
An Insight into Vaginal Microbiome Techniques

 , ,
, ,  ,
,
Background
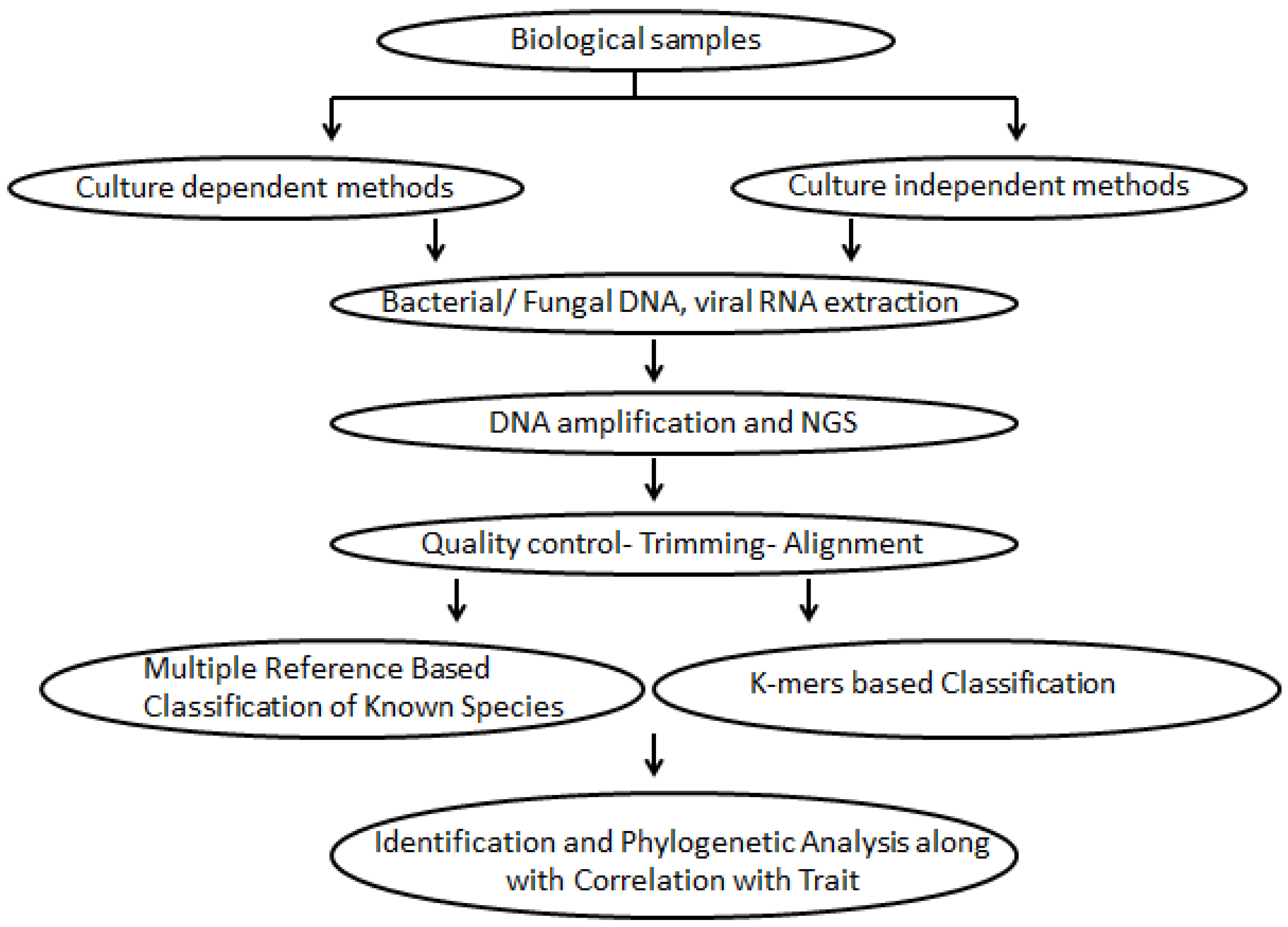
1. Introduction
2. Considerations for Sample Collection
2.1. Sample Collection Methods and Storage
2.2. Sample Metadata
2.2.1. Culture-Dependent Characterization of the Vaginal Microbiome
2.2.2. Culture-Independent Methods
2.2.3. DNA Extraction of the Vaginal Microbiome
2.2.4. Choice of Universal PCR Primers
3. Sequencing Methodologies
4. Bioinformatics Data Analysis Tools
4.1. Pre-Processing and Signal Extraction
4.2. Analysis Tools for Targeted Amplicon Data
4.3. Operational Taxonomic Units (OTU) Grouping
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Kapil, A.; Sood, S.; Sengupta, S. Female reproductive tract microbiome in gynecological health and problems. J. Reprod. Health Med. 2016, 2, S48–S54. [Google Scholar] [CrossRef]
- Muhleisen, A.L.; Herbst-Kralovetz, M.M. Menopause and the vaginal microbiome. Maturitas 2016, 91, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, A.; Kumar, R.; Cliver, S.P.; Zhi, D.; Szychowski, J.M.; Abramovici, A.; Biggio, J.R.; Lefkowitz, E.J.; Morrow, C.; Edwards, R.K. Vaginal microbiota in pregnancy: Evaluation based on vaginal flora, birth outcome, and race. Am. J. Perinatol. 2016, 33, 401. [Google Scholar]
- Taha, T.E.; Hoover, D.R.; Dallabetta, G.A.; Kumwenda, N.I.; Mtimavalye, L.A.; Yang, L.-P.; Liomba, G.N.; Broadhead, R.L.; Chiphangwi, J.D.; Miotti, P.G. Bacterial vaginosis and disturbances of vaginal flora: Association with increased acquisition of HIV. Aids 1998, 12, 1699–1706. [Google Scholar] [CrossRef]
- Sha, B.E.; Zariffard, M.R.; Wang, Q.J.; Chen, H.Y.; Bremer, J.; Cohen, M.H.; Spear, G.T. Female genital-tract HIV load correlates inversely with Lactobacillus species but positively with bacterial vaginosis and Mycoplasma hominis. J. Infect. Dis. 2005, 191, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Van de Wijgert, J.H.; Morrison, C.S.; Cornelisse, P.G.; Munjoma, M.; Moncada, J.; Awio, P.; Wang, J.; Van der Pol, B.; Chipato, T.; Salata, R.A. Bacterial vaginosis and vaginal yeast, but not vaginal cleansing, increase HIV-1 acquisition in African women. J. Acquir. Immune Defic. Syndr. 2008, 48, 203–210. [Google Scholar] [CrossRef]
- Hay, P.E. Bacterial vaginosis and miscarriage. Curr. Opin. Infect. Dis. 2004, 17, 41–44. [Google Scholar] [CrossRef]
- Ralph, S.; Rutherford, A.; Wilson, J. Influence of bacterial vaginosis on conception and miscarriage in the first trimester: Cohort study. BMJ 1999, 319, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Llahi-Camp, J.; Rai, R.; Ison, C.; Regan, L.; Taylor-Robinson, D. Association of bacterial vaginosis with a history of second trimester miscarriage. Hum. Reprod. 1996, 11, 1575–1578. [Google Scholar] [CrossRef] [Green Version]
- Hay, P.E.; Lamont, R.F.; Taylor-Robinson, D.; Morgan, D.J.; Ison, C.; Pearson, J. Abnormal bacterial colonisation of the genital tract and subsequent preterm delivery and late miscarriage. BMJ 1994, 308, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Larsson, P.-G.; Platz-Christensen, J.-J.; Dalaker, K.; Eriksson, K.; Fåhraeus, L.; Irminger, K.; Jerve, F.; Stray-Pedersen, B.; Wölner-Hanssen, P. Treatment with 2% clindamycin vaginal cream prior to first trimester surgical abortion to reduce signs of postoperative infection: A prospective, double-blinded, placebo-controlled, multicenter study. Acta Obstet. Gynecol. Scand. Suppl. 2000, 79, 390–396. [Google Scholar] [CrossRef]
- Işik, G.; Demirezen, Ş.; Dönmez, H.G.; Beksaç, M.S. Bacterial vaginosis in association with spontaneous abortion and recurrent pregnancy losses. J. Cytol. 2016, 33, 135. [Google Scholar]
