
Interplay between Mitochondrial Protein Import and Respiratory Complexes Assembly in Neuronal Health and Degeneration

 , , and
, , and
Abstract
1. Introduction
2. Protein Translocation
2.1. Crossing the Outer Membrane
2.2. Biogenesis of OMM Proteins
2.2.1. Insertion of β-Barrel Proteins in the OMM
2.2.2. Incorporation of α-Helical Anchors in the OMM
2.3. Co- and Post-Translational Translocation
2.4. Staying in the Intermembrane Space—The Disulfide Relay System
2.5. Crossing or Insertion in the Inner Membrane
2.5.1. TIM23 Complex (Presequence Pathway)
2.5.2. TIM22 Complex (Carrier Pathway)
2.5.3. Oxa1
3. The Respiratory Chain and Supercomplexes
3.1. Respiratory Complexes Assembly
3.1.1. CI Assembly
3.1.2. CII Assembly
3.1.3. CIII Assembly
3.1.4. CIV Assembly
3.1.5. CV Assembly
4. Pathologies with Underlying Mitochondrial Import Defects
4.1. Mitochondrial Diseases
4.2. Alzheimer’s Disease
4.3. Parkinson’s Disease
4.4. Huntington’s Disease
4.5. Amyotrophic Lateral Sclerosis
5. Repair Pathways
5.1. UPRmt
5.2. UPRam
5.3. mPOS
5.4. mitoCPR
5.5. mitoTAD
6. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endoc. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, J.A.; Payne, R.M. Mitochondrial protein import and human health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 509–523. [Google Scholar] [CrossRef]
- Briston, T.; Hicks, A.R. Mitochondrial dysfunction and neurodegenerative proteinopathies: Mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 2018, 46, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.D.; Palmer, C.S.; Stojanovski, D. Mitochondrial diseases caused by dysfunctional mitochondrial protein import. Biochem. Soc. Trans. 2018, 46, 1225–1238. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Tucker, K.; Park, E. Cryo-EM structure of the mitochondrial protein-import channel TOM complex at near-atomic resolution. Nat. Struct. Mol. Biol. 2019, 26, 1158–1166. [Google Scholar] [CrossRef]
- Sollner, T.; Griffiths, G.; Pfaller, R.; Pfanner, N.; Neupert, W. Mom19, an import receptor for mitochondrial precursor proteins. Cell 1989, 59, 1061–1070. [Google Scholar] [CrossRef]
- Moczko, M.; Dietmeier, K.; Sollner, T.; Segui, B.; Steger, H.F.; Neupert, W.; Pfanner, N. Identification of the mitochondrial receptor complex in saccharomyces-cerevisiae. FEBS Lett. 1992, 310, 265–268. [Google Scholar] [CrossRef]
- Hines, V.; Brandt, A.; Griffiths, G.; Horstmann, H.; Brutsch, H.; Schatz, G. Protein import into yeast mitochondria is accelerated by the outer-membrane protein mas70. Embo J. 1990, 9, 3191–3200. [Google Scholar] [CrossRef]
- Sollner, T.; Pfaller, R.; Griffiths, G.; Pfanner, N.; Neupert, W. A mitochondrial import receptor for the adp/atp carrier. Cell 1990, 62, 107–115. [Google Scholar] [CrossRef]
- Chacinska, A.; Lind, M.; Frazier, A.E.; Dudek, J.; Meisinger, C.; Geissler, A.; Sickmann, A.; Meyer, H.E.; Truscott, K.N.; Guiard, B.; et al. Mitochondrial presequence translocase: Switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 2005, 120, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Van Wilpe, S.; Ryan, M.T.; Hill, K.; Maarse, A.C.; Meisinger, C.; Brix, J.; Dekker, P.J.; Moczko, M.; Wagner, R.; Meijer, M.; et al. Tom22 is a multifunctional organizer of the mitochondrial preprotein translocase. Nature 1999, 401, 485–489. [Google Scholar] [CrossRef]
- Lithgow, T.; Junne, T.; Suda, K.; Gratzer, S.; Schatz, G. The mitochondrial outer membrane protein Mas22p is essential for protein import and viability of yeast. Proc. Natl. Acad. Sci. USA 1994, 91, 11973–11977. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, H.; Shiota, T.; Ishizaka, N.; Kawano, S.; Tamura, Y.; Tan, K.S.; Imai, K.; Motono, C.; Hirokawa, T.; Taki, K.; et al. Porin Associates with Tom22 to Regulate the Mitochondrial Protein Gate Assembly. Mol. Cell 2019, 73, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Grevel, A.; Becker, T. Porins as helpers in mitochondrial protein translocation. Biol. Chem. 2020, 401, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Harbauer, A.B.; Opalinska, M.; Gerbeth, C.; Herman, J.S.; Rao, S.; Schonfisch, B.; Guiard, B.; Schmidt, O.; Pfanner, N.; Meisinger, C. Cell cycle-dependent regulation of mitochondrial preprotein translocase. Science 2014, 346, 1109–1113. [Google Scholar] [CrossRef]
- Kunkele, K.P.; Heins, S.; Dembowski, M.; Nargang, F.E.; Benz, R.; Thieffry, M.; Walz, J.; Lill, R.; Nussberger, S.; Neupert, W. The preprotein translocation channel of the outer membrane of mitochondria. Cell 1998, 93, 1009–1019. [Google Scholar] [CrossRef]
- Shiota, T.; Imai, K.; Qiu, J.; Hewitt, V.L.; Tan, K.; Shen, H.H.; Sakiyama, N.; Fukasawa, Y.; Hayat, S.; Kamiya, M.; et al. Molecular architecture of the active mitochondrial protein gate. Science 2015, 349, 1544–1548. [Google Scholar] [CrossRef]
- Model, K.; Prinz, T.; Ruiz, T.; Radermacher, M.; Krimmer, T.; Kuhlbrandt, W.; Pfanner, N.; Meisinger, C. Protein translocase of the outer mitochondrial membrane: Role of import receptors in the structural organization of the TOM complex. J. Mol. Biol. 2002, 316, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Model, K.; Meisinger, C.; Kuhlbrandt, W. Cryo-Electron Microscopy Structure of a Yeast Mitochondrial Preprotein Translocase. J. Mol. Biol. 2008, 383, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Bausewein, T.; Mills, D.J.; Langer, J.D.; Nitschke, B.; Nussberger, S.; Kuhlbrandt, W. Cryo-EM Structure of the TOM Core Complex from Neurospora crassa. Cell 2017, 170, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Moczko, M.; Bomer, U.; Kubrich, M.; Zufall, N.; Honlinger, A.; Pfanner, N. The intermembrane space domain of mitochondrial Tom22 functions as a trans binding site for properties with N-terminal targeting sequences. Mol. Cell. Biol. 1997, 17, 6574–6584. [Google Scholar] [CrossRef] [PubMed]
- Harner, M.; Neupert, W.; Deponte, M. Lateral release of proteins from the TOM complex into the outer membrane of mitochondria. Embo J. 2011, 30, 3232–3241. [Google Scholar] [CrossRef]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.X.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef]
- Jacoupy, M.; Hamon-Keromen, E.; Ordureau, A.; Erpapazoglou, Z.; Coge, F.; Corvol, J.C.; Nosjean, O.; la Cour, C.M.; Millan, M.J.; Boutin, J.A.; et al. The PINK1 kinase-driven ubiquitin ligase Parkin promotes mitochondrial protein import through the presequence pathway in living cells. Sci. Rep. 2019, 9, 11829. [Google Scholar] [CrossRef]
- Phu, L.; Rose, C.M.; Tea, J.S.; Wall, C.E.; Verschueren, E.; Cheung, T.K.; Kirkpatrick, D.S.; Bingol, B. Dynamic Regulation of Mitochondrial Import by the Ubiquitin System. Mol. Cell 2020, 77, 1107–1123. [Google Scholar] [CrossRef]
- Ordureau, A.; Paulo, J.A.; Zhang, J.C.; An, H.; Swatek, K.N.; Cannon, J.R.; Wan, Q.Q.; Komander, D.; Harper, J.W. Global Landscape and Dynamics of Parkin and USP30-Dependent Ubiquitylomes in iNeurons during Mitophagic Signaling. Mol. Cell 2020, 77, 1124–1142. [Google Scholar] [CrossRef]
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The Protein Import Machinery of Mitochondria-A Regulatory Hub in Metabolism, Stress, and Disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef]
- Paschen, S.A.; Waizenegger, T.; Stan, T.; Preuss, M.; Cyrklaff, M.; Hell, K.; Rapaport, D.; Neupert, W. Evolutionary conservation of biogenesis of beta-barrel membrane proteins. Nature 2003, 426, 862–866. [Google Scholar] [CrossRef]
- Kutik, S.; Stojanovski, D.; Becker, L.; Becker, T.; Meinecke, M.; Kruger, V.; Prinz, C.; Meisinger, C.; Guiard, B.; Wagner, R.; et al. Dissecting membrane insertion of mitochondrial beta-barrel proteins. Cell 2008, 132, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Voegtle, F.N.; Stojanovski, D.; Meisinger, C. Sorting and assembly of mitochondrial outer membrane proteins. Biochim. Biophys. Acta-Bioenerg. 2008, 1777, 557–563. [Google Scholar] [CrossRef]
- Diederichs, K.A.; Ni, X.D.; Rollauer, S.E.; Botos, I.; Tan, X.F.; King, M.S.; Kunji, E.R.S.; Jiang, J.S.; Buchanan, S.K. Structural insight into mitochondrial beta-barrel outer membrane protein biogenesis. Nat. Commun. 2020, 11, 3290. [Google Scholar] [CrossRef] [PubMed]
- Wenz, L.S.; Ellenrieder, L.; Qiu, J.; Bohnert, M.; Zufall, N.; van der Laan, M.; Pfanner, N.; Wiedemann, N.; Becker, T. Sam37 is crucial for formation of the mitochondrial TOM-SAM supercomplex, thereby promoting beta-barrel biogenesis. J. Cell Biol. 2015, 210, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Meisinger, C.; Rissler, M.; Chacinska, A.; Szklarz, L.K.S.; Milenkovic, D.; Kozjak, V.; Schonfisch, B.; Lohaus, C.; Meyer, H.E.; Yaffe, M.P.; et al. The mitochondrial morphology protein Mdm10 functions in assembly of the preprotein translocase of the outer membrane. Dev. Cell 2004, 7, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Doan, K.N.; Grevel, A.; Martensson, C.U.; Ellenrieder, L.; Thornton, N.; Wenz, L.S.; Opalinski, L.; Guiard, B.; Pfanner, N.; Becker, T. The Mitochondrial Import Complex MIM Functions as Main Translocase for alpha-Helical Outer Membrane Proteins. Cell Rep. 2020, 31, 107567. [Google Scholar] [CrossRef]
- Becker, T.; Wenz, L.S.; Kruger, V.; Lehmann, W.; Muller, J.M.; Goroncy, L.; Zufall, N.; Lithgow, T.; Guiard, B.; Chacinska, A.; et al. The mitochondrial import protein Mim1 promotes biogenesis of multispanning outer membrane proteins. J. Cell Biol. 2011, 194, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Krumpe, K.; Frumkin, I.; Herzig, Y.; Rimon, N.; Ozbalci, C.; Brugger, B.; Rapaport, D.; Schuldiner, M. Ergosterol content specifies targeting of tail-anchored proteins to mitochondrial outer membranes. Mol. Biol. Cell 2012, 23, 3927–3935. [Google Scholar] [CrossRef]
- Kruger, V.; Becker, T.; Becker, L.; Montilla-Martinez, M.; Ellenrieder, L.; Vogtle, F.N.; Meyer, H.E.; Ryan, M.T.; Wiedemann, N.; Warscheid, B.; et al. Identification of new channels by systematic analysis of the mitochondrial outer membrane. J. Cell Biol. 2017, 216, 3485–3495. [Google Scholar] [CrossRef]
- Dimmer, K.S.; Papic, D.; Schumann, B.; Sperl, D.; Krumpe, K.; Walther, D.M.; Rapaport, D. A crucial role for Mim2 in the biogenesis of mitochondrial outer membrane proteins. J. Cell Sci. 2012, 125, 3464–3473. [Google Scholar] [CrossRef]
- Wenz, L.S.; Opalinski, L.; Schuler, M.H.; Ellenrieder, L.; Ieva, R.; Bottinger, L.; Qiu, J.; van der Laan, M.; Wiedemann, N.; Guiard, B.; et al. The presequence pathway is involved in protein sorting to the mitochondrial outer membrane. Embo Rep. 2014, 15, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Sinzel, M.; Tan, T.; Wendling, P.; Kalbacher, H.; Ozbalci, C.; Chelius, X.; Westermann, B.; Brugger, B.; Rapaport, D.; Dimmer, K.S. Mcp3 is a novel mitochondrial outer membrane protein that follows a unique IMP-dependent biogenesis pathway. Embo Rep. 2016, 17, 965–981. [Google Scholar] [CrossRef] [PubMed]
