
Whole Exome Sequencing Identifies a Novel Homozygous Missense Mutation in the CSB Protein-Encoding ERCC6 Gene in a Taiwanese Boy with Cockayne Syndrome

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Purification of Genomic DNA from Isolated Human Blood Leukocytes
2.3. WES, Sanger Sequencing, and Database Interpretation
2.4. Multiple Sequence Alignment of CSB
2.5. Predictive Three-Dimensional Models of CSB
2.6. Evaluating the Effect of the Change of Mutation Energy Caused by Mutations on the Stability of CSB
3. Results
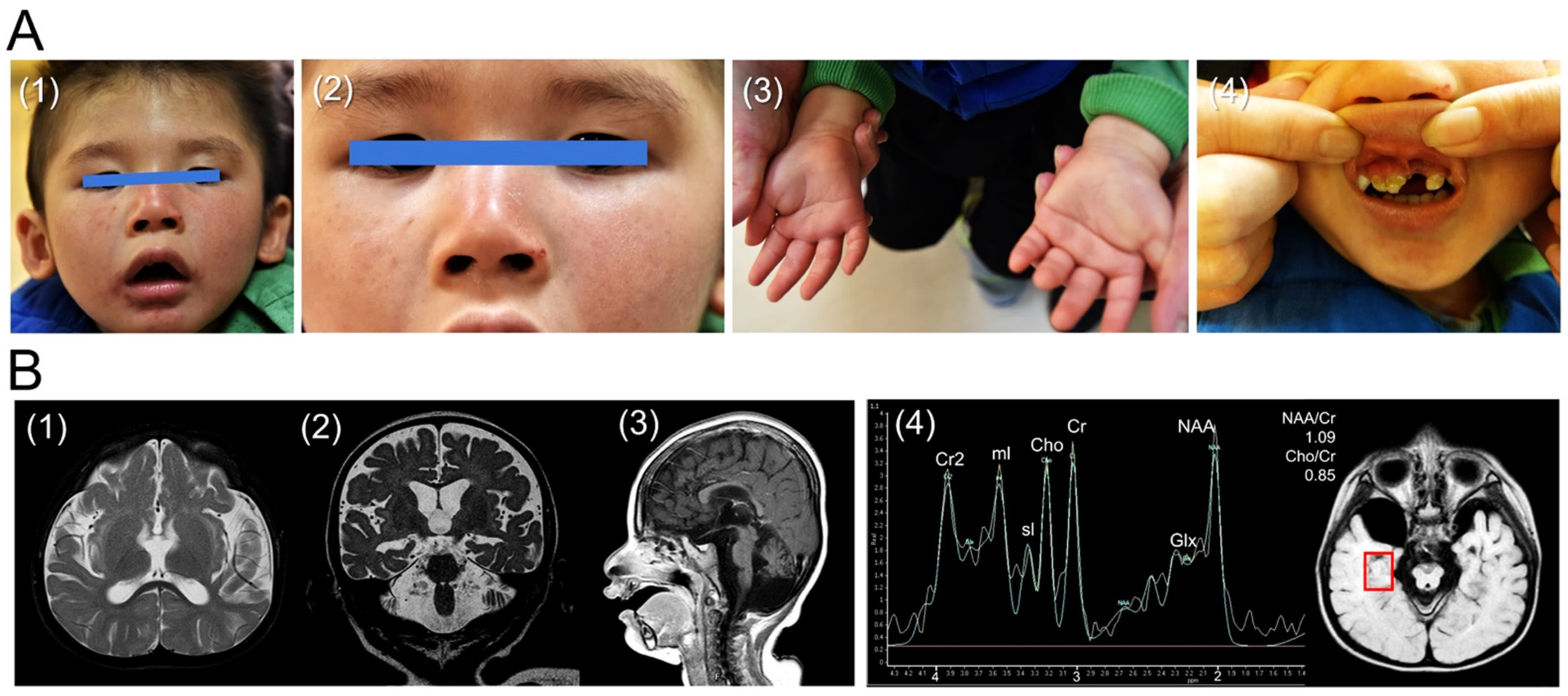
3.1. Case Presentation
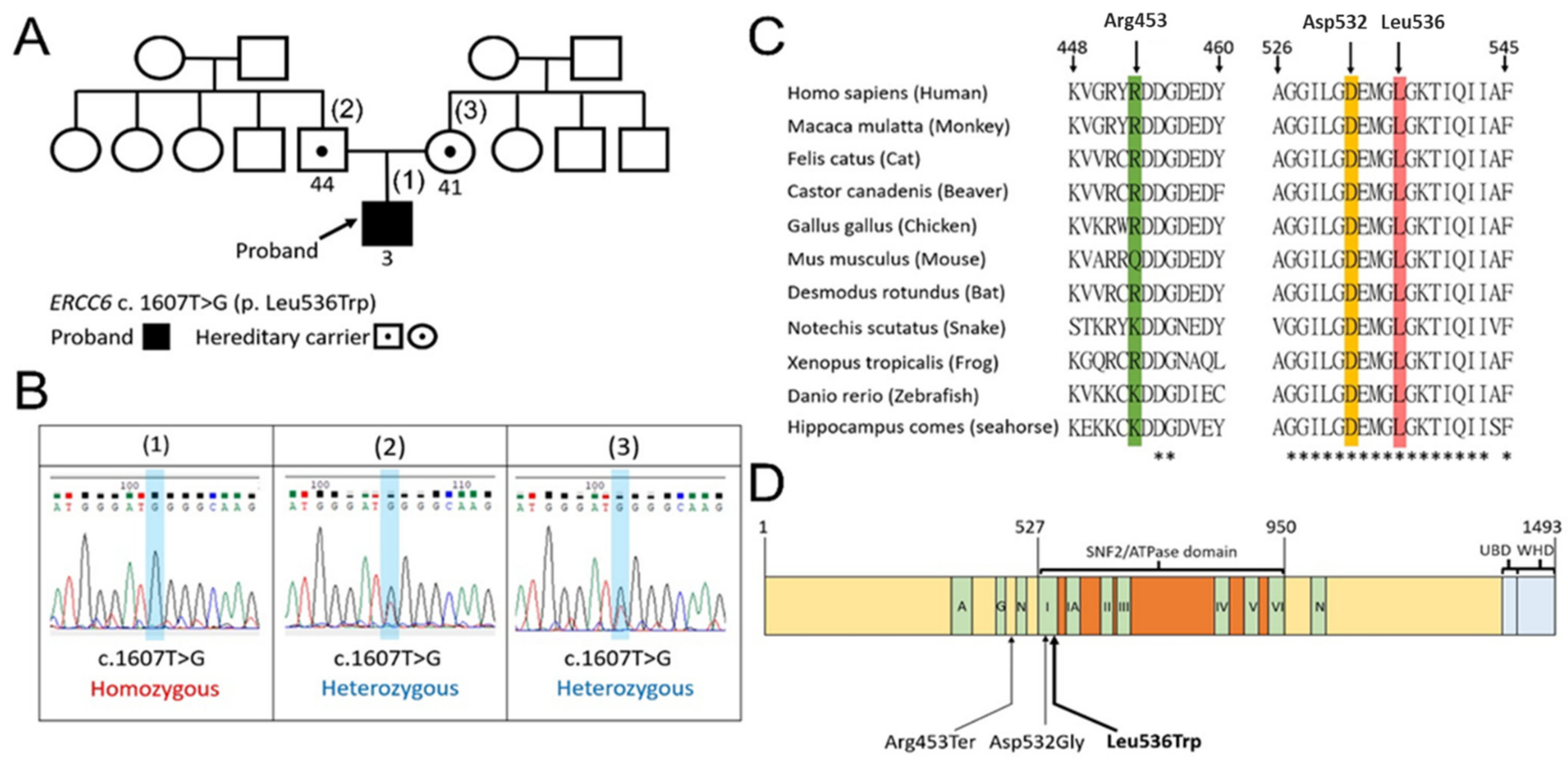
3.2. The Family Pedigree, Sanger Sequencing, the Conservation of the Amino Acid Sequences, and Domain Structure of CSB Protein
3.3. Predictive Three-Dimensional Models of CSB Protein
3.4. Computational Assessment of the Impact of CS-Associated Mutations on the Stability of CSB
4. Discussion
4.1. The First Reported Homozygous Mutation Identified in the ERCC6 Gene (c.1607T>G, p.Leu536Trp)
4.2. Computational Modeling Results Strongly Suggest That a Homozygous Mutation in the ERCC6 Gene (c.1607T>G, p.Leu536Trp) Is Potentially a True Pathogenic Variant in Clinical Context
4.3. A Founder Effect of the ERCC6 Gene Mutation (c.1607T>G, p.Leu536Trp) in the Southeast Coast of East ASIA
4.4. The Role of CSB in Neurodegeneration
4.5. Mutations in the ATPase Motif of CSB Inactivate Its ATPase Activity and Ultimately Its Function
4.6. Potential Treatments for CS Patients
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Laugel, V.; Dalloz, C.; Durand, M.; Sauvanaud, F.; Kristensen, U.; Vincent, M.C.; Pasquier, L.; Odent, S.; Cormier-Daire, V.; Gener, B.; et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2010, 31, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, M.; Stevnsner, T.; Modin, C.; Martensen, P.M.; Brosh, R.M., Jr.; Bohr, V.A. Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res. 2003, 31, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Stevnsner, T.; Muftuoglu, M.; Aamann, M.D.; Bohr, V.A. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech. Ageing Dev. 2008, 129, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Vessoni, A.T.; Guerra, C.C.C.; Kajitani, G.S.; Nascimento, L.L.S.; Garcia, C.C.M. Cockayne Syndrome: The many challenges and approaches to understand a multifaceted disease. Genet. Mol. Biol. 2020, 43, e20190085. [Google Scholar] [CrossRef]