- Ziaei, S.; Sadrkhanlu, M.; Moeini, A.; Faghihzadeh, S. Effect of bacterial vaginosis on premature rupture of membranes and related complications in pregnant women with a gestational age of 37–42 weeks. Gynecol. Obstet. Investig. 2006, 61, 135–138. [Google Scholar] [CrossRef]
- Gibbs, R.S. Chorioamnionitis and bacterial vaginosis. Am. J. Obstet. Gynecol. 1993, 169, 460–462. [Google Scholar] [CrossRef]
- Martius, J.; Eschenbach, D. The role of bacterial vaginosis as a cause of amniotic fluid infection, chorioamnionitis and prematurity—A review. Arch. Gynecol. Obstet. 1990, 247, 1. [Google Scholar] [CrossRef]
- Pretorius, C.; Jagatt, A.; Lamont, R.F. The relationship between periodontal disease, bacterial vaginosis, and preterm birth. J. Perinat. Med. 2007, 35, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Sanu, O.; Lamont, R.F. Periodontal disease and bacterial vaginosis as genetic and environmental markers for the risk of spontaneous preterm labor and preterm birth. J. Matern. Fetal Neonatal Med. 2011, 24, 1476–1485. [Google Scholar] [CrossRef]
- Zaman, U.; Qureshi, S.A.; Shah, I.; Nazir, A.; Fatima, S. Common risk factors of spontaneous preterm labor with intact fetal membranes. Pak. J. Physiol. 2019, 15, 10–12. [Google Scholar]
- Jacobsson, B.; Pernevi, P.; Chidekel, L.; JörgenPlatz-Christensen, J. Bacterial vaginosis in early pregnancy may predispose for preterm birth and postpartum endometritis. Acta Obstet. Gynecol. Scand. 2002, 81, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Faro, S. Postpartum endometritis. Clin. Perinatol. 2005, 32, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Mimee, M.; Citorik, R.J.; Lu, T.K. Microbiome therapeutics—Advances and challenges. Adv. Drug Deliv. Rev. 2016, 105, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brubaker, L.; Wolfe, A.J. The new world of the urinary microbiota in women. Am. J. Obstet. Gynecol. 2015, 213, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Kong, J.; Liu, F.; Zhu, H.; Chen, X.; Wang, Y.; Li, L.; Nelson, K.E.; Xia, Y.; Xiang, C. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genom. 2010, 11, 488. [Google Scholar] [CrossRef] [Green Version]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-induced changes in the intestinal microbiota and disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Tyson, G.W.; DeLong, E.F. Metatranscriptomics reveals unique microbial small RNAs in the ocean’s water column. Nature 2009, 459, 266–269. [Google Scholar] [CrossRef]
- Maron, P.-A.; Ranjard, L.; Mougel, C.; Lemanceau, P. Metaproteomics: A new approach for studying functional microbial ecology. Microb. Ecol. 2007, 53, 486–493. [Google Scholar] [CrossRef]
- Clayton, T.A.; Baker, D.; Lindon, J.C.; Everett, J.R.; Nicholson, J.K. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 14728–14733. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, S.; Kalliala, I.; Nieminen, P.; Salonen, A. Comparative analysis of vaginal microbiota sampling using 16S rRNA gene analysis. PLoS ONE 2017, 12, e0181477. [Google Scholar] [CrossRef] [Green Version]
- Van Der Pol, W.J.; Kumar, R.; Morrow, C.D.; Blanchard, E.E.; Taylor, C.M.; Martin, D.H.; Lefkowitz, E.J.; Muzny, C.A. In silico and experimental evaluation of primer sets for species-level resolution of the vaginal microbiota using 16S ribosomal RNA gene sequencing. J. Infect. Dis. 2019, 219, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Cobo, T.; Vergara, A.; Collado, M.C.; Casals-Pascual, C.; Herreros, E.; Bosch, J.; Sánchez-García, A.B.; López-Parellada, R.; Ponce, J.; Gratacós, E. Characterization of vaginal microbiota in women with preterm labor with intra-amniotic inflammation. Sci. Rep. 2019, 9, 18963. [Google Scholar] [CrossRef] [Green Version]