- Mokranjac, D.; Neupert, W. Energetics of protein translocation into mitochondria. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 758–762. [Google Scholar] [CrossRef]
- Komiya, T.; Rospert, S.; Schatz, G.; Mihara, K. Binding of mitochondrial precursor proteins to the cytoplasmic domains of the import receptors Tom70 and Tom20 is determined by cytoplasmic chaperones. Embo J. 1997, 16, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Esaki, M.; Kanamori, T.; Tamura, Y.; Nishikawa, S.; Endo, T. Tim50 is a subunit of the TIM23 complex that links protein translocation across the outer and inner mitochondrial membranes. Cell 2002, 111, 519–528. [Google Scholar] [CrossRef]
- Komiya, T.; Rospert, S.; Koehler, C.; Looser, R.; Schatz, G.; Mihara, K. Interaction of mitochondrial targeting signals with acidic receptor domains along the protein import pathway: Evidence for the ‘acid chain’ hypothesis. Embo J. 1998, 17, 3886–3898. [Google Scholar] [CrossRef]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial protein import: Common principles and physiological networks. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 274–285. [Google Scholar] [CrossRef]
- Garcia, M.; Delaveau, T.; Goussard, S.; Jacq, C. Mitochondrial presequence and open reading frame mediate asymmetric localization of messenger RNA. Embo Rep. 2010, 11, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Margeot, A.; Blugeon, C.; Sylvestre, J.; Vialette, S.; Jacq, C.; Corral-Debrinski, M. In Saccharomyces cerevisiae, ATP2 mRNA sorting to the vicinity of mitochondria is essential for respiratory function. Embo J. 2002, 21, 6893–6904. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Blugeon, C.; Jacq, C. In yeast, the 3’ untranslated region or the presequence of ATM1 is required for the exclusive localization of its mRNA to the vicinity of mitochondria. Mol Cell Biol 2000, 20, 7881–7892. [Google Scholar] [CrossRef]
- George, R.; Walsh, P.; Beddoe, T.; Lithgow, T. The nascent polypeptide-associated complex (NAC) promotes interaction of ribosomes with the mitochondrial surface in vivo. FEBS Lett. 2002, 516, 213–216. [Google Scholar] [CrossRef]
- MacKenzie, J.A.; Payne, R.M. Ribosomes specifically bind to mammalian mitochondria via protease-sensitive proteins on the outer membrane. J. Biol. Chem. 2004, 279, 9803–9810. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.; Eaglesfield, R.; Tokatlidis, K. The mitochondrial intermembrane space: The most constricted mitochondrial sub-compartment with the largest variety of protein import pathways. Open Biol. 2021, 11, 210002. [Google Scholar] [CrossRef]
- Peleh, V.; Cordat, E.; Herrmann, J.M. Mia40 is a trans-site receptor that drives protein import into the mitochondrial intermembrane space by hydrophobic substrate binding. eLife 2016, 5, e16177. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Pfannschmidt, S.; Wiedemann, N.; Kozjak, V.; Szklarz, L.K.S.; Schulze-Specking, A.; Truscott, K.N.; Guiard, B.; Meisinger, C.; Pfanner, N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. Embo J. 2004, 23, 3735–3746. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Cefaro, C.; Ciofi-Baffoni, S.; Gallo, A.; Martinelli, M.; Sideris, D.P.; Katrakili, N.; Tokatlidis, K. MIA40 is an oxidoreductase that catalyzes oxidative protein folding in mitochondria. Nat. Struct. Mol. Biol. 2009, 16, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, C.; Arena, G.; Nedara, K.; Edwards, R.; Brenner, C.; Tokatlidis, K.; Modjtahedi, N. AIF meets the CHCHD4/Mia40-dependent mitochondrial import pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165746. [Google Scholar] [CrossRef] [PubMed]
- Bien, M.; Longen, S.; Wagener, N.; Chwalla, I.; Herrmann, J.M.; Riemer, J. Mitochondrial Disulfide Bond Formation Is Driven by Intersubunit Electron Transfer in Erv1 and Proofread by Glutathione. Mol. Cell 2010, 37, 516–528. [Google Scholar] [CrossRef]
- Allen, S.; Balabanidou, V.; Sideris, D.P.; Lisowsky, T.; Tokatlidis, K. Erv1 mediates the Mia40-dependent protein import pathway and provides a functional link to the respiratory chain by shuttling electrons to cytochrome c. J. Mol. Biol. 2005, 353, 937–944. [Google Scholar] [CrossRef]
- Neupert, W. Protein import into mitochondria. Annu. Rev. Biochem. 1997, 66, 863–917. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Hess, S.; Boos, F.; Woellhaf, M.W.; Godel, S.; Jung, M.; Muhlhaus, T.; Herrmann, J.M. Tom70 enhances mitochondrial preprotein import efficiency by binding to internal targeting sequences. J. Cell Biol. 2018, 217, 1369–1382. [Google Scholar] [CrossRef]
- Demishtein-Zohary, K.; Azem, A. The TIM23 mitochondrial protein import complex: Function and dysfunction. Cell Tissue Res. 2017, 367, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Maarse, A.C.; Blom, J.; Keil, P.; Pfanner, N.; Meijer, M. Identification of the essential yeast protein mim17, an integral mitochondrial inner membrane-protein involved in protein import. FEBS Lett. 1994, 349, 215–221. [Google Scholar] [CrossRef]
- Dekker, P.J.T.; Keil, P.; Rassow, J.; Maarse, A.C.; Pfanner, N.; Meijer, M. Identification of mim23, a putative component of the protein import machinery of the mitochondrial inner membrane. FEBS Lett. 1993, 330, 66–70. [Google Scholar] [CrossRef]
- Geissler, A.; Chacinska, A.; Truscott, K.N.; Wiedemann, N.; Brandner, K.; Sickmann, A.; Meyer, H.E.; Meisinger, C.; Pfanner, N.; Rehling, P. The mitochondrial presequence translocase: An essential role of Tim50 in directing preproteins to the import channel. Cell 2002, 111, 507–518. [Google Scholar] [CrossRef]
- Gebert, M.; Schrempp, S.G.; Mehnert, C.S.; Heisswolf, A.K.; Oeljeklaus, S.; Ieva, R.; Bohnert, M.; von der Malsburg, K.; Wiese, S.; Kleinschroth, T.; et al. Mgr2 promotes coupling of the mitochondrial presequence translocase to partner complexes. J. Cell Biol. 2012, 197, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Maarse, A.C.; Blom, J.; Grivell, L.A.; Meijer, M. Mpi1, an essential gene encoding a mitochondrial-membrane protein, is possibly involved in protein import into yeast mitochondria. Embo J. 1992, 11, 3619–3628. [Google Scholar] [CrossRef]
- Kang, P.J.; Ostermann, J.; Shilling, J.; Neupert, W.; Craig, E.A.; Pfanner, N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature 1990, 348, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Frazier, A.E.; Dudek, J.; Guiard, B.; Voos, W.; Li, Y.F.; Lind, M.; Meisinger, C.; Geissler, A.; Sickmann, A.; Meyer, H.E.; et al. Pam16 has an essential role in the mitochondrial protein import motor. Nat. Struct. Mol. Biol. 2004, 11, 226–233. [Google Scholar] [CrossRef]
- Truscott, K.N.; Voos, W.; Frazier, A.E.; Lind, M.; Li, Y.F.; Geissler, A.; Dudek, J.; Muller, H.; Sickmann, A.; Meyer, H.E.; et al. A J-protein is an essential subunit of the presequence translocase-associated protein import motor of mitochondria. J. Cell Biol. 2003, 163, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, M.; Chacinska, A.; Lind, M.; Perschil, I.; Sickmann, A.; Meyer, H.E.; Guiard, B.; Meisinger, C.; Pfanner, N.; Rehling, P. Pam17 is required for architecture and translocation activity of the mitochondrial protein import motor. Mol. Cell. Biol. 2005, 25, 7449–7458. [Google Scholar] [CrossRef] [PubMed]
- Laloraya, S.; Gambill, B.D.; Craig, E.A. A role for a eukaryotic grpe-related protein, mge1p, in protein translocation. Proc. Natl. Acad. Sci. USA 1994, 91, 6481–6485. [Google Scholar] [CrossRef]
- Tamura, Y.; Harada, Y.; Shiota, T.; Yamano, K.; Watanabe, K.; Yokota, M.; Yamamoto, H.; Sesaki, H.; Endo, T. Tim23-Tim50 pair coordinates functions of translocators and motor proteins in mitochondrial protein import. J. Cell Biol. 2009, 184, 129–141. [Google Scholar] [CrossRef]
- Schwartz, M.P.; Matouschek, A. The dimensions of the protein import channels in the outer and inner mitochondrial membranes. Proc. Natl. Acad. Sci. USA 1999, 96, 13086–13090. [Google Scholar] [CrossRef] [PubMed]
- Truscott, K.N.; Kovermann, P.; Geissler, A.; Merlin, A.; Meijer, M.; Driessen, A.J.M.; Rassow, J.; Pfanner, N.; Wagner, R. A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat. Struct. Biol. 2001, 8, 1074–1082. [Google Scholar] [CrossRef]
- Martinez-Caballero, S.; Grigoriev, S.M.; Herrmann, J.M.; Campo, M.L.; Kinnally, K.W. Tim17p regulates the twin pore structure and voltage gating of the mitochondrial protein import complex TIM23. J. Biol. Chem. 2007, 282, 3584–3593. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Rehling, P.; Guiard, B.; Frazier, A.E.; Schulze-Specking, A.; Pfanner, N.; Voos, W.; Meisinger, C. Mitochondrial translocation contact sites: Separation of dynamic and stabilizing elements in formation of a TOM-TIM-preprotein supercomplex. Embo J. 2003, 22, 5370–5381. [Google Scholar] [CrossRef]
- Waegemann, K.; Popov-Celeketic, D.; Neupert, W.; Azem, A.; Mokranjac, D. Cooperation of TOM and TIM23 complexes during translocation of proteins into mitochondria. J Mol. Biol. 2015, 427, 1075–1084. [Google Scholar] [CrossRef]
- Shiota, T.; Mabuchi, H.; Tanaka-Yamano, S.; Yamano, K.; Endo, T. In vivo protein-interaction mapping of a mitochondrial translocator protein Tom22 at work. Proc. Natl. Acad. Sci. USA 2011, 108, 15179–15183. [Google Scholar] [CrossRef]
- Niemi, N.M.; Wilson, G.M.; Overmyer, K.A.; Vogtle, F.N.; Myketin, L.; Lohman, D.C.; Schueler, K.L.; Attie, A.D.; Meisinger, C.; Coon, J.J.; et al. Pptc7 is an essential phosphatase for promoting mammalian mitochondrial metabolism and biogenesis. Nat. Commun. 2019, 10, 3197. [Google Scholar] [CrossRef]
- Marom, M.; Dayan, D.; Demishtein-Zohary, K.; Mokranjac, D.; Neupert, W.; Azem, A. Direct Interaction of Mitochondrial Targeting Presequences with Purified Components of the TIM23 Protein Complex. J. Biol. Chem. 2011, 286, 43809–43815. [Google Scholar] [CrossRef] [PubMed]
- Ting, S.Y.; Yan, N.L.; Schilke, B.A.; Craig, E.A. Dual interaction of scaffold protein Tim44 of mitochondrial import motor with channel-forming translocase subunit Tim23. eLife 2017, 6, e23609. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W.; Brunner, M. The protein import motor of mitochondria. Nat. Rev. Mol. Cell Biol. 2002, 3, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Gruhler, A.; Arnold, I.; Seytter, T.; Guiard, B.; Schwarz, E.; Neupert, W.; Stuart, R.A. N-terminal hydrophobic sorting signals of preproteins confer mitochondrial hsp70 independence for import into mitochondria. J. Biol. Chem. 1997, 272, 17410–17415. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, M.; Wiedemann, N.; Mick, D.U.; Guiard, B.; Rehling, P.; Pfanner, N. A role for Tim21 in membrane-potential-dependent preprotein sorting in mitochondria. Curr. Biol. 2006, 16, 2271–2276. [Google Scholar] [CrossRef]