- Yu, S.; Chen, L.; Ye, L.; Fei, L.; Tang, W.; Tian, Y.; Geng, Q.; Yi, X.; Xie, J. Identification of two missense mutations of ERCC6 in three Chinese sisters with Cockayne syndrome by whole exome sequencing. PLoS ONE 2014, 9, e113914. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, L.; Wu, M.; Liu, H.; Cai, Y.; Sheng, H.; Li, X.; Cheng, J.; Li, D.; Huang, Y. Clinical and molecular analysis of two Chinese siblings with Cockayne syndrome. Zhonghua Er Ke Za Zhi 2016, 54, 56–60. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Adjeroh, D.; Bell, T.; Mukherjee, A. The Burrows-Wheeler Transform: Data Compression, Suffix Arrays, and Pattern Matching; Springer: Boston, MA, USA, 2008. [Google Scholar]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Coutsias, E.A.; Seok, C.; Dill, K.A. Using quaternions to calculate RMSD. J. Comput. Chem. 2004, 25, 1849–1857. [Google Scholar] [CrossRef]
- Takahashi, T.S.; Sato, Y.; Yamagata, A.; Goto-Ito, S.; Saijo, M.; Fukai, S. Structural basis of ubiquitin recognition by the winged-helix domain of Cockayne syndrome group B protein. Nucleic Acids Res. 2019, 47, 3784–3794. [Google Scholar] [CrossRef] [Green Version]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment; Release 2017; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Safriel, Y.; Pol-Rodriguez, M.; Novotny, E.J.; Rothman, D.L.; Fulbright, R.K. Reference values for long echo time MR spectroscopy in healthy adults. AJNR Am. J. Neuroradiol. 2005, 26, 1439–1445. [Google Scholar]
- Cecil, K.M.; Jones, B.V. Magnetic resonance spectroscopy of the pediatric brain. Top. Magn. Reson. Imaging 2001, 12, 435–452. [Google Scholar] [CrossRef]
- Filippi, C.G.; Uluğ, A.M.; Deck, M.D.; Zimmerman, R.D.; Heier, L.A. Developmental delay in children: Assessment with proton MR spectroscopy. AJNR Am. J. Neuroradiol. 2002, 23, 882–888. [Google Scholar]