- Blostein, F.; Gelaye, B.; Sanchez, S.E.; Williams, M.A.; Foxman, B. Vaginal microbiome diversity and preterm birth: Results of a nested case–control study in Peru. Ann. Epidemiol. 2020, 41, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Mendz, G.; Kaakoush, N.; Quinlivan, J. New techniques to characterise the vaginal microbiome in pregnancy. AIMS Microbiol. 2016, 2, 55–68. [Google Scholar] [CrossRef]
- Brusa, T.; Canzi, E.; Pacini, N.; Zanchi, R.; Ferrari, A. Oxygen tolerance of anaerobic bacteria isolated from human feces. Curr. Microbiol. 1989, 19, 39–43. [Google Scholar] [CrossRef]
- Lindau, S.T.; Hoffmann, J.N.; Lundeen, K.; Jaszczak, A.; McClintock, M.K.; Jordan, J.A. Vaginal self-swab specimen collection in a home-based survey of older women: Methods and applications. J. Gerontol. B Psychol. Sci. Soc. Sci. 2009, 64, i106–i118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, G.; Gajer, P.; Nandy, M.; Ma, B.; Yang, H.; Sakamoto, J.; Blanchard, M.H.; Ravel, J.; Brotman, R.M. Comparison of storage conditions for human vaginal microbiome studies. PLoS ONE 2012, 7, e36934. [Google Scholar] [CrossRef] [Green Version]
- Uchihashi, M.; Bergin, I.; Bassis, C.; Hashway, S.; Chai, D.; Bell, J. Influence of age, reproductive cycling status, and menstruation on the vaginal microbiome in baboons (Papio anubis). Am. J. Primatol. 2015, 77, 563–578. [Google Scholar] [CrossRef] [Green Version]
- Farage, M.A.; Miller, K.W.; Sobel, J.D. Dynamics of the vaginal ecosystem—Hormonal influences. Infect. Dis. Res. Treat. 2010, 3, IDRT-S3903. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [Green Version]
- Song, S.D.; Acharya, K.D.; Zhu, J.E.; Deveney, C.M.; Walther-Antonio, M.R.; Tetel, M.J.; Chia, N. Daily Vaginal Microbiota Fluctuations Associated with Natural Hormonal Cycle, Contraceptives, Diet, and Exercise. Msphere 2020, 5, e00593-20. [Google Scholar] [CrossRef] [PubMed]
- Brotman, R.M.; He, X.; Gajer, P.; Fadrosh, D.; Sharma, E.; Mongodin, E.F.; Ravel, J.; Glover, E.D.; Rath, J.M. Association between cigarette smoking and the vaginal microbiota: A pilot study. BMC Infect. Dis. 2014, 14, 471. [Google Scholar] [CrossRef] [Green Version]
- Fosch, S.E.; Ficoseco, C.A.; Marchesi, A.; Cocucci, S.; Nader-Macias, M.E.; Perazzi, B.E. Contraception: Influence on vaginal microbiota and identification of vaginal lactobacilli using MALDI-TOF MS and 16S rDNA sequencing. Open Microbiol. J. 2018, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Gajer, P.; Brotman, R.M.; Bai, G.; Sakamoto, J.; Schütte, U.M.; Zhong, X.; Koenig, S.S.; Fu, L.; Ma, Z.S.; Zhou, X. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 2012, 4, 132ra52. [Google Scholar] [CrossRef] [Green Version]
- Amegashie, C.P.; Gilbert, N.M.; Peipert, J.F.; Allsworth, J.E.; Lewis, W.G.; Lewis, A.L. Relationship between Nugent score and vaginal epithelial exfoliation. PLoS ONE 2017, 12, e0177797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, M.L.; Onderdonk, A.B.; Microbiology and Prematurity Study Group. Nugent score related to vaginal culture in pregnant women. Obstet. Gynecol. 2001, 98, 79–84. [Google Scholar]
- Green, K.A.; Zarek, S.M.; Catherino, W.H. Gynecologic health and disease in relation to the microbiome of the female reproductive tract. Fertil. Steril. 2015, 104, 1351–1357. [Google Scholar] [CrossRef] [Green Version]
- Nugent, R.P.; Krohn, M.A.; Hillier, S.L. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 1991, 29, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Sirota, I.; Zarek, S.M.; Segars, J.H. In Potential influence of the microbiome on infertility and assisted reproductive technology. In Seminars in Reproductive Medicine; NIH: New York, NY, USA, 2014; p. 35. [Google Scholar]