- Albrecht, R.; Rehling, P.; Chacinska, A.; Brix, J.; Cadamuro, S.A.; Volkmer, R.; Guiard, B.; Pfanner, N.; Zeth, K. The Tim21 binding domain connects the preprotein translocases of both mitochondrial membranes. Embo Rep. 2006, 7, 1233–1238. [Google Scholar] [CrossRef]
- Mokranjac, D.; Popov-Celeketic, D.; Hell, K.; Neupert, W. Role of Tim21 in mitochondrial translocation contact sites. J. Biol. Chem. 2005, 280, 23437–23440. [Google Scholar] [CrossRef] [PubMed]
- Ieva, R.; Schrempp, S.G.; Opalinski, L.; Wollweber, F.; Hoss, P.; Heisswolf, A.K.; Gebert, M.; Zhang, Y.; Guiard, B.; Rospert, S.; et al. Mgr2 Functions as Lateral Gatekeeper for Preprotein Sorting in the Mitochondrial Inner Membrane. Mol. Cell 2014, 56, 641–652. [Google Scholar] [CrossRef]
- Endres, M.; Neupert, W.; Brunner, M. Transport of the ADP ATP carrier of mitochondria from the TOM complex to the TIM22.54 complex. Embo J. 1999, 18, 3214–3221. [Google Scholar] [CrossRef]
- Curran, S.P.; Leuenberger, D.; Oppliger, W.; Koehler, C.M. The Tim9p-Tim10p complex binds to the transmembrane domains of the ADP/ATP carrier. Embo J. 2002, 21, 942–953. [Google Scholar] [CrossRef]
- Rehling, P.; Model, K.; Brandner, K.; Kovermann, P.; Sickmann, A.; Meyer, H.E.; Kuhlbrandt, W.; Wagner, R.; Truscott, K.N.; Pfanner, N. Protein insertion into the mitochondrial inner membrane by a twin-pore translocase. Science 2003, 299, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Brix, J.; Rudiger, S.; Bukau, B.; Schneider-Mergener, J.; Pfanner, N. Distribution of binding sequences for the mitochondrial import receptors Tom20, Tom22, and Tom70 in a presequence-carrying preprotein and a non-cleavable preprotein. J. Biol. Chem. 1999, 274, 16522–16530. [Google Scholar] [CrossRef] [PubMed]
- Curran, S.P.; Leuenberger, D.; Schmidt, E.; Koehler, C.M. The role of the Tim8p-Tim13p complex in a conserved import pathway for mitochondrial polytopic inner membrane proteins. J. Cell Biol. 2002, 158, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.B.; Wang, Q.; Guan, Z.Y.; Wu, Y.; Shen, C.C.; Hong, S.X.; Cao, J.B.; Zhang, X.; Yan, C.Y.; Yin, P. Cryo-EM structure of the human mitochondrial translocase TIM22 complex. Cell Res. 2021, 31, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ou, X.; Wang, X.; Sun, D.; Zhou, X.; Wu, X.; Li, Q.; Li, L. Structure of the mitochondrial TIM22 complex from yeast. Cell Res 2021, 31, 366–368. [Google Scholar] [CrossRef]
- Callegari, S.; Richter, F.; Chojnacka, K.; Jans, D.C.; Lorenzi, I.; Pacheu-Grau, D.; Jakobs, S.; Lenz, C.; Urlaub, H.; Dudek, J.; et al. TIM29 is a subunit of the human carrier translocase required for protein transport. Febs Lett. 2016, 590, 4147–4158. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.L.; Baker, M.J.; Liem, M.; Louber, J.; McKenzie, M.; Atukorala, I.; Ang, C.S.; Keerthikumar, S.; Mathivanan, S.; Stojanovski, D. Tim29 is a novel subunit of the human TIM22 translocase and is involved in complex assembly and stability. Elife 2016, 5, e17463. [Google Scholar] [CrossRef]
- Vukotic, M.; Nolte, H.; Konig, T.; Saita, S.; Ananjew, M.; Kruger, M.; Tatsuta, T.; Langer, T. Acylglycerol Kinase Mutated in Sengers Syndrome Is a Subunit of the TIM22 Protein Translocase in Mitochondria. Mol. Cell 2017, 67, 471–483. [Google Scholar] [CrossRef]
- Kang, Y.L.; Stroud, D.A.; Baker, M.J.; De Souza, D.P.; Frazier, A.E.; Liem, M.; Tull, D.; Mathivanan, S.; McConville, M.J.; Thorburn, D.R.; et al. Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 2017, 67, 457–470. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. In Annual Review of Biochemistry; Kornberg, R.D., Ed.; Annual Reviews: Palo Alto, CA, USA, 2017; Volume 86, pp. 685–714. [Google Scholar]
- Rehling, P.; Brandner, K.; Pfanner, N. Mitochondrial import and the twin-pore translocase. Nat. Rev. Mol. Cell Biol. 2004, 5, 519–530. [Google Scholar] [CrossRef]
- Wu, Y.K.; Sha, B.D. Crystal structure of yeast mitochondrial outer membrane translocon member Tom70p. Nat. Struct. Mol. Biol. 2006, 13, 589–593. [Google Scholar] [CrossRef]
- Young, J.C.; Hoogenraad, N.J.; Hartl, F.U. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 2003, 112, 41–50. [Google Scholar] [CrossRef]
- Bhangoo, M.K.; Tzankov, S.; Fan, A.C.Y.; Dejgaard, K.; Thomas, D.Y.; Young, J.C. Multiple 40-kDa heat-shock protein chaperones function in Tom70-dependent mitochondrial import. Mol. Biol. Cell 2007, 18, 3414–3428. [Google Scholar] [CrossRef]
- Backes, S.; Bykov, Y.S.; Räschle, M.; Zhou, J.; Lenhard, S.; Krämer, L.; Mühlhaus, T.; Bibi, C.; Jann, C.; Smith, J.D.; et al. The mitochondrial surface receptor Tom70 protects the cytosol against mitoprotein-induced stress. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N.; Ryan, M.T. The three modules of ADP/ATP carrier cooperate in receptor recruitment and translocation into mitochondria. Embo J. 2001, 20, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Ellenrieder, L.; Dieterle, M.P.; Doan, K.N.; Martensson, C.U.; Floerchinger, A.; Campo, M.L.; Pfanner, N.; Becker, T. Dual Role of Mitochondrial Porin in Metabolite Transport across the Outer Membrane and Protein Transfer to the Inner Membrane. Mol. Cell 2019, 73, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Callegari, S.; Muller, T.; Schulz, C.; Lenz, C.; Jans, D.C.; Wissel, M.; Opazo, F.; Rizzoli, S.O.; Jakobs, S.; Urlaub, H.; et al. A MICOS-TIM22 Association Promotes Carrier Import into Human Mitochondria. J. Mol. Biol. 2019, 431, 2835–2851. [Google Scholar] [CrossRef]
- Rampelt, H.; Sucec, I.; Bersch, B.; Horten, P.; Perschil, I.; Martinou, J.C.; van der Laan, M.; Wiedemann, N.; Schanda, P.; Pfanner, N. The mitochondrial carrier pathway transports non-canonical substrates with an odd number of transmembrane segments. BMC Biol. 2020, 18, 2. [Google Scholar] [CrossRef]
- Jackson, T.D.; Hock, D.H.; Fujihara, K.M.; Palmer, C.S.; Frazier, A.E.; Low, Y.C.; Kang, Y.; Ang, C.-S.; Clemons, N.J.; Thorburn, D.R.; et al. The TIM22 complex mediates the import of Sideroflexins and is required for efficient mitochondrial one-carbon metabolism. Mol. Biol. Cell 2021, 32, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Luirink, J.; Samuelsson, T.; de Gier, J.W. YidC/Oxa1p/Alb3: Evolutionarily conserved mediators of membrane protein assembly. FEBS Lett. 2001, 501, 1–5. [Google Scholar] [CrossRef]
- Bonnefoy, N.; Chalvet, F.; Hamel, P.; Slonimski, P.P.; Dujardin, G. OXA1, a saccharomyces-cerecconserved from prokaryotes to eukaryotes controls cytochrome-oxidase biogenesis. J. Mol. Biol. 1994, 239, 201–212. [Google Scholar] [CrossRef]
- Herrmann, J.M.; Neupert, W.; Stuart, R.A. Insertion into the mitochondrial inner membrane of a polytopic protein, the nuclear-encoded Oxa1p. Embo J. 1997, 16, 2217–2226. [Google Scholar] [CrossRef]
- Nargang, F.E.; Preuss, M.; Neupert, W.; Herrmann, J.M. The oxal protein forms a homooligomeric complex and is an essential part of the mitochondrial export translocase in Neurospora crassa. J. Biol. Chem. 2002, 277, 12846–12853. [Google Scholar] [CrossRef] [PubMed]
- Stiburek, L.; Fornuskova, D.; Wenchich, L.; Pejznochova, M.; Hansikova, H.; Zeman, J. Knockdown of human Oxa1l impairs the biogenesis of F1Fo-ATP synthase and NADH: Ubiquinone oxidoreductase. J. Mol. Biol. 2007, 374, 506–516. [Google Scholar] [CrossRef]
- Haque, M.E.; Elmore, K.B.; Tripathy, A.; Koc, H.; Koc, E.C.; Spremulli, L.L. Properties of the C-terminal Tail of Human Mitochondrial Inner Membrane Protein Oxa1L and Its Interactions with Mammalian Mitochondrial Ribosomes. J. Biol. Chem. 2010, 285, 28353–28362. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Andrell, J.; Choi, A.; Richter, U.; Maiti, P.; Best, R.B.; Barrientos, A.; Battersby, B.J.; Amunts, A. Mechanism of membrane-tethered mitochondrial protein synthesis. Science 2021, 371, 846–849. [Google Scholar] [CrossRef]
- Stuart, R.A. Insertion of proteins into the inner membrane of mitochondria: The role of the Oxa1 complex. Biochim. Biophys. Acta Mol. Cell Res. 2002, 1592, 79–87. [Google Scholar] [CrossRef]
- Bohnert, M.; Rehling, P.; Guiard, B.; Herrmann, J.M.; Pfanner, N.; van der Laan, M. Cooperation of Stop-Transfer and Conservative Sorting Mechanisms in Mitochondrial Protein Transport. Curr. Biol. 2010, 20, 1227–1232. [Google Scholar] [CrossRef]
- Anghel, S.A.; McGilvray, P.T.; Hegde, R.S.; Keenan, R.J. Identification of Oxa1 Homologs Operating in the Eukaryotic Endoplasmic Reticulum. Cell Rep. 2017, 21, 3708–3716. [Google Scholar] [CrossRef]
- Pleiner, T.; Tomaleri, G.P.; Januszyk, K.; Inglis, A.J.; Hazu, M.; Voorhees, R.M. Structural basis for membrane insertion by the human ER membrane protein complex. Science 2020, 369, 433–436. [Google Scholar] [CrossRef]
- Bai, L.; You, Q.L.; Feng, X.; Kovach, A.; Li, H.L. Structure of the ER membrane complex, a transmembrane-domain insertase. Nature 2020, 584, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Chitwood, P.J.; Juszkiewicz, S.; Guna, A.; Shao, S.C.; Hegde, R.S. EMC Is Required to Initiate Accurate Membrane Protein Topogenesis. Cell 2018, 175, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Rojo, E.E.; Stuart, R.A.; Neupert, W. Conservative sorting of f-0-atpase subunit-9—export from matrix requires delta-ph across inner membrane and matrix atp. Embo J. 1995, 14, 3445–3451. [Google Scholar] [CrossRef] [PubMed]
- Hell, K.; Neupert, W.; Stuart, R.A. Oxa1p acts as a general membrane insertion machinery for proteins encoded by mitochondrial DNA. Embo J. 2001, 20, 1281–1288. [Google Scholar] [CrossRef]
- Herrmann, J.M.; Bonnefoy, N. Protein export across the inner membrane of mitochondria—The nature of translocated domains determines the dependence on the Oxa1 translocase. J. Biol. Chem. 2004, 279, 2507–2512. [Google Scholar] [CrossRef]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. Embo J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Bayona-Bafaluy, M.P.; Fernandez-Silva, P.; Moreno-Loshuertos, R.; Perez-Martos, A.; Bruno, C.; Moraes, C.T.; Enriquez, J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- Diaz, F.; Fukui, H.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol. Cell Biol. 2006, 26, 4872–4881. [Google Scholar] [CrossRef]
- Protasoni, M.; Pérez-Pérez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Peñas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III-mediated maturation of human mitochondrial complexes I and IV. Embo J. 2020, 39, e102817. [Google Scholar] [CrossRef]
- Blakely, E.L.; Mitchell, A.L.; Fisher, N.; Meunier, B.; Nijtmans, L.G.; Schaefer, A.M.; Jackson, M.J.; Turnbull, D.M.; Taylor, R.W. A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast. FEBS J. 2005, 272, 3583–3592. [Google Scholar] [CrossRef] [PubMed]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Lamantea, E.; Carrara, F.; Mariotti, C.; Morandi, L.; Tiranti, V.; Zeviani, M. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes I and III. Neuromuscul. Disord. NMD 2002, 12, 49–52. [Google Scholar] [CrossRef]
