, A.; Murphy, G.L. Pigments, light penetration and photosynthetic activity in the multilayered microbial mats of Great Sippewissett Salt Marsh, Massachusetts. FEMS Microbiol. Ecol. 1987, 45, 365–376. [Google Scholar] [CrossRef][Green Version]
- Lynum, C.A.; Bulseco, A.N.; Dunphy, C.M.; Osborne, S.M.; Vineis, J.H.; Bowen, J.L. Microbial community response to a passive salt marsh restoration. Estuaries Coasts 2020, 43, 1439–1455. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | SM2A | SM2B | SM2C | SM2E |
|---|---|---|---|---|
| GpI/GpIX | 3.9 | 13.9 | 12.3 | 2.3 |
| Bacillariophyta | 17.1 | 3.4 | 6.4 | 16.8 |
| Rubribacterium | <2 | 4.8 | 5.0 | <2 |
| Pseudoalteromonas | 14.1 | n.d. | n.d. | <2 |
| Parcubacteria | 7.3 | n.d. | <2 | <1 |
| Psychroflexus | 6.3 | <1 | <1 | <2 |
| Sulfurimonas | <2 | <2 | <2 | 16.3 |
| Arcobacter | <2 | n.d. | n.d. | 5 |
| Thiomicrospira | <2 | <2 | <1 | 3.5 |
| Halochromatium | <1 | <2 | <2 | 2.1 |
| Salinivibrio | 2.1 | n.d. | <1 | <1 |
| Unclassified | 12.4 | 31.4 | 27.3 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athen, S.R.; Dubey, S.; Kyndt, J.A. The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment. Life 2021, 11, 446. https://doi.org/10.3390/life11050446
Athen SR, Dubey S, Kyndt JA. The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment. Life. 2021; 11(5):446. https://doi.org/10.3390/life11050446
Chicago/Turabian StyleAthen, Sierra R., Shivangi Dubey, and John A. Kyndt. 2021. "The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment" Life 11, no. 5: 446. https://doi.org/10.3390/life11050446
APA StyleAthen, S. R., Dubey, S., & Kyndt, J. A. (2021). The Eastern Nebraska Salt Marsh Microbiome Is Well Adapted to an Alkaline and Extreme Saline Environment. Life, 11(5), 446. https://doi.org/10.3390/life11050446