- Pace, N.R. A molecular view of microbial diversity and the biosphere. Science 1997, 276, 734–740. [Google Scholar] [CrossRef]
- Cui, L.; Morris, A.; Ghedin, E. The human mycobiome in health and disease. Genome Med. 2013, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Thatcher, S.A. DNA/RNA preparation for molecular detection. Clin. Chem. 2015, 61, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.-W.; Li, M.; Ma, L.-C.; Wei, F.-W. A widely applicable protocol for DNA isolation from fecal samples. Biochem. Genet. 2006, 44, 494. [Google Scholar] [CrossRef] [PubMed]
- Eychner, A.M.; Lebo, R.J.; Elkins, K.M. Comparison of proteases in DNA extraction via quantitative polymerase chain reaction. Anal. Biochem. 2015, 478, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Remm, M.; Frasquilho, S.; Betsou, F.; Mathieson, W. How severely is DNA quantification hampered by RNA co-extraction? Biopreserv. Biobank. 2015, 13, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K. Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol. 2001, 56. [Google Scholar] [CrossRef]
- Yuan, S.; Cohen, D.B.; Ravel, J.; Abdo, Z.; Forney, L.J. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS ONE 2012, 7, e33865. [Google Scholar] [CrossRef] [Green Version]
- Street, T.L.; Barker, L.; Sanderson, N.D.; Kavanagh, J.; Hoosdally, S.; Cole, K.; Newnham, R.; Selvaratnam, M.; Andersson, M.; Llewelyn, M.J. Optimizing DNA Extraction Methods for Nanopore Sequencing of Neisseria gonorrhoeae Directly from Urine Samples. J. Clin. Microbiol. 2020, 58, e01822-19. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.L.; Meyer, A.; Johnson, P.J.; Ericsson, A.C. Comparative evaluation of DNA extraction methods from feces of multiple host species for downstream next-generation sequencing. PLoS ONE 2015, 10, e0143334. [Google Scholar]
- Corcoll, N.; Österlund, T.; Sinclair, L.; Eiler, A.; Kristiansson, E.; Backhaus, T.; Eriksson, K.M. Comparison of four DNA extraction methods for comprehensive assessment of 16S rRNA bacterial diversity in marine biofilms using high-throughput sequencing. FEMS Microbiol. Lett. 2017, 364, fnx139. [Google Scholar] [CrossRef] [Green Version]
- Yergeau, E.; Michel, C.; Tremblay, J.; Niemi, A.; King, T.L.; Wyglinski, J.; Lee, K.; Greer, C.W. Metagenomic survey of the taxonomic and functional microbial communities of seawater and sea ice from the Canadian Arctic. Sci. Rep. 2017, 7, 42242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Liang, P.; Wang, B.; Fang, J.; Lang, J.; Tian, G.; Jiang, J.; Zhu, T.F. Optimized DNA extraction and metagenomic sequencing of airborne microbial communities. Nat. Protoc. 2015, 10, 768–779. [Google Scholar] [CrossRef]
- Mattei, V.; Murugesan, S.; Al Hashmi, M.; Mathew, R.; James, N.; Singh, P.; Lakshmanan, A.; Terranegra, A.; Al Khodor, S.; Tomei, S. Evaluation of methods for the extraction of microbial DNA from vaginal swabs used for microbiome studies. Front. Cell. Infect. Microbiol. 2019, 9, 197. [Google Scholar] [CrossRef]
- Gill, C.; van de Wijgert, J.H.; Blow, F.; Darby, A.C. Evaluation of lysis methods for the extraction of bacterial DNA for analysis of the vaginal microbiota. PLoS ONE 2016, 11, e0163148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriakose, S.; Sabeena, S.; Damodaran, B.; Ravishankar, N.; Ramachandran, A.; Ameen, N. Comparison of two self-sampling methods for Human Papillomavirus (HPV) DNA testing among women with high prevalence rates. J. Med. Virol. 2020, 92, 3884–3888. [Google Scholar] [CrossRef]