- Moreno-Lastres, D.; Fontanesi, F.; Garcia-Consuegra, I.; Martin, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 2012, 15, 324–335. [Google Scholar] [CrossRef]
- Wiedemann, N.; van der Laan, M.; Hutu, D.P.; Rehling, P.; Pfanner, N. Sorting switch of mitochondrial presequence translocase involves coupling of motor module to respiratory chain. J. Cell Biol. 2007, 179, 1115–1122. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef]
- Fontanesi, F.; Soto, I.C.; Horn, D.; Barrientos, A. Mss51 and Ssc1 facilitate translational regulation of cytochrome c oxidase biogenesis. Mol. Cell Biol. 2010, 30, 245–259. [Google Scholar] [CrossRef]
- Böttinger, L.; Guiard, B.; Oeljeklaus, S.; Kulawiak, B.; Zufall, N.; Wiedemann, N.; Warscheid, B.; van der Laan, M.; Becker, T. A complex of Cox4 and mitochondrial Hsp70 plays an important role in the assembly of the cytochrome c oxidase. Mol. Biol. Cell 2013, 24, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Rieger, B.; Junge, W.; Busch, K.B. Lateral pH gradient between OXPHOS complex IV and F0F1 ATP-synthase in folded mitochondrial membranes. Nat. Commun. 2014, 5, 3103. [Google Scholar] [CrossRef]
- Couvillion, M.T.; Soto, I.C.; Shipkovenska, G.; Churchman, L.S. Synchronized mitochondrial and cytosolic translation programs. Nature 2016, 533, 499–503. [Google Scholar] [CrossRef]
- Herrmann, J.M.; Woellhaf, M.W.; Bonnefoy, N. Control of protein synthesis in yeast mitochondria: The concept of translational activators. Biochim. Biophys. Acta 2013, 1833, 286–294. [Google Scholar] [CrossRef]
- Ott, M.; Amunts, A.; Brown, A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu. Rev. Biochem. 2016, 85, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Dennerlein, S.; Wang, C.; Rehling, P. Plasticity of Mitochondrial Translation. Trends Cell Biol. 2017, 27, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Timón-Gómez, A.N.E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef]
- Gruschke, S.; Römpler, K.; Hildenbeutel, M.; Kehrein, K.; Kühl, I.; Bonnefoy, N.; Ott, M. The Cbp3-Cbp6 complex coordinates cytochrome b synthesis with bc(1) complex assembly in yeast mitochondria. J. Cell Biol. 2012, 199, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Hildenbeutel, M.; Hegg, E.L.; Stephan, K.; Gruschke, S.; Meunier, B.; Ott, M. Assembly factors monitor sequential hemylation of cytochrome b to regulate mitochondrial translation. J. Cell Biol. 2014, 205, 511–524. [Google Scholar] [CrossRef]
- García-Villegas, R.; Camacho-Villasana, Y.; Shingú-Vázquez, M.; Cabrera-Orefice, A.; Uribe-Carvajal, S.; Fox, T.D.; Pérez-Martínez, X. The Cox1 C-terminal domain is a central regulator of cytochrome c oxidase biogenesis in yeast mitochondria. J. Biol. Chem. 2017, 292, 10912–10925. [Google Scholar] [CrossRef]
- Perez-Martinez, X.; Butler, C.A.; Shingu-Vazquez, M.; Fox, T.D. Dual functions of Mss51 couple synthesis of Cox1 to assembly of cytochrome c oxidase in Saccharomyces cerevisiae mitochondria. Mol. Biol Cell 2009, 20, 4371–4380. [Google Scholar] [CrossRef]
- Tavares-Carreón, F.; Camacho-Villasana, Y.; Zamudio-Ochoa, A.; Shingú-Vázquez, M.; Torres-Larios, A.; Pérez-Martínez, X. The pentatricopeptide repeats present in Pet309 are necessary for translation but not for stability of the mitochondrial COX1 mRNA in yeast. J. Biol. Chem. 2008, 283, 1472–1479. [Google Scholar] [CrossRef]
- Zamudio-Ochoa, A.; Camacho-Villasana, Y.; García-Guerrero, A.E.; Pérez-Martínez, X. The Pet309 pentatricopeptide repeat motifs mediate efficient binding to the mitochondrial COX1 transcript in yeast. RNA Biol. 2014, 11, 953–967. [Google Scholar] [CrossRef]
- Godard, F.; Tetaud, E.; Duvezin-Caubet, S.; di Rago, J.P. A genetic screen targeted on the FO component of mitochondrial ATP synthase in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 18181–18189. [Google Scholar] [CrossRef]
- Helfenbein, K.G.; Ellis, T.P.; Dieckmann, C.L.; Tzagoloff, A. ATP22, a nuclear gene required for expression of the F0 sector of mitochondrial ATPase in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 19751–19756. [Google Scholar] [CrossRef]
- Kellems, R.E.; Allison, V.F.; Butow, R.A. Cytoplasmic type 80S ribosomes associated with yeast mitochondria. IV. Attachment of ribosomes to the outer membrane of isolated mitochondria. J. Cell Biol. 1975, 65, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ades, I.Z.; Butow, R.A. The transport of proteins into yeast mitochondria. Kinetics and pools. J. Biol. Chem. 1980, 255, 9925–9935. [Google Scholar] [CrossRef]
- Suissa, M.; Schatz, G. Import of proteins into mitochondria. Translatable mRNAs for imported mitochondrial proteins are present in free as well as mitochondria-bound cytoplasmic polysomes. J. Biol. Chem. 1982, 257, 13048–13055. [Google Scholar] [CrossRef]
- Hwang, S.T.; Wachter, C.; Schatz, G. Protein import into the yeast mitochondrial matrix. A new translocation intermediate between the two mitochondrial membranes. J. Biol. Chem. 1991, 266, 21083–21089. [Google Scholar] [CrossRef]
- Garcia, M.D.X.; Devaux, F.; Singer, R.H.; Jacq, C. Yeast Mitochondrial Transcriptomics; Humana Press: Totowa, NJ, USA, 2007; Volume 372. [Google Scholar]
- Marc, P.; Margeot, A.; Devaux, F.; Blugeon, C.; Corral-Debrinski, M.; Jacq, C. Genome-wide analysis of mRNAs targeted to yeast mitochondria. EMBO Rep. 2002, 3, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Darzacq, X.; Delaveau, T.; Jourdren, L.; Singer, R.H.; Jacq, C. Mitochondria-associated yeast mRNAs and the biogenesis of molecular complexes. Mol. Biol. Cell 2007, 18, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Saint-Georges, Y.; Garcia, M.; Delaveau, T.; Jourdren, L.; Le Crom, S.; Lemoine, S.; Tanty, V.; Devaux, F.; Jacq, C. Yeast mitochondrial biogenesis: A role for the PUF RNA-binding protein Puf3p in mRNA localization. PLoS ONE 2008, 3, e2293. [Google Scholar] [CrossRef] [PubMed]
- Gadir, N.; Haim-Vilmovsky, L.; Kraut-Cohen, J.; Gerst, J.E. Localization of mRNAs coding for mitochondrial proteins in the yeast Saccharomyces cerevisiae. RNA 2011, 17, 1551–1565. [Google Scholar] [CrossRef]
- Fox, T. Mitochondrial Protein Synthesis, Import, and Assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; Selker, J.M.; Capaldi, R.A. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 2003, 546, 355–358. [Google Scholar] [CrossRef]
- Vogel, F.; Bornhövd, C.; Neupert, W.; Reichert, A.S. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 2006, 175, 237–247. [Google Scholar] [CrossRef]
- Stoldt, S.; Wenzel, D.; Kehrein, K.; Riedel, D.; Ott, M.; Jakobs, S. Spatial orchestration of mitochondrial translation and OXPHOS complex assembly. Nat. Cell Biol. 2018, 20, 528–534. [Google Scholar] [CrossRef]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brethes, D.; di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. Embo J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Interface mobility between monomers in dimeric bovine ATP synthase participates in the ultrastructure of inner mitochondrial membranes. Proc. Natl. Acad. Sci. USA 2021, 118, e2021012118. [Google Scholar] [CrossRef]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1604–1616. [Google Scholar] [CrossRef]
- Stram, A.R.; Payne, R.M. Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell. Mol. Life Sci. 2016, 73, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Rodriguez, M.; Chatzi, A.; Tokatlidis, K. Iron-sulfur clusters: From metals through mitochondria biogenesis to disease. J. Biol. Inorg. Chem. 2018, 23, 509–520. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nature 2014, 515, 80–84. [Google Scholar] [CrossRef]
- Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Post-translational modifications near the quinone binding site of mammalian complex I. J. Biol. Chem. 2013, 288, 24799–24808. [Google Scholar] [CrossRef] [PubMed]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Muellner-Wong, L.; Reljic, B.; Sharpe, A.J.; Jackson, T.D.; Beilharz, T.H.; Stojanovski, D.; Lazarou, M.; Stroud, D.A.; Ryan, M.T. Dissecting the Roles of Mitochondrial Complex I Intermediate Assembly Complex Factors in the Biogenesis of Complex I. Cell Rep. 2020, 31, 107541. [Google Scholar] [CrossRef]
- Andrews, B.; Carroll, J.; Ding, S.J.; Fearnley, I.M.; Walker, J.E. Assembly factors for the membrane arm of human complex I. Proc. Natl. Acad. Sci. USA 2013, 110, 18934–18939. [Google Scholar] [CrossRef] [PubMed]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef]
- Claros, M.G.; Vincens, P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996, 241, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, R.; Wanschers, B.F.J.; Nabuurs, S.B.; Nouws, J.; Nijtmans, L.G.; Huynen, M.A. NDUFB7 and NDUFA8 are located at the intermembrane surface of complex I. FEBS Lett. 2011, 585, 737–743. [Google Scholar] [CrossRef]
- Friederich, M.W.; Erdogan, A.J.; Coughlin, C.R.; Elos, M.T.; Jiang, H.; O’Rourke, C.P.; Lovell, M.A.; Wartchow, E.; Gowan, K.; Chatfield, K.C.; et al. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum. Mol. Genet. 2017, 26, 702–716. [Google Scholar] [CrossRef]
- Sánchez-Caballero, L.; Ruzzenente, B.; Bianchi, L.; Assouline, Z.; Barcia, G.; Metodiev, M.D.; Rio, M.; Funalot, B.; van den Brand, M.A.; Guerrero-Castillo, S.; et al. Mutations in Complex I Assembly Factor TMEM126B Result in Muscle Weakness and Isolated Complex I Deficiency. Am. J. Hum. Genet. 2016, 99, 208–216. [Google Scholar] [CrossRef]
- Zarsky, V.; Dolezal, P. Evolution of the Tim17 protein family. Biol. Direct 2016, 11, 54. [Google Scholar] [CrossRef]
- Wang, Y.; Carrie, C.; Giraud, E.; Elhafez, D.; Narsai, R.; Duncan, O.; Whelan, J.; Murcha, M.W. Dual Location of the Mitochondrial Preprotein Transporters B14.7 and Tim23-2 in Complex I and the TIM17:23 Complex in Arabidopsis Links Mitochondrial Activity and Biogenesis. Plant Cell 2012, 24, 2675–2695. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Netz, D.J.; Kerscher, S.; Elsasser, H.P.; Wittig, I.; Balk, J.; Brandt, U.; Lill, R. Human ind1, an iron-sulfur cluster assembly factor for respiratory complex I. Mol. Cell Biol. 2009, 29, 6059–6073. [Google Scholar] [CrossRef] [PubMed]
- Bych, K.; Kerscher, S.; Netz, D.J.; Pierik, A.J.; Zwicker, K.; Huynen, M.A.; Lill, R.; Brandt, U.; Balk, J. The iron-sulphur protein Ind1 is required for effective complex I assembly. Embo J. 2008, 27, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Van Vranken, J.; Na, U.; Winge, D.R.; Rutter, J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 168–180. [Google Scholar] [CrossRef]