- Koob, M.; Laugel, V.; Durand, M.; Fothergill, H.; Dalloz, C.; Sauvanaud, F.; Dollfus, H.; Namer, I.J.; Dietemann, J.L. Neuroimaging in Cockayne syndrome. AJNR Am. J. Neuroradiol. 2010, 31, 1623–1630. [Google Scholar] [CrossRef] [Green Version]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Davidson, J.W. The Island of Formosa Past and Present: History, People, Resources and Commercial Prospects, Tea Camphor, Sugar, Gold, Coal, Sulphur, Economical Plants, and Other Productions; MacMillan: London, UK, 1903. [Google Scholar]
- Venema, J.; Mullenders, L.H.; Natarajan, A.T.; van Zeeland, A.A.; Mayne, L.V. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 4707–4711. [Google Scholar] [CrossRef] [Green Version]
- Licht, C.L.; Stevnsner, T.; Bohr, V.A. Cockayne syndrome group B cellular and biochemical functions. Am. J. Hum. Genet. 2003, 73, 1217–1239. [Google Scholar] [CrossRef] [Green Version]
- De Waard, H.; de Wit, J.; Gorgels, T.G.; van den Aardweg, G.; Andressoo, J.O.; Vermeij, M.; van Steeg, H.; Hoeijmakers, J.H.; van der Horst, G.T. Cell type-specific hypersensitivity to oxidative damage in CSB and XPA mice. DNA Repair 2003, 2, 13–25. [Google Scholar] [CrossRef]
- Maddukuri, L.; Speina, E.; Christiansen, M.; Dudzińska, D.; Zaim, J.; Obtułowicz, T.; Kabaczyk, S.; Komisarski, M.; Bukowy, Z.; Szczegielniak, J.; et al. Cockayne syndrome group B protein is engaged in processing of DNA adducts of lipid peroxidation product trans-4-hydroxy-2-nonenal. Mutat. Res. 2009, 666, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Tuo, J.; Jaruga, P.; Rodriguez, H.; Bohr, V.A.; Dizdaroglu, M. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. 2003, 17, 668–674. [Google Scholar] [CrossRef]
- Citterio, E.; Rademakers, S.; van der Horst, G.T.; van Gool, A.J.; Hoeijmakers, J.H.; Vermeulen, W. Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J. Biol. Chem. 1998, 273, 11844–11851. [Google Scholar] [CrossRef] [Green Version]