- Hernandes, C.; Silveira, P.; Rodrigues Sereia, A.F.; Christoff, A.P.; Mendes, H.; Valter de Oliveira, L.F.; Podgaec, S. Microbiome Profile of Deep Endometriosis Patients: Comparison of Vaginal Fluid, Endometrium and Lesion. Diagnostics 2020, 10, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, N.R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 1985, 82, 6955–6959. [Google Scholar] [CrossRef] [Green Version]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, F.; Laiola, M.; Blaiotta, G.; Ercolini, D. Different amplicon targets for sequencing-based studies of fungal diversity. Appl. Environ. Microbiol. 2017, 83, e00905-17. [Google Scholar] [CrossRef] [Green Version]
- Motooka, D.; Fujimoto, K.; Tanaka, R.; Yaguchi, T.; Gotoh, K.; Maeda, Y.; Furuta, Y.; Kurakawa, T.; Goto, N.; Yasunaga, T. Fungal ITS1 deep-sequencing strategies to reconstruct the composition of a 26-species community and evaluation of the gut mycobiota of healthy Japanese individuals. Front. Microbiol. 2017, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Barb, J.J.; Oler, A.J.; Kim, H.-S.; Chalmers, N.; Wallen, G.R.; Cashion, A.; Munson, P.J.; Ames, N.J. Development of an analysis pipeline characterizing multiple hypervariable regions of 16S rRNA using mock samples. PLoS ONE 2016, 11, e0148047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.S.; Brooker, M.R.; Dowd, S.E.; Camerlengo, T. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS ONE 2011, 6, e20956. [Google Scholar] [CrossRef] [PubMed]
- Gottschick, C.; Deng, Z.-L.; Vital, M.; Masur, C.; Abels, C.; Pieper, D.H.; Wagner-Döbler, I. The urinary microbiota of men and women and its changes in women during bacterial vaginosis and antibiotic treatment. Microbiome 2017, 5, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouts, D.E.; Pieper, R.; Szpakowski, S.; Pohl, H.; Knoblach, S.; Suh, M.-J.; Huang, S.-T.; Ljungberg, I.; Sprague, B.M.; Lucas, S.K. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. Front. Microbiol. 2012, 10, 174. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.A.; Brown, R.; Williams, J.; White, P.; Jacobson, S.K.; Marchesi, J.; Drake, M.J. The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front. Cell. Infect. Microbiol. 2013, 3, 41. [Google Scholar] [CrossRef] [Green Version]
- Graspeuntner, S.; Loeper, N.; Künzel, S.; Baines, J.F.; Rupp, J. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci. Rep. 2018, 8, 9678. [Google Scholar] [CrossRef]
- Liu, Z.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 2008, 36, e120. [Google Scholar] [CrossRef] [Green Version]
- Mizrahi-Man, O.; Davenport, E.R.; Gilad, Y. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: Evaluation of effective study designs. PLoS ONE 2013, 8, e53608. [Google Scholar]
- Frank, J.A.; Reich, C.I.; Sharma, S.; Weisbaum, J.S.; Wilson, B.A.; Olsen, G.J. Critical Evaluation of Two Commonly-Used Primers for Amplification of Bacterial 16S rRNA Genes. Appl. Environ. Microbiol. 2008, 74, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, J.; Olofsson, M.; MONSTEIN, H.-J. Classification, identification and subtyping of bacteria based on pyrosequencing and signature matching of 16S rDNA fragments. Commentary. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2007, 115, 668–679. [Google Scholar]
- Liu, Z.; Lozupone, C.; Hamady, M.; Bushman, F.D.; Knight, R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007, 35, e120. [Google Scholar] [CrossRef] [Green Version]
- Prince, A.L.; Chu, D.M.; Seferovic, M.D.; Antony, K.M.; Ma, J.; Aagaard, K.M. The perinatal microbiome and pregnancy: Moving beyond the vaginal microbiome. Cold Spring Harb. Perspect. Med. 2015, 5, a023051. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef] [Green Version]