- Hao, H.-X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.-P.; Kunst, H.; Devilee, P.; Cremers, C.W.R.J.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef]
- Cecchini, G. Function and structure of complex II of the respiratory chain. Annu Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef]
- Maio, N.; Ghezzi, D.; Verrigni, D.; Rizza, T.; Bertini, E.; Martinelli, D.; Zeviani, M.; Singh, A.; Carrozzo, R.; Rouault, T.A. Disease-Causing SDHAF1 Mutations Impair Transfer of Fe-S Clusters to SDHB. Cell Metab. 2016, 23, 292–302. [Google Scholar] [CrossRef]
- Na, U.; Yu, W.; Cox, J.; Bricker, D.K.; Brockmann, K.; Rutter, J.; Thummel, C.S.; Winge, D.R. The LYR factors SDHAF1 and SDHAF3 mediate maturation of the iron-sulfur subunit of succinate dehydrogenase. Cell Metab. 2014, 20, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Gebert, N.; Gebert, M.; Oeljeklaus, S.; von der Malsburg, K.; Stroud, D.A.; Kulawiak, B.; Wirth, C.; Zahedi, R.P.; Dolezal, P.; Wiese, S.; et al. Dual Function of Sdh3 in the Respiratory Chain and TIM22 Protein Translocase of the Mitochondrial Inner Membrane. Mol. Cell 2011, 44, 811–818. [Google Scholar] [CrossRef]
- Schagger, H.; Link, T.A.; Engel, W.D.; von Jagow, G. Isolation of the eleven protein subunits of the bc1 complex from beef heart. Methods Enzymol. 1986, 126, 224–237. [Google Scholar]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 1998, 281, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Mai, N.; Oláhová, M.; Scialó, F.; Formosa, L.E.; Stroud, D.A.; Garrett, M.; Lax, N.Z.; Robertson, F.M.; Jou, C.; et al. OXA1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol. Med. 2018, 10, e9060. [Google Scholar] [CrossRef]
- Cogliati, S.; Lorenzi, I.; Rigoni, G.; Caicci, F.; Soriano, M.E. Regulation of Mitochondria! Electron Transport Chain Assembly. J. Mol. Biol. 2018, 430, 4849–4873. [Google Scholar] [CrossRef] [PubMed]
- Ndi, M.; Marin-Buera, L.; Salvatori, R.; Singh, A.P.; Ott, M. Biogenesis of the bc1 Complex of the Mitochondrial Respiratory Chain. J. Mol. Biol. 2018, 430, 3892–3905. [Google Scholar] [CrossRef] [PubMed]
- Tucker, E.J.; Wanschers, B.F.; Szklarczyk, R.; Mountford, H.S.; Wijeyeratne, X.W.; van den Brand, M.A.; Leenders, A.M.; Rodenburg, R.J.; Reljic, B.; Compton, A.G.; et al. Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 2013, 9, e1004034. [Google Scholar] [CrossRef] [PubMed]
- Wanschers, B.F.J.; Szklarczyk, R.; van den Brand, M.A.M.; Jonckheere, A.; Suijskens, J.; Smeets, R.; Rodenburg, R.J.; Stephan, K.; Helland, I.B.; Elkamil, A.; et al. A mutation in the human CBP4 ortholog UQCC3 impairs complex III assembly, activity and cytochrome b stability. Hum. Mol. Genet. 2014, 23, 6356–6365. [Google Scholar] [CrossRef]
- Zara, V.; Palmisano, I.; Conte, L.; Trumpower, B.L. Further insights into the assembly of the yeast cytochrome bc1 complex based on analysis of single and double deletion mutants lacking supernumerary subunits and cytochrome b. Eur. J. Biochem. 2004, 271, 1209–1218. [Google Scholar] [CrossRef]
- Zara, V.; Conte, L.; Trumpower, B.L. Identification and characterization of cytochrome bc(1) subcomplexes in mitochondria from yeast with single and double deletions of genes encoding cytochrome bc(1) subunits. FEBS J. 2007, 274, 4526–4539. [Google Scholar] [CrossRef]
- Zara, V.; Conte, L.; Trumpower, B.L. Biogenesis of the yeast cytochrome bc1 complex. Biochim. Biophys. Acta 2009, 1793, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Zara, V.; Conte, L.; Trumpower, B.L. Evidence that the assembly of the yeast cytochrome bc1 complex involves the formation of a large core structure in the inner mitochondrial membrane. FEBS J. 2009, 276, 1900–1914. [Google Scholar] [CrossRef]
- Stephan, K.; Ott, M. Timing of dimerization of the bc(1) complex during mitochondrial respiratory chain assembly. Biochim. Et Biophys. Acta. Bioenerg. 2020, 1861, 148177. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Yu, C.A.; Kim, H.; Xia, J.Z.; Kachurin, A.M.; Zhang, L.; Yu, L.; Deisenhofer, J. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science 1997, 277, 60–66. [Google Scholar] [CrossRef]
- Sadler, I.; Suda, K.; Schatz, G.; Kaudewitz, F.; Haid, A. Sequencing of the nuclear gene for the yeast cytochrome c1 precursor reveals an unusually complex amino-terminal presequence. Embo J. 1984, 3, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Arnold, I.; Folsch, H.; Neupert, W.; Stuart, R.A. Two distinct and independent mitochondrial targeting signals function in the sorting of an inner membrane protein, cytochrome c1. J. Biol. Chem. 1998, 273, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Wachter, C.; Schatz, G.; Glick, B.S. Role of atp in the intramitochondrial sorting of cytochrome-C(1) and the adenine-nucleotide translocator. Embo J. 1992, 11, 4787–4794. [Google Scholar] [CrossRef]
- Zollner, A.; Rödel, G.; Haid, A. Molecular cloning and characterization of the Saccharomyces cerevisiae CYT2 gene encoding cytochrome-c1–heme lyase. FEBS J. 1992, 207, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, A.; Brandli, A.W.; Pesold-Hurt, B.; Blank, D.; Schatz, G. Transport of proteins to the mitochondrial intermembrane space: The ‘matrix-targeting’ and the ‘sorting’ domains in the cytochrome c1 presequence. Embo J. 1987, 6, 2433–2439. [Google Scholar] [CrossRef]
- Phillips, J.; Schmitt, M.E.; Brown, T.A.; Beckmann, J.D.; Trumpower, B.L. Isolation and characterization of QCR9, a nuclear gene encoding the 7.3-kDa subunit 9 of the Saccharomyces cerevisiae ubiquinol-cytochrome c oxidoreductase complex. An intron-containing gene with a conserved sequence occurring in the intron of COX4. J. Biol. Chem. 1990, 265, 20813–20821. [Google Scholar] [CrossRef]
- Brandt, U.; Uribe, S.; Schägger, H.; Trumpower, B.L. Isolation and characterization of QCR10, the nuclear gene encoding the 8.5-kDa subunit 10 of the Saccharomyces cerevisiae cytochrome bc1 complex. J. Biol. Chem. 1994, 269, 12947–12953. [Google Scholar] [CrossRef]
- Wasilewski, M.; Chojnacka, K.; Chacinska, A. Protein trafficking at the crossroads to mitochondria. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 125–137. [Google Scholar] [CrossRef]
- Fu, W.; Japa, S.; Beattie, D.S. Import of the iron-sulfur protein of the cytochrome b.c1 complex into yeast mitochondria. J. Biol. Chem. 1990, 265, 16541–16547. [Google Scholar] [CrossRef]
- Wagener, N.; Ackermann, M.; Funes, S.; Neupert, W. A pathway of protein translocation in mitochondria mediated by the AAA-ATPase Bcs1. Mol. Cell. 2011, 44, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Kater, L.; Wagener, N.; Berninghausen, O.; Becker, T.; Neupert, W.; Beckmann, R. Structure of the Bcs1 AAA-ATPase suggests an airlock-like translocation mechanism for folded proteins. Nat. Struct. Mol. Biol. 2020, 27, 142–149. [Google Scholar] [CrossRef]
- Tang, W.K.; Borgnia, M.J.; Hsu, A.L.; Esser, L.; Fox, T.; de Val, N.; Xia, D. Structures of AAA protein translocase Bcs1 suggest translocation mechanism of a folded protein. Nat. Struct. Mol. Biol. 2020, 27, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.Z.; Smith, P.M.; Fox, J.L.; Khalimonchuk, O.; Winge, D.R. Late-Stage Maturation of the Rieske Fe/S Protein: Mzm1 Stabilizes Rip1 but Does Not Facilitate Its Translocation by the AAA ATPase Bcs1. Mol. Cell. Biol. 2012, 32, 4400–4409. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Lobo, T.; Fox, J.L.; Zeviani, M.; Winge, D.R.; Fernandez-Vizarra, E. LYRM7/MZM1L is a UQCRFS1 chaperone involved in the last steps of mitochondrial Complex III assembly in human cells. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U.; Yu, L.; Yu, C.A.; Trumpower, B.L. The mitochondrial targeting presequence of the Rieske iron-sulfur protein is processed in a single step after insertion into the cytochrome bc1 complex in mammals and retained as a subunit in the complex. J. Biol. Chem. 1993, 268, 8387–8390. [Google Scholar] [CrossRef]
- Bottani, E.; Cerutti, R.; Harbour, M.E.; Ravaglia, S.; Dogan, S.A.; Giordano, C.; Fearnley, I.M.; D’Amati, G.; Viscomi, C.; Fernandez-Vizarra, E.; et al. TTC19 Plays a Husbandry Role on UQCRFS1 Turnover in the Biogenesis of Mitochondrial Respiratory Complex III. Mol. Cell 2017, 67, 96–105. [Google Scholar] [CrossRef]
- Ghezzi, D.; Arzuffi, P.; Zordan, M.; Da Re, C.; Lamperti, C.; Benna, C.; D’Adamo, P.; Diodato, D.; Costa, R.; Mariotti, C.; et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat. Genet. 2011, 43, 259–263. [Google Scholar] [CrossRef]
- Capaldi, R.A. Structure and function of cytochrome c oxidase. Annu. Rev. Biochem. 1990, 59, 569–596. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, S.H.M.E.; Guerrero-Castillo, S.; Signes, A.; Ding, S.; Fearnley, I.M.; Taylor, R.W.; Tiranti, V.; Arnold, S.; Fernandez-Vizarra, E.; Zeviani, M. MR-1S Interacts with PET100 and PET117 in Module-Based Assembly of Human Cytochrome c Oxidase. Cell Rep. 2017, 18, 1727–1738. [Google Scholar] [CrossRef]
- Hayashi, T.; Asano, Y.; Shintani, Y.; Aoyama, H.; Kioka, H.; Tsukamoto, O.; Hikita, M.; Shinzawa-Itoh, K.; Takafuji, K.; Higo, S.; et al. Higd1a is a positive regulator of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 1553–1558. [Google Scholar] [CrossRef]
- Mick, D.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 15, 1528–1541. [Google Scholar] [CrossRef] [PubMed]
- Dennerlein, S.; Oeljeklaus, S.; Jans, D.; Hellwig, C.; Bareth, B.; Jakobs, S.; Deckers, M.; Warscheid, B.; Rehling, P. MITRAC7 Acts as a COX1-Specific Chaperone and Reveals a Checkpoint during Cytochrome c Oxidase Assembly. Cell Rep. 2015, 12, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, R.; Wanschers, B.F.; Cuypers, T.D.; Esseling, J.J.; Riemersma, M.; van den Brand, M.A.; Gloerich, J.; Lasonder, E.; van den Heuvel, L.P.; Nijtmans, L.G.; et al. Iterative orthology prediction uncovers new mitochondrial proteins and identifies C12orf62 as the human ortholog of COX14, a protein involved in the assembly of cytochrome c oxidase. Genome Biol. 2012, 13, R12. [Google Scholar] [CrossRef] [PubMed]
- Clemente, P.; Peralta, S.; Cruz-Bermudez, A.; Echevarría, L.; Fontanesi, F.; Barrientos, A.; Fernandez-Moreno, M.A.; Garesse, R. hCOA3 stabilizes cytochrome c oxidase 1 (COX1) and promotes cytochrome c oxidase assembly in human mitochondria. J. Biol. Chem. 2013, 288, 8321–8331. [Google Scholar] [CrossRef] [PubMed]
- Mick, D.; Vukotic, M.; Piechura, H.; Meyer, H.E.; Warscheid, B.; Deckers, M.; Rehling, P. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J. Cell Biol. 2010, 191, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Leary, S.C.; Guercin, G.H.; Agar, J.N.; Horvath, R.; Kennaway, N.G.; Harding, C.O.; Jaksch, M.; Shoubridge, E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef]