- Muftuoglu, M.; Selzer, R.; Tuo, J.; Brosh, R.M., Jr.; Bohr, V.A. Phenotypic consequences of mutations in the conserved motifs of the putative helicase domain of the human Cockayne syndrome group B gene. Gene 2002, 283, 27–40. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.F.C.; Bozek, K.; Herholz, M.; Trifunovic, A.; Rieckher, M.; Schumacher, B. A C. elegans model for neurodegeneration in Cockayne syndrome. Nucleic Acids Res. 2020, 48, 10973–10985. [Google Scholar] [CrossRef]
- Van Wyhe, R.D.; Emery, C.V.; Williamson, R.A. Cochlear implantation in pediatric patients with Cockayne Syndrome. Int. J. Pediatr. Otorhinolaryngol. 2018, 106, 64–67. [Google Scholar] [CrossRef]
- Neilan, E.G.; Delgado, M.R.; Donovan, M.A.; Kim, S.Y.; Jou, R.L.; Wu, B.L.; Kang, P.B. Response of motor complications in Cockayne syndrome to carbidopa-levodopa. Arch. Neurol. 2008, 65, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Ramamoorthy, M.; Sykora, P.; Maynard, S.; Lin, P.C.; Minor, R.K.; Wilson, D.M., 3rd; Cooper, M.; Spencer, R.; de Cabo, R.; et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012, 209, 855–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bové, J.; Martínez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Dello Russo, C.; Lisi, L.; Feinstein, D.L.; Navarra, P. mTOR kinase, a key player in the regulation of glial functions: Relevance for the therapy of multiple sclerosis. Glia 2013, 61, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Cenni, V.; Lattanzi, G. Potential therapeutic effects of the MTOR inhibitors for preventing ageing and progeria-related disorders. Br. J. Clin. Pharmacol. 2016, 82, 1229–1244. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | I-TASSER | AlphaFold | ||
|---|---|---|---|---|
| Mutation | Mutation Energy (kcal/mol) | Predicted Effect | Mutation Energy (kcal/mol) | Predicted Effect |
| Asp532→Gly532 | 3.56 | Destabilizing | 5.8 | Destabilizing |
| Leu536→Trp536 | 0.18 | Neutral | 0.98 | Destabilizing |
| Asp532→Gly532+ Leu536→Trp536 | 4.01 | Destabilizing | 6.99 | Destabilizing |
| Zhonghua Er Ke Za Zhi (Zhizi Zhou, 2016) [8] | PLoS One (Yu et al., 2014) [7] | Our Patient | |||
|---|---|---|---|---|---|
| Case Number | 2 | 3 | 1 | ||
| Mutation | Compound heterozygous | Compound heterozygous | Homozygous | ||
| GRCh37 | Chr10:50732119 | Chr10:50708662 | Chr10:50708674 | Chr10:50708662 | Chr10:50708662 |