- Fettweis, J.M.; Serrano, M.G.; Sheth, N.U.; Mayer, C.M.; Glascock, A.L.; Brooks, J.P.; Jefferson, K.K.; Buck, G.A.; Consortium, V.M. Species-level classification of the vaginal microbiome. BMC Genom. 2012, 13, S17. [Google Scholar] [CrossRef] [Green Version]
- Hummelen, R.; Fernandes, A.D.; Macklaim, J.M.; Dickson, R.J.; Changalucha, J.; Gloor, G.B.; Reid, G. Deep sequencing of the vaginal microbiota of women with HIV. PLoS ONE 2010, 5, e12078. [Google Scholar] [CrossRef] [Green Version]
- Ghartey, J.P.; Smith, B.C.; Chen, Z.; Buckley, N.; Lo, Y.; Ratner, A.J.; Herold, B.C.; Burk, R.D. Lactobacillus crispatus dominant vaginal microbiome is associated with inhibitory activity of female genital tract secretions against Escherichia coli. PLoS ONE 2014, 9, e96659. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuczynski, J.; Lauber, C.L.; Walters, W.A.; Parfrey, L.W.; Clemente, J.C.; Gevers, D.; Knight, R. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 2012, 13, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; DiGiulio, D.B.; Goltsman, D.S.A.; Sun, C.L.; Costello, E.K.; Jeganathan, P.; Biggio, J.R.; Wong, R.J.; Druzin, M.L.; Shaw, G.M. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl. Acad. Sci. USA 2017, 114, 9966–9971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quince, C.; Lanzén, A.; Curtis, T.P.; Davenport, R.J.; Hall, N.; Head, I.M.; Read, L.F.; Sloan, W.T. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods 2009, 6, 639–641. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef]
- Huse, S.M.; Huber, J.A.; Morrison, H.G.; Sogin, M.L.; Welch, D.M. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007, 8, R143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas-White, K.J.; Hilt, E.E.; Fok, C.; Pearce, M.M.; Mueller, E.R.; Kliethermes, S.; Jacobs, K.; Zilliox, M.J.; Brincat, C.; Price, T.K. Incontinence medication response relates to the female urinary microbiota. Int. Urogynecol. J. 2016, 27, 723–733. [Google Scholar] [CrossRef]
- Pearce, M.M.; Hilt, E.E.; Rosenfeld, A.B.; Zilliox, M.J.; Thomas-White, K.; Fok, C.; Kliethermes, S.; Schreckenberger, P.C.; Brubaker, L.; Gai, X. The female urinary microbiome: A comparison of women with and without urgency urinary incontinence. MBio 2014, 5, e01283-14. [Google Scholar] [CrossRef] [Green Version]
- Pearce, M.M.; Zilliox, M.J.; Rosenfeld, A.B.; Thomas-White, K.J.; Richter, H.E.; Nager, C.W.; Visco, A.G.; Nygaard, I.E.; Barber, M.D.; Schaffer, J. The female urinary microbiome in urgency urinary incontinence. Am. J. Obstet. Gynecol. 2015, 213, 347.e1–347.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.A.; Kwon, E.J.; Choi, S.J.; Hwang, H.S.; Choi, S.K.; Lee, S.M.; Kim, Y.J. Vaginal microbiome profiles of pregnant women in Korea using a 16S metagenomics approach. Am. J. Reprod. Immunol. 2019, 82, e13124. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, Y.; Wei, X.; Zhu, J.; Wang, X.; Xie, X.; Lu, W. The Alterations of Vaginal Microbiome in HPV16 Infection as Identified by Shotgun Metagenomic Sequencing. Front. Cell. Infect. Microbiol. 2020, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Fettweis, J.M.; Serrano, M.G.; Brooks, J.P.; Edwards, D.J.; Girerd, P.H.; Parikh, H.I.; Huang, B.; Arodz, T.J.; Edupuganti, L.; Glascock, A.L. The vaginal microbiome and preterm birth. Nat. Med. 2019, 25, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, K.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.-A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Van den Veyver, I.; Milosavljevic, A. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Nikita, L.; Galuppi, M.; Lamont, R.F.; Chaemsaithong, P.; Miranda, J. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2014, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.G.; Parikh, H.I.; Brooks, J.P.; Edwards, D.J.; Arodz, T.J.; Edupuganti, L.; Huang, B.; Girerd, P.H.; Bokhari, Y.A.; Bradley, S.P. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat. Med. 2019, 25, 1001–1011. [Google Scholar] [CrossRef]