- Diaz, F.; Thomas, C.K.; Garcia, S.; Hernandez, D.; Moraes, C.T. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum. Mol. Genet. 2005, 14, 2737–2748. [Google Scholar] [CrossRef]
- Hiser, L.; Di Valentin, M.; Hamer, A.G.; Hosler, J.P. Cox11p is required for stable formation of the Cu(B) and magnesium centers of cytochrome c oxidase. J. Biol. Chem. 2000, 275, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Glerum, D.; Shtanko, A.; Tzagoloff, A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 1996, 271, 14504–14509. [Google Scholar] [CrossRef]
- Bode, M.; Woellhaf, M.W.; Bohnert, M.; van der Laan, M.; Sommer, F.; Jung, M.; Zimmermann, R.; Schroda, M.; Herrmann, J.M. Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol. Biol. Cell 2015, 26, 2385–2401. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, N.; Racca, S.; Gras, D.E.; Gonzalez, D.H.; Welchen, E. The Complexity of Mitochondrial Complex IV: An Update of Cytochrome c Oxidase Biogenesis in Plants. Int. J. Mol. Sci. 2018, 19, 662. [Google Scholar] [CrossRef] [PubMed]
- Bourens, M.; Boulet, A.; Leary, S.C.; Barrientos, A. Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet. 2014, 23, 2901–2913. [Google Scholar] [CrossRef]
- Lorenzi, I.; Oeljeklaus, S.; Aich, A.; Ronsör, C.; Callegari, S.; Dudek, J.; Warscheid, B.; Dennerlein, S.; Rehling, P. The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Sasarman, F.; Nishimura, T.; Shoubridge, E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009, 18, 2230–2240. [Google Scholar] [CrossRef]
- Leary, S.; Cobine, P.A.; Kaufman, B.A.; Guercin, G.H.; Mattman, A.; Palaty, J.; Lockitch, G.; Winge, D.R.; Rustin, P.; Horvath, R.; et al. The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab. 2007, 5, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.; Kaufman, B.A.; Pellecchia, G.; Guercin, G.H.; Mattman, A.; Jaksch, M.; Shoubridge, E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 2004, 13, 1839–1848. [Google Scholar] [CrossRef]
- Stroud, D.; Maher, M.J.; Lindau, C.; Vögtle, F.N.; Frazier, A.E.; Surgenor, E.; Mountford, H.; Singh, A.P.; Bonas, M.; Oeljeklaus, S.; et al. COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 2015, 24, 5404–5415. [Google Scholar] [CrossRef]
- Pacheu-Grau, D.; Bareth, B.; Dudek, J.; Juris, L.; Vögtle, F.N.; Wissel, M.; Leary, S.C.; Dennerlein, S.; Rehling, P.; Deckers, M. Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell Metab. 2015, 21, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Aich, A.; Wang, C.; Chowdhury, A.; Ronsör, C.; Pacheu-Grau, D.; Richter-Dennerlein, R.; Dennerlein, S.; Rehling, P. COX16 promotes COX2 metallation and assembly during respiratory complex IV biogenesis. eLife 2018, 7, e32572. [Google Scholar] [CrossRef] [PubMed]
- Cerqua, C.; Morbidoni, V.; Desbats, M.A.; Doimo, M.; Frasson, C.; Sacconi, S.; Baldoin, M.C.; Sartori, G.; Basso, G.; Salviati, L.; et al. COX16 is required for assembly of cytochrome c oxidase in human cells and is involved in copper delivery to COX2. Biochim. Biophys. Acta 2018, 1859, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef]
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 1994, 370, 621–628. [Google Scholar] [CrossRef]
- Wittig, I.; Schagger, H. Structural organization of mitochondrial ATP synthase. Biochim. Biophys. Acta 2008, 1777, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, S.H.; Tzagoloff, A. Identification of two nuclear genes (ATP11, ATP12) required for assembly of the yeast F1-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 4986–4990. [Google Scholar] [CrossRef]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef]
- Dyer, M.R.; Walker, J.E. Sequences of members of the human gene family for the c subunit of mitochondrial ATP synthase. Biochem. J. 1993, 293 Pt 1, 51–64. [Google Scholar] [CrossRef]
- Yan, W.L.; Lerner, T.J.; Haines, J.L.; Gusella, J.F. Sequence analysis and mapping of a novel human mitochondrial ATP synthase subunit 9 cDNA (ATP5G3). Genomics 1994, 24, 375–377. [Google Scholar] [CrossRef]
- Van Bloois, E.; Haan, G.J.; de Gier, J.W.; Oudega, B.; Luirink, J. F1F0 ATP synthase subunit c is targeted by the SRP to YidC in the E. coli inner membrane. FEBS Lett. 2004, 576, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Kolli, R.; Soll, J.; Carrie, C. Plant Mitochondrial Inner Membrane Protein Insertion. Int. J. Mol. Sci. 2018, 19, 641. [Google Scholar] [CrossRef]
- Bahri, H.; Buratto, J.; Rojo, M.; Dompierre, J.P.; Salin, B.; Blancard, C.; Cuvellier, S.; Rose, M.; Ben Ammar Elgaaied, A.; Tetaud, E.; et al. TMEM70 forms oligomeric scaffolds within mitochondrial cristae promoting in situ assembly of mammalian ATP synthase proton channel. Biochim. Biophys. Acta. Mol. Cell Res. 2021, 1868, 118942. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. TMEM70 and TMEM242 help to assemble the rotor ring of human ATP synthase and interact with assembly factors for complex I. Proc. Natl. Acad. Sci. USA 2021, 118, e2100558118. [Google Scholar] [CrossRef] [PubMed]
- Ahting, U.; Floss, T.; Uez, N.; Schneider-Lohmar, I.; Becker, L.; Kling, E.; Iuso, A.; Bender, A.; de Angelis, M.H.; Gailus-Durner, V.; et al. Neurological phenotype and reduced lifespan in heterozygous Tim23 knockout mice, the first mouse model of defective mitochondrial import. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Sirk, D.; Zhu, Z.; Wadia, J.S.; Shulyakova, N.; Phan, N.; Fong, J.; Mills, L.R. Chronic exposure to sub-lethal beta-amyloid (A beta) inhibits the import of nuclear-encoded proteins to mitochondria in differentiated PC12 cells. J. Neurochem. 2007, 103, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Et Biophys. Acta-Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.N.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.Y.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Jiang, H.B.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.P.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. Alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef]
- Bender, A.; Desplats, P.; Spencer, B.; Rockenstein, E.; Adame, A.; Elstner, M.; Laub, C.; Mueller, S.; Koob, A.O.; Mante, M.; et al. TOM40 Mediates Mitochondrial Dysfunction Induced by alpha-Synuclein Accumulation in Parkinson’s Disease. PLoS ONE 2013, 8, e62277. [Google Scholar] [CrossRef]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.C.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef]
- Napoli, E.; Wong, S.; Hung, C.; Ross-Inta, C.; Bomdica, P.; Giulivi, C. Defective mitochondrial disulfide relay system, altered mitochondrial morphology and function in Huntingtons disease. Hum. Mol. Genet. 2013, 22, 989–1004. [Google Scholar] [CrossRef]
- Fischer, L.R.; Igoudjil, A.; Magrane, J.; Li, Y.J.; Hansen, J.M.; Manfredi, G.; Glass, J.D. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain 2011, 134, 196–209. [Google Scholar] [CrossRef]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J. Neurosci. 2005, 25, 2463–2470. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef]
- Li, Q.A.; Vande Velde, C.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.Q.; Chun, S.J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef] [PubMed]
- Brischigliaro, M.; Zeviani, M. Cytochrome c oxidase deficiency. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. In Mitochondrial Diseases; Garone, C., Minczuk, M., Eds.; Portland Press Ltd: London, UK, 2018; Volume 62, pp. 271–286. [Google Scholar]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Sazanov, L.A. Mammalian Mitochondrial Complex I Structure and Disease-Causing Mutations. Trends Cell Biol. 2018, 28, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Shahrour, M.A.; Staretz-Chacham, O.; Dayan, D.; Stephen, J.; Weech, A.; Damseh, N.; Pri Chen, H.; Edvardson, S.; Mazaheri, S.; Saada, A.; et al. Mitochondrial epileptic encephalopathy, 3-methylglutaconic aciduria and variable complex V deficiency associated with TIMM50 mutations. Clin. Genet. 2017, 91, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Melchionda, L.; Burlina, A.; Robinson, A.J.; Ghezzi, D.; Zeviani, M. Mutations in TIMM50 compromise cell survival in OxPhos-dependent metabolic conditions. EMBO Mol. Med. 2018, 10, e8698. [Google Scholar] [CrossRef]
- Tort, F.; Ugarteburu, O.; Texidó, L.; Gea-Sorlí, S.; García-Villoria, J.; Ferrer-Cortès, X.; Arias, Á.; Matalonga, L.; Gort, L.; Ferrer, I.; et al. Mutations in TIMM50 cause severe mitochondrial dysfunction by targeting key aspects of mitochondrial physiology. Hum. Mutat. 2019, 40, 1700–1712. [Google Scholar] [CrossRef]
- Roesch, K.; Curran, S.P.; Tranebjaerg, L.; Koehler, C.M. Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum. Mol. Genet. 2002, 11, 477–486. [Google Scholar] [CrossRef]
- Pacheu-Grau, D.; Callegari, S.; Emperador, S.; Thompson, K.; Aich, A.; Topol, S.E.; Spencer, E.G.; McFarland, R.; Ruiz-Pesini, E.; Torkamani, A.; et al. Mutations of the mitochondrial carrier translocase channel subunit TIM22 cause early-onset mitochondrial myopathy. Hum. Mol. Genet. 2018, 27, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Du, M.; Xie, J.; Luo, T.; Zhou, Y.; Zhang, K.; Li, J.; Chen, D.; Xu, P.; Jia, M.; et al. Mutations in TOMM70 lead to multi-OXPHOS deficiencies and cause severe anemia, lactic acidosis, and developmental delay. J. Hum. Genet. 2020, 65, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Koo, E.H. Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 2011, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Mann, V.M.; Cooper, J.M.; Daniel, S.E.; Srai, K.; Jenner, P.; Marsden, C.D.; Schapira, A.H.V. Complex-i, iron, and ferritin in parkinsons-disease substantia-nigra. Ann. Neurol. 1994, 36, 876–881. [Google Scholar] [CrossRef]
- Bindoff, L.A.; Birchmachin, M.A.; Cartlidge, N.E.F.; Parker, W.D.; Turnbull, D.M. Respiratory-chain abnormalities in skeletal-muscle from patients with parkinsons-disease. J. Neurol. Sci. 1991, 104, 203–208. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Parkinsonism Relat. Disord. 1999, 5, 139–143. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.F.; Flanagan, L.; Desilva, H.A.R.; Kittel, A.; Saitoh, T. The precursor protein of non-a-beta component of alzheimers-disease amyloid is a presynaptic protein of the central-nervous-system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett 1994, 345, 27–32. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Song, D.D.; Shults, C.W.; Sisk, A.; Rockenstein, E.; Masliah, E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp. Neurol. 2004, 186, 158–172. [Google Scholar] [CrossRef]