| dbSNP | rs121917902 | rs774175886 | rs752712823 | rs774175886 | rs774175886 |
| DNA change | c.1357C>T | c.1607T > G | c.1595A>G | c.1607T>G | c.1607T>G |
| Amino acid change | p.Arg453Ter | p.Leu536Trp | p.Asp532Gly | p.Leu536Trp | p.Leu536Trp |
| Allele frequency (Taiwan) 1 | NA | 0.001318 | NA | 0.001318 | 0.001318 |
| Allele frequency (Global) 2 | 0/478 (ALFA3 Project) | 0.00002628 (GenomAD v3.1.1) | 0.000003983 (GenomAD v2.1.1) | 0.00002628 (GenomAD v3.1.1) | 0.00002628 (GenomAD v3.1.1) |
| Allele frequency (East Asian) 3 | NA | 0.0002627 (GenomAD v3.1.1) | NA | 0.0002627 (GenomAD v3.1.1) | 0.0002627 (GenomAD v3.1.1) |
| ACMG Classification | Pathogenic | Likely Pathogenic | Uncertain Significance | Likely Pathogenic | Likely Pathogenic |
| PMID | 26791926 | 25463447 | Non-Applicable | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-M.; Yang, J.-H.; Lee, H.-J.; Lin, Y.-P.; Tsai, L.-P.; Hsu, C.-S.; Luxton, G.W.G.; Hu, C.-F. Whole Exome Sequencing Identifies a Novel Homozygous Missense Mutation in the CSB Protein-Encoding ERCC6 Gene in a Taiwanese Boy with Cockayne Syndrome. Life 2021, 11, 1230. https://doi.org/10.3390/life11111230
Lin C-M, Yang J-H, Lee H-J, Lin Y-P, Tsai L-P, Hsu C-S, Luxton GWG, Hu C-F. Whole Exome Sequencing Identifies a Novel Homozygous Missense Mutation in the CSB Protein-Encoding ERCC6 Gene in a Taiwanese Boy with Cockayne Syndrome. Life. 2021; 11(11):1230. https://doi.org/10.3390/life11111230
Chicago/Turabian StyleLin, Ching-Ming, Jay-How Yang, Hwei-Jen Lee, Yu-Pang Lin, Li-Ping Tsai, Chih-Sin Hsu, G. W. Gant Luxton, and Chih-Fen Hu. 2021. "Whole Exome Sequencing Identifies a Novel Homozygous Missense Mutation in the CSB Protein-Encoding ERCC6 Gene in a Taiwanese Boy with Cockayne Syndrome" Life 11, no. 11: 1230. https://doi.org/10.3390/life11111230
APA StyleLin, C.-M., Yang, J.-H., Lee, H.-J., Lin, Y.-P., Tsai, L.-P., Hsu, C.-S., Luxton, G. W. G., & Hu, C.-F. (2021). Whole Exome Sequencing Identifies a Novel Homozygous Missense Mutation in the CSB Protein-Encoding ERCC6 Gene in a Taiwanese Boy with Cockayne Syndrome. Life, 11(11), 1230. https://doi.org/10.3390/life11111230