- Anukam, K.; Agbakoba, N.; Okoli, A.; Oguejiofor, C. Vaginal bacteriome of Nigerian women in health and disease: A study with 16S rRNA metagenomics. Trop. J. Obstet. Gynaecol. 2019, 36, 96–104. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Brooks, J.P.; Serrano, M.G.; Sheth, N.U.; Girerd, P.H.; Edwards, D.J.; Strauss, J.F., III; Jefferson, K.K.; Buck, G.A.; Consortium, V.M. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiology 2014, 160 Pt 10, 2272. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, D.A.; Chandiramani, M.; Lee, Y.S.; Kindinger, L.; Smith, A.; Angelopoulos, N.; Lehne, B.; Arulkumaran, S.; Brown, R.; Teoh, T.G. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep. 2015, 5, 8988. [Google Scholar] [CrossRef] [Green Version]
- Hyman, R.W.; Fukushima, M.; Jiang, H.; Fung, E.; Rand, L.; Johnson, B.; Vo, K.C.; Caughey, A.B.; Hilton, J.F.; Davis, R.W. Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci. 2014, 21, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther-António, M.R.; Jeraldo, P.; Miller, M.E.B.; Yeoman, C.J.; Nelson, K.E.; Wilson, B.A.; White, B.A.; Chia, N.; Creedon, D.J. Pregnancy’s stronghold on the vaginal microbiome. PLoS ONE 2014, 9, e98514. [Google Scholar]
- Stout, M.J.; Zhou, Y.; Wylie, K.M.; Tarr, P.I.; Macones, G.A.; Tuuli, M.G. Early pregnancy vaginal microbiome trends and preterm birth. Am. J. Obstet. Gynecol. 2017, 217, 356.e1–356.e18. [Google Scholar] [CrossRef]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; MacIntyre, D.; Lee, Y.; Smith, A.; Marchesi, J.R.; Lehne, B.; Bhatia, R.; Lyons, D.; Paraskevaidis, E.; Li, J. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 2015, 5, 16865. [Google Scholar] [CrossRef] [Green Version]
- Hummelen, R.; Macklaim, J.M.; Bisanz, J.E.; Hammond, J.-A.; McMillan, A.; Vongsa, R.; Koenig, D.; Gloor, G.B.; Reid, G. Vaginal microbiome and epithelial gene array in post-menopausal women with moderate to severe dryness. PLoS ONE 2011, 6, e26602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [Green Version]
- Fettweis, J.M. A new era of the vaginal microbiome: Advances using next-generation sequencing. Chem. Biodivers. 2012, 9, 965–976. [Google Scholar] [CrossRef]
- Huang, B. The changing landscape of the vaginal microbiome. Clin. Lab. Med. 2014, 34, 747–761. [Google Scholar] [CrossRef] [Green Version]
- Köser, C.U.; Ellington, M.J.; Peacock, S.J. Whole-genome sequencing to control antimicrobial resistance. Trends Genet. 2014, 30, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; McLaren, M.; Callahan, B. Understanding and interpreting community sequencing measurements of the vaginal microbiome. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 139–146. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gołębiewski, M.; Deja-Sikora, E.; Cichosz, M.; Tretyn, A.; Wróbel, B. 16S rDNA pyrosequencing analysis of bacterial community in heavy metals polluted soils. Microb. Ecol. 2014, 67, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Mysara, M.; Njima, M.; Leys, N.; Raes, J.; Monsieurs, P. From reads to operational taxonomic units: An ensemble processing pipeline for MiSeq amplicon sequencing data. Gigascience 2017, 6, giw017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quince, C.; Lanzen, A.; Davenport, R.J.; Turnbaugh, P.J. Removing noise from pyrosequenced amplicons. BMC Bioinform. 2011, 12, 38. [Google Scholar] [CrossRef]
- Rappé, M.S.; Giovannoni, S.J. The uncultured microbial majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westcott, S.L.; Schloss, P.D. De novo clustering methods outperform reference-based methods for assigning 16S rRNA gene sequences to operational taxonomic units. PeerJ 2015, 3, e1487. [Google Scholar] [CrossRef] [PubMed]
- Navas-Molina, J.A.; Peralta-Sánchez, J.M.; González, A.; McMurdie, P.J.; Vázquez-Baeza, Y.; Xu, Z.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J. Advancing our understanding of the human microbiome using QIIME. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 531, pp. 371–444. [Google Scholar]
- Kopylova, E.; Navas-Molina, J.A.; Mercier, C.; Xu, Z.Z.; Mahé, F.; He, Y.; Zhou, H.-W.; Rognes, T.; Caporaso, J.G.; Knight, R. Open-source sequence clustering methods improve the state of the art. MSystems 2016, 1, e00003-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rideout, J.R.; He, Y.; Navas-Molina, J.A.; Walters, W.A.; Ursell, L.K.; Gibbons, S.M.; Chase, J.; McDonald, D.; Gonzalez, A.; Robbins-Pianka, A. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2014, 2, e545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Xu, Z.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A. Deblur rapidly resolves single-nucleotide community sequence patterns. MSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv 2016, 1, 081257. [Google Scholar]
- Eren, A.M.; Morrison, H.G.; Lescault, P.J.; Reveillaud, J.; Vineis, J.H.; Sogin, M.L. Minimum entropy decomposition: Unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J. 2015, 9, 968–979. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Met. 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Huse, S.M.; Mark Welch, D.B.; Voorhis, A.; Shipunova, A.; Morrison, H.G.; Eren, A.M.; Sogin, M.L. VAMPS: A website for visualization and analysis of microbial population structures. BMC Bioinform. 2014, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, V.M.; Ivanova, N.N.; Szeto, E.; Palaniappan, K.; Chu, K.; Dalevi, D.; Chen, I.-M.A.; Grechkin, Y.; Dubchak, I.; Anderson, I. IMG/M: A data management and analysis system for metagenomes. Nucleic Acids Res. 2007, 36 (Suppl. 1), D534–D538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buza, T.M.; Tonui, T.; Stomeo, F.; Tiambo, C.; Katani, R.; Schilling, M.; Lyimo, B.; Gwakisa, P.; Cattadori, I.M.; Buza, J.; et al. iMAP: An integrated bioinformatics and visualization pipeline for microbiome data analysis. BMC Bioinform. 2019, 20, 374. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.R.; Liu, B.; Delcher, A.L.; Pop, M.; Salzberg, S.L. Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res. 2012, 40, e9. [Google Scholar] [CrossRef]

{kind=link}
| Serial No. | Study | Study Aim | Sample No. | Sample Site | Technique | Primer Used | Analysis Software/Tools Used | Findings |
|---|---|---|---|---|---|---|---|---|
| 1 | [37] | Investigation of the effect of storage conditions on the vaginal microbiota | N = 8 | Mid-vagina | 454 Life Sciences FLX sequencing(Pyrosequencing) | V1–V2 27F 338R | QIIME software UCLUST software | At ultra-low temperatures (−80 °C) or storage for one week at (−20 °C) prior to storage at (−80 °C) for four weeks, no significant changes were observed when compared to non-frozen samples. |
| 2 | [89] | Characterization of the vaginal microbiota of women with preterm labor (PTL) and preterm pre-labor rupture of membranes (PPROM) | N = 65 | Posterior fornix | Illumina 16S rRNA gene