- Li, W.W.; Yang, R.; Guo, J.C.; Ren, H.M.; Zha, X.L.; Cheng, J.S.; Cai, D.F. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 2007, 18, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int. J. Biochem. Cell Biol. 2009, 41, 2015–2024. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, Paraquat, and Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser(65). Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Stevens, D.A.; Kang, S.U.; Jiang, H.S.; Lee, Y.I.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Bertolin, G.; Jacoupy, M.; Traver, S.; Ferrando-Miguel, R.; Saint Georges, T.; Grenier, K.; Ardila-Osorio, H.; Muriel, M.P.; Takahashi, H.; Lees, A.J.; et al. Parkin maintains mitochondrial levels of the protective Parkinson’s disease-related enzyme 17-beta hydroxysteroid dehydrogenase type 10. Cell Death Differ. 2015, 22, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.; Wu, Z.H.; Klinkenberg, M.; Sun, Y.P.; Auburger, G.; Guo, S.; Lu, B.W. PINK1 and Parkin Control Localized Translation of Respiratory Chain Component mRNAs on Mitochondria Outer Membrane. Cell Metab. 2015, 21, 95–108. [Google Scholar] [CrossRef]
- Macdonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntingtons-disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Li, H.; Li, S.H.; Johnston, H.; Shelbourne, P.F.; Li, X.J. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat. Genet. 2000, 25, 385–389. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Li, S.H.; Wang, C.E.; Li, H.; Wang, J.J.; Rong, J.; Xu, X.S.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008, 28, 2783–2792. [Google Scholar] [CrossRef]
- Yu, Z.X.; Li, S.H.; Evans, J.; Pillarisetti, A.; Li, H.; Li, X.J. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington’s disease. J. Neurosci. 2003, 23, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Benchoua, A.; Trioulier, Y.; Zala, D.; Gaillard, M.C.; Lefort, N.; Dufour, N.; Saudou, F.; Elalouf, J.M.; Hirsch, E.; Hantraye, P.; et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated Huntingtin. Mol. Biol. Cell 2006, 17, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Brennan, W.A.; Bird, E.D.; Aprille, J.R. Regional mitochondrial respiratory activity in huntingtons-disease brain. J. Neurochem. 1985, 44, 1948–1950. [Google Scholar] [CrossRef]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Beal, M.F.; Manfredi, G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar] [CrossRef]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef] [PubMed]
- Lisowsky, T. Erv1 is involved in the cell-division cycle and the maintenance of mitochondrial genomes in saccharomyces-cerevisiae. Curr. Genet. 1994, 26, 15–20. [Google Scholar] [CrossRef]
- Becher, D.; Kricke, J.; Stein, G.; Lisowsky, T. A mutant for the yeast scERV1 gene displays a new defect in mitochondrial morphology and distribution. Yeast 1999, 15, 1171–1181. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Ronchi, D.; Lodi, T.; Fassone, E.; Tigano, M.; Lamperti, C.; Corti, S.; Bordoni, A.; Fortunato, F.; Nizzardo, M.; et al. The Mitochondrial Disulfide Relay System Protein GFER Is Mutated in Autosomal-Recessive Myopathy with Cataract and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2009, 84, 594–604. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.K.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Gu, M.; Gash, M.T.; Mann, V.M.; JavoyAgid, F.; Cooper, J.M.; Schapira, A.H.V. Mitochondrial defect in Huntington’s disease on caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef]
- Browne, S.E.; Beal, M.F. The energetics of Huntington’s disease. Neurochem. Res. 2004, 29, 531–546. [Google Scholar] [CrossRef]
- Valentine, J.S.; Doucette, P.A.; Potter, S.Z. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem 2005, 74, 563–593. [Google Scholar] [CrossRef] [PubMed]
- Carri, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg. Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef]
- Cozzolino, M.; Rossi, S.; Mirra, A.; Carri, M.T. Mitochondrial dynamism and the pathogenesis of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2015, 9, 31. [Google Scholar] [CrossRef]
- Crugnola, V.; Lamperti, C.; Lucchini, V.; Ronchi, D.; Peverelli, L.; Prelle, A.; Sciacco, M.; Bordoni, A.; Fassone, E.; Fortunato, F.; et al. Mitochondrial Respiratory Chain Dysfunction in Muscle From Patients With Amyotrophic Lateral Sclerosis. Arch. Neurol. 2010, 67, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Corti, S.; Donadoni, C.; Ronchi, D.; Bordoni, A.; Fortunato, F.; Santoro, D.; Del Bo, R.; Lucchini, V.; Crugnola, V.; Papadimitriou, D.; et al. Amyotrophic lateral sclerosis linked to a novel SOD1 mutation with muscle mitochondrial dysfunction. J. Neurol. Sci. 2009, 276, 170–174. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The Next (Neurode) Generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Sturtz, L.A.; Diekert, K.; Jensen, L.T.; Lill, R.; Culotta, V.C. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria—A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 2001, 276, 38084–38089. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver—Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Hervias, I.; Beal, M.F.; Manfredi, G. Mitochondrial dysfunction and amyotrophic lateral sclerosis. Muscle Nerve 2006, 33, 598–608. [Google Scholar] [CrossRef]
- Lehmer, C.; Schludi, M.H.; Ransom, L.; Greiling, J.; Junghanel, M.; Exner, N.; Riemenschneider, H.; van der Zee, J.; Van Broeckhoven, C.; Weydt, P.; et al. A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol. Med. 2018, 10, e8558. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546.e539. [Google Scholar] [CrossRef]
- Baker, M.J.; Palmer, C.S.; Stojanovski, D. Mitochondrial protein quality control in health and disease. Br. J. Pharm. 2014, 171, 1870–1889. [Google Scholar] [CrossRef]
- Samluk, L.; Chroscicki, P.; Chacinska, A. Mitochondrial protein import stress and signaling. Curr. Opin. Physiol. 2018, 3, 41–48. [Google Scholar] [CrossRef]
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, D.; Sorrentino, V.; Auwerx, J. Cytosolic Proteostasis Networks of the Mitochondrial Stress Response. Trends Biochem Sci 2017, 42, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and Nuclear Accumulation of the Transcription Factor ATFS-1 Promotes OXPHOS Recovery during the UPRmt. Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The Matrix Peptide Exporter HAF-1 Signals a Mitochondrial UPR by Activating the Transcription Factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.G.; Schneid, S.; Schwarz, M.; Rackles, E.; Fischer, C.; Haeussler, S.; Regmi, S.G.; Yeroslaviz, A.; Habermann, B.; Mokranjac, D.; et al. Compromised Mitochondrial Protein Import Acts as a Signal for UPRmt. Cell Rep. 2019, 28, 1659–1669. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 2017, 6, e30952. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Labbadia, J.; Brielmann, R.M.; Neto, M.F.; Lin, Y.F.; Haynes, C.M.; Morimoto, R.I. Mitochondrial Stress Restores the Heat Shock Response and Prevents Proteostasis Collapse during Aging. Cell Rep. 2017, 21, 1481–1494. [Google Scholar] [CrossRef]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci Rep. 2017, 7, 16441. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C-elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef]
- St Martin, J.L.; Klucken, J.; Outeiro, T.F.; Nguyen, P.; Keller-McGandy, C.; Cantuti-Castelvetri, I.; Grammatopoulos, T.N.; Standaert, D.G.; Hyman, B.T.; McLean, P.J. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J. Neurochem. 2007, 100, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M.; Szegezdi, E.; Quigney, D.J.; Samali, A. Hsp27 inhibits 6-hydroxydopamine-induced cytochrome c release and apoptosis in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 327, 801–810. [Google Scholar] [CrossRef]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 reduces alpha-synuclein aggregation and toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef]
- Riar, A.K.; Burstein, S.R.; Palomo, G.M.; Arreguin, A.; Manfredi, G.; Germain, D. Sex specific activation of the ER alpha axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum. Mol. Genet. 2017, 26, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; Van Remmen, H. Metabolic and Stress Response Changes Precede Disease Onset in the Spinal Cord of Mutant SOD1 ALS Mice. Front. Neurosci. 2019, 13, 487. [Google Scholar] [CrossRef]
- Shen, Y.; Ding, M.; Xie, Z.H.; Liu, X.T.; Yang, H.; Jin, S.Q.; Xu, S.L.; Zhu, Z.Y.; Wang, Y.; Wang, D.W.; et al. Activation of Mitochondrial Unfolded Protein Response in SHSY5Y Expressing APP Cells and APP/PS1 Mice. Front. Cell. Neurosci. 2020, 13, 568. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for Mitochondrial UPR Gene Activation in Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 610–614. [Google Scholar] [CrossRef]
- Regitz, C.; Fitzenberger, E.; Mahn, F.L.; Dussling, L.M.; Wenzel, U. Resveratrol reduces amyloid-beta (A beta(1-42))-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur. J. Nutr. 2016, 55, 741–747. [Google Scholar] [CrossRef]
- Poveda-Huertes, D.; Matic, S.; Marada, A.; Habernig, L.; Licheva, M.; Myketin, L.; Gilsbach, R.; Tosal-Castano, S.; Papinski, D.; Mulica, P.; et al. An Early mtUPR: Redistribution of the Nuclear Transcription Factor Rox1 to Mitochondria Protects against Intramitochondrial Proteotoxic Aggregates. Mol. Cell 2020, 77, 180–188. [Google Scholar] [CrossRef]
- Wrobel, L.; Topf, U.; Bragoszewski, P.; Wiese, S.; Sztolsztener, M.E.; Oeljeklaus, S.; Varabyova, A.; Lirski, M.; Chroscicki, P.; Mroczek, S.; et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 2015, 524, 485–488. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Callegari, S.; Dennerlein, S. Sensing the Stress: A Role for the UPRmt and UPRam in the Quality Control of Mitochondria. Front. Cell. Dev. Biol. 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.N.; Upadhya, S.C. Role of ubiquitin-proteasome-mediated proteolysis in nervous system disease. Biochim. Biophys. Acta Gene Regul. Mech. 2011, 1809, 128–140. [Google Scholar] [CrossRef]
- Dennissen, F.J.A.; Kholod, N.; van Leeuwen, F.W. The ubiquitin proteasome system in neurodegenerative diseases: Culprit, accomplice or victim? Prog. Neurobiol. 2012, 96, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated With Dysfunction of the Ubiquitin-Proteasome System. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Chen, X.J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 2015, 524, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Gerbasi, V.R.; Link, A.J. The myotonic dystrophy type 2 protein ZNF9 is part of an ITAF complex that promotes cap-independent translation. Mol. Cell. Proteom. 2007, 6, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Granneman, S.; Thoms, M.; Manikas, R.G.; Tollervey, D.; Hurt, E. Coupled GTPase and remodelling ATPase activities form a checkpoint for ribosome export. Nature 2014, 505, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.D.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Dachsel, J.C.; Vilarino-Guell, C.; Lincoln, S.J.; Lepretre, F.; Hulihan, M.M.; Kachergus, J.; Milnerwood, A.J.; Tapia, L.; Song, M.S.; et al. Translation Initiator EIF4G1 Mutations in Familial Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 398–406. [Google Scholar] [CrossRef]
- Coyne, L.P.; Chen, X.J. mPOS is a novel mitochondrial trigger of cell death—Implications for neurodegeneration. FEBS Lett. 2018, 592, 759–775. [Google Scholar] [CrossRef]
- Weidberg, H.; Amon, A. MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science 2018, 360, eaan4146. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.Z.; Luo, X.L.; Huang, Z.; Chen, L.X. MitoCPR: A novel protective mechanism in response to mitochondrial protein import stress. Acta Biochim. Biophys. Sin. 2018, 50, 1072–1074. [Google Scholar] [CrossRef]
- Martensson, C.U.; Priesnitz, C.; Song, J.Y.; Ellenrieder, L.; Doan, K.N.; Boos, F.; Floerchinger, A.; Zufall, N.; Oeljeklaus, S.; Warscheid, B.; et al. Mitochondrial protein translocation-associated degradation. Nature 2019, 569, 679–683. [Google Scholar] [CrossRef]
- Neuber, O.; Jarosch, E.; Volkwein, C.; Walter, J.; Sommer, T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, C.; Buchberger, A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.C.; Allen, W.J.; Watkins, D.W.; Buddrus, L.; Noone, D.; Liu, X.; Richardson, A.P.; Chacinska, A.; Collinson, I. A High-Resolution Luminescent Assay for Rapid and Continuous Monitoring of Protein Translocation across Biological Membranes. J. Mol. Biol. 2019, 431, 1689–1699. [Google Scholar] [CrossRef]




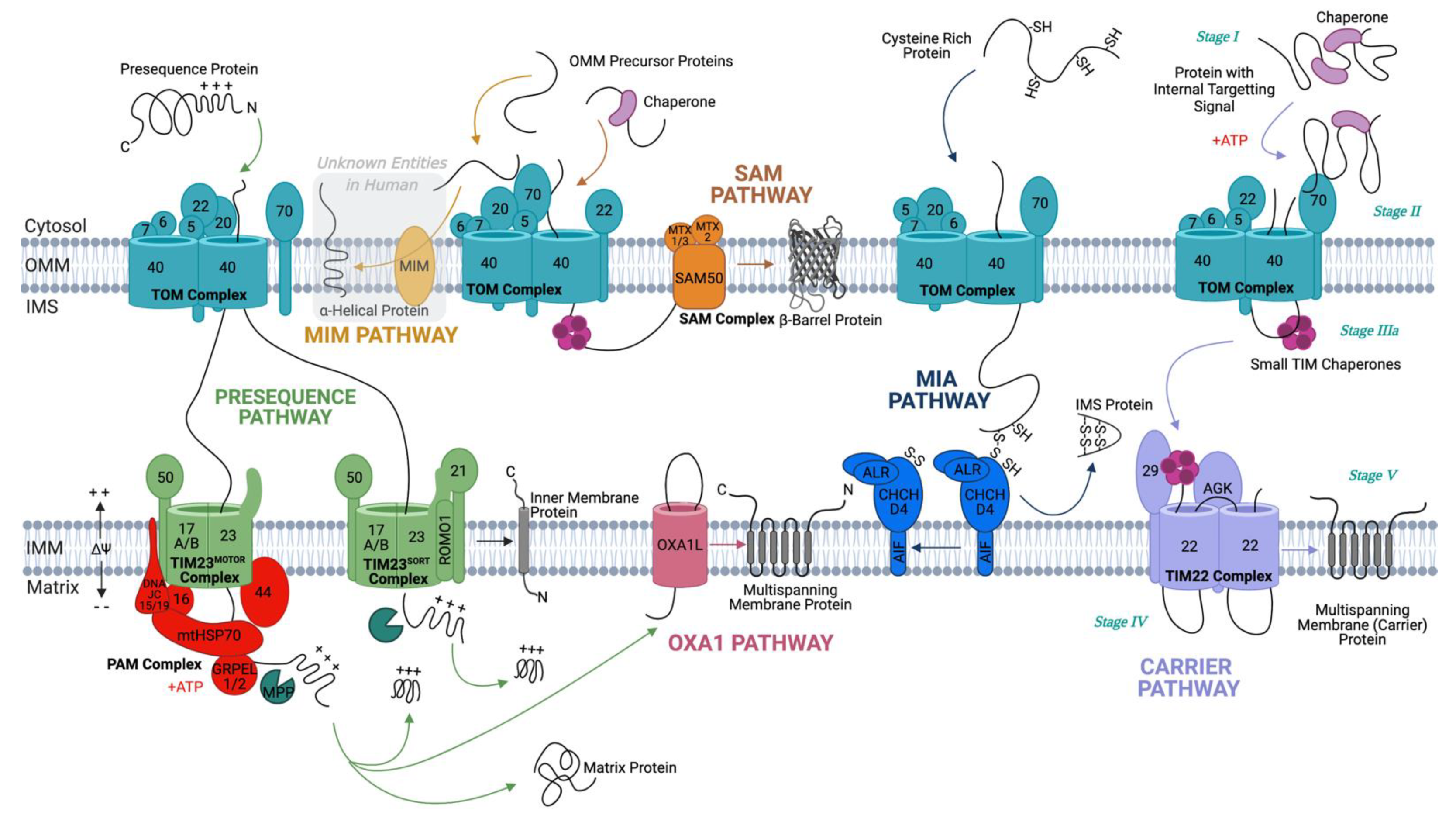{kind=link}
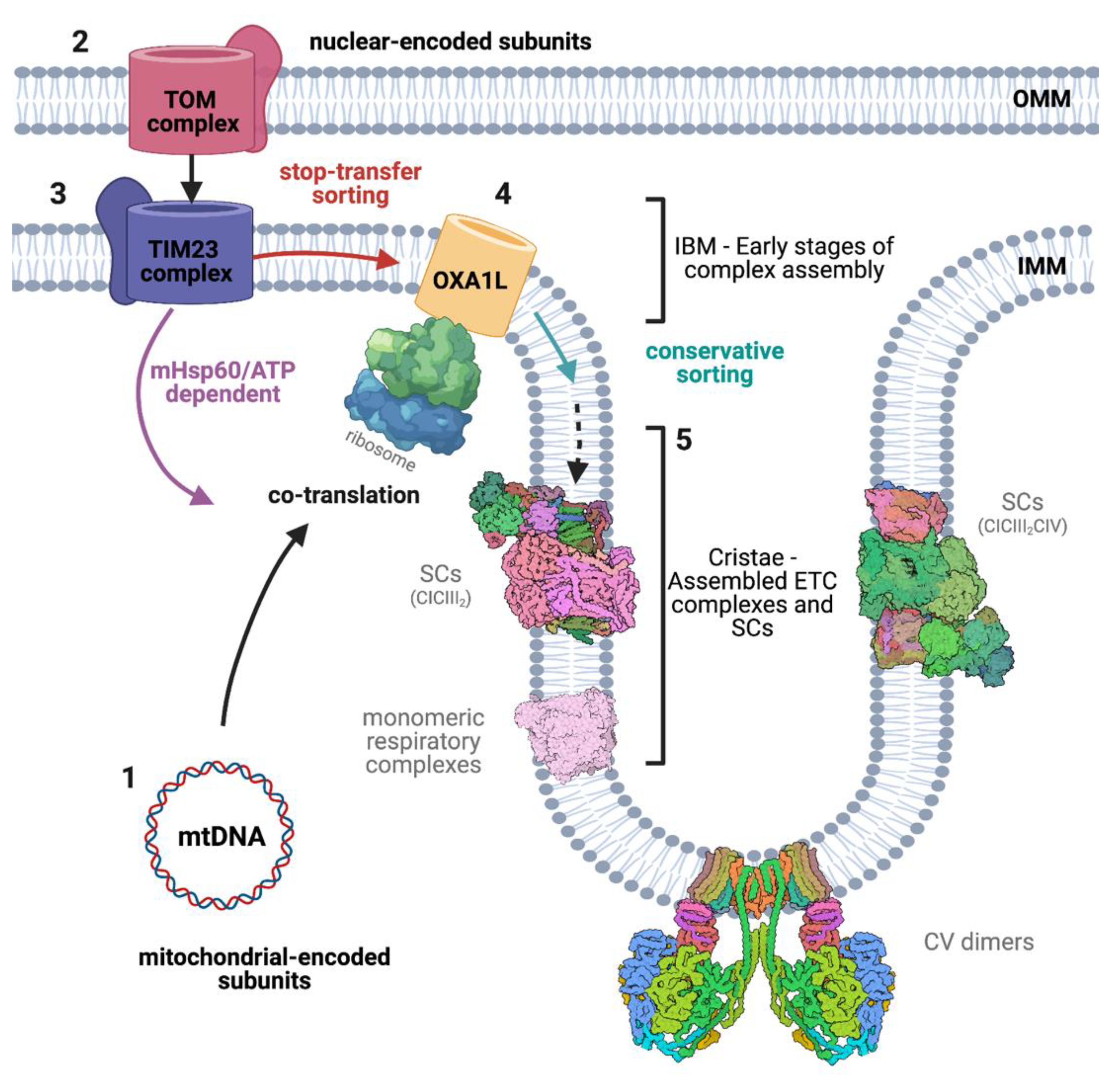{kind=link}
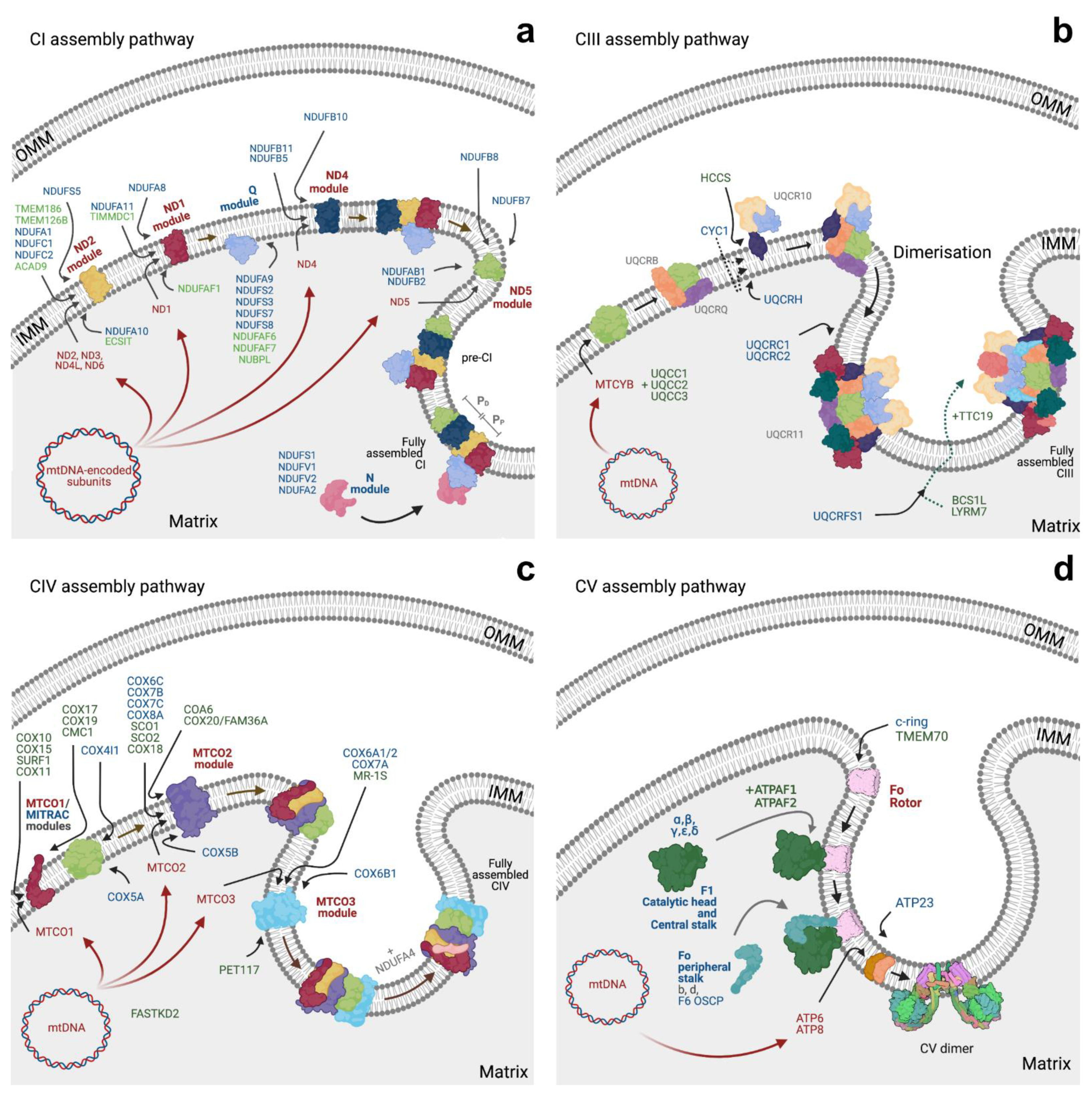{kind=link}
| Pathway | Complex | Subunit (Mammalian) | Yeast Homolog | Main Function | Topology | |
|---|---|---|---|---|---|---|
| TOM | TOM-Holo Complex | Core complex | TOM40 and TOM40L | Tom40 | Channel protein | β-barrel (19 β strands) and one N-terminal α-helical segment located inside pore |
| TOM22 | Tom22 | Receptor protein. Located at the dimer interface between TOM40 pores. | α-helical (single TMD); Cin-Nout | |||
| TOM5 | Tom5 | Complex assembly/stability | α-helical (single TMD); Cin-Nout | |||
| TOM6 | Tom6 | Complex assembly/stability | α-helical (single TMD); Cin-Nout | |||
| TOM7 | Tom7 | Complex assembly/stability | α-helical (single TMD); Cin-Nout | |||
| Receptors | TOM70 | Tom70 | Receptor for carrier precursors | α-helical (single TMD); N-terminally inserted | ||
| TOM20 | Tom20 | Receptor for presequence precursors | α-helical (single TMD); N-terminally inserted | |||
| SAM | SAM Complex | SAM50 | Sam50 | Core subunit responsible for β-barrel protein insertion | β-barrel (16 β-strands) | |
| MTX1 and MTX3 | Sam37 | Accessory subunit | N/A | |||
| MTX2 | Sam35 | Accessory subunit | N/A | |||
| MIM | MIM Complex | Unknown | Mim1 | Biogenesis of α-helical OMM proteins | -- | |
| Unknown | Mim2 | Biogenesis of α-helical OMM proteins | -- | |||
| MIA | MIA Complex | CHCHD4 | Mia40 | Oxidoreductase | Helix-loop-helix attached to a flexible helical arm | |
| ALR | Erv1 | Reoxidises Mia40 | α-helical (a1-5) bundle | |||
| Cytochrome C/ETC | Cytochrome C/ETC | Final electron acceptor | Class I of the c type cytochrome | |||
| AIF | - | Anchors CHCHD4 to the IMM | One C-terminal TMD; Nin, Cout | |||
| TIM23/Presequence | TIM23SORT Complex | TIM21 | Tim21 | Recognition/direction of precursor proteins to TIM23 | α-helical (single TMD) with a large IMS domain; Nin-Cout | |
| ROMO1 | Mgr2 | Lateral release of proteins into the IMM | Two α-helical TMDs, joined by a basic loop | |||
| TIM23MOTOR Complex | TIM17A/B | Tim17 | Channel forming | 4 TMDs and a small IMS domain | ||
| TIM23 | Tim23 | Channel forming | Multiple TMDs, and IMS exposed hydrophilic domain | |||
| TIM50 | Tim50 | Receptor Protein | Single TMD, large IMS exposed C-terminal domain | |||
| PAM Complex | TIM44 | Tim44 | Scaffold for complex & binding emerging precursor | Peripheral membrane protein on matrix side | ||
| mtHSP70 (Mortalin) | SSC1 (mtHsp70) | ATPase | β-sheet and α-helical domains | |||
| DNAJC15 and DNAJC19 | Pam18 (Tim14) | Stimulates ATPase activity of mHsp70 | Single α-helical TMD, with large C-terminal matrix domain and small N-terminal IMS domain | |||
| TIM16 | Pam16 (Tim16) | Inhibits Pam18 stimulatory effect on ATPase activity of mHsp70 | Three α-helices forming an antiparallel hairpin | |||
| GrpEL1/2 | Mge1 | Regeneration of mtHSP70 | Long N-terminal α-helical region, small helical bundle region, and a C-teminal β-sheet domain | |||
| Unknown | Pam17 | Binds precursor:chaperone complex in matrix | -- | |||
| TIM22/Carrier | TIM22 Complex | TIM22 | Tim22 | Channel | 4 TMs that form a curved surface; IMS-facing N-helix | |
| TIM29 | - | Scaffold | Matrix-facing N-helix, single TM and an IMS domain | |||
| AGK | - | Assembly and function | N-terminally inserted with an IMS α/β motif | |||
| TIM9 | Tim9 | Chaperone | Donut-shaped hexamer structure | |||
| TIM10A | Tim10 | Chaperone | ||||
| TIM10B | Tim12 | Chaperone | ||||
| - | Tim54 | Holds chaperone ring in tilted conformation | N-terminally inserted with an IMS α/β motif | |||
| - | Tim18 | Docking platform for chaperones | 3 TMs and an amphipathic helix on the IMS side | |||
| - | Sdh3 | Docking platform for chaperones | 3 TMs and an amphipathic helix on the IMS side | |||
| OXA Pathway | OXA Complex | OXA1L | OXA1 | Insertion of mtDNA encoded proteins and N-terminal insertion of nuclear encoded proteins into the IMM | 5 TM helices and a large internal C-terminal domain; Nout-Cin | |
| Pathology | Import Defect(s) | Known Consequence(s) | Model Organism/System | Reference |
|---|---|---|---|---|
| Alzheimer’s Disease | APP accumulation in Tom40 and Tim23 channels, with higher levels in AD susceptible brain regions. | Inhibition of import of CIV 4 and 5b, and subsequent reduction in CIV activity, leading to increased ROS. | Human AD brains. | [260] |
| Chronic, sub-lethal Aβ exposure induces a significant reduction in mitochondrial protein import. | Reduction in Δψ, altered mitochondrial morphology, and increased ROS production. | PC12 cells. | [261] | |
| Tau accumulation in OMM and IMS, and interactions between N-terminal Tau fragment with OPA1 and Mfn1. | N/A | HEK293T cells, HeLa cells. | [262,263] | |
| Parkinson’s Disease | α-syn localises to and accumulates within mitochondria, mediated by a cryptic non-canonical MTS, in an ATP and Δψ dependent manner | N/A | Human dopaminergic neuronal cultures, PD brains. | [264] |
| A53T version of α-syn is imported more efficiently than wildtype variant. | May account for faster development of cellular abnormalities seen in cells expressing the A53T version of α-syn compared to the wildtype. | Human dopaminergic neuronal cultures, PD brains, A53T mutant alpha-synuclein-inducible PC12 cell lines. | [265,266] | |
| Mitochondrial α-syn accumulates at IMM and interacts with CI. | Reduction in CI activity, increase in ROS production, inducing oxidative stress. | Human dopaminergic neuronal cultures, PD brains, rat SN neurons, human neuroblastoma cell line (SK-N-MC cells). | [266] | |
| S129 phosphorylated α-syn binds tightly to Tom20, inducing loss in Tom20-Tom22 interaction. | Impaired protein import, loss of Δψ, reduced respiratory capacity, and increased oxidative stress. Rescued by in vivo knockdown of endogenous α-syn, and by in vitro Tom20 overexpression. | SH-SY5Y cells and dopaminergic neurons from SN of post-mortem PD patient brains. | [267] | |
| Tom40 downregulation, corresponding with α-syn accumulation in PD brains. | N/A | Midbrain of PD patients and α-syn transgenic mice. | [268] | |
| Excessively low levels of mitochondrial import in cells from PINK1- and PARK2-linked PD patients. | N/A Import defects reversed by phosphomimetic ubiquitin in cells with residual Parkin activity. | Cells from PINK1- and PARK2-linked PD patients. | [27] | |
| Huntington’s Disease | Disease variant Htt localises to mitochondria and directly interacts with the TIM23 complex. | Inhibited import and subsequent respiratory dysfunction, triggering cell death, rescued by TIM23 overexpression. | Isolated mitochondria from human HD brains, primary neurons expressing Htt variant, forebrain synaptosomal mitochondria in HD mice at early stages of HD. | [269] |
| Dysfunctions in MIA pathway associated with mutant Htt: reduced levels and ratio of Erv1 and Mia40. | Reduced import of MIA pathway precursors, CIV assembly defects, deficient respiration, alterations in mtDNA, altered mitochondrial morphology. | Neuronal cell lines. | [270] | |
| Amyotrophic Lateral Sclerosis | Variants of SOD1 accumulate in IMS, matrix, and OMM, and interact with OMM proteins. | Excessive ROS production, mitochondrial dysfunction, and toxic effects on the cells rescued by selective IMS targeting of wildtype SOD1. | Transgenic mouse models, spinal cord mitochondria. | [271,272,273] |
| Increased levels of TOM subunits Tom20, Tom22, and Tom40. Overall reduction in import efficiency by 30%. | Changes in CI related protein expression levels. | Rat spinal cord of ALS-linked variant SOD1G93A. | [274] | |
| Novel CHCHD10 mutant, Q108P, discovered in a patient with rapidly progressing ALS, almost completely abolishes its import. | Reduced mitochondrial respiratory capacity, an effect that is rescued by Mia40 overexpression. | HeLa cells and primary rat embryonic neurons transduced with genomic DNA from a young ALS patient. | [275] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Needs, H.I.; Protasoni, M.; Henley, J.M.; Prudent, J.; Collinson, I.; Pereira, G.C. Interplay between Mitochondrial Protein Import and Respiratory Complexes Assembly in Neuronal Health and Degeneration. Life 2021, 11, 432. https://doi.org/10.3390/life11050432
Needs HI, Protasoni M, Henley JM, Prudent J, Collinson I, Pereira GC. Interplay between Mitochondrial Protein Import and Respiratory Complexes Assembly in Neuronal Health and Degeneration. Life. 2021; 11(5):432. https://doi.org/10.3390/life11050432
Chicago/Turabian StyleNeeds, Hope I., Margherita Protasoni, Jeremy M. Henley, Julien Prudent, Ian Collinson, and Gonçalo C. Pereira. 2021. "Interplay between Mitochondrial Protein Import and Respiratory Complexes Assembly in Neuronal Health and Degeneration" Life 11, no. 5: 432. https://doi.org/10.3390/life11050432
APA StyleNeeds, H. I., Protasoni, M., Henley, J. M., Prudent, J., Collinson, I., & Pereira, G. C. (2021). Interplay between Mitochondrial Protein Import and Respiratory Complexes Assembly in Neuronal Health and Degeneration. Life, 11(5), 432. https://doi.org/10.3390/life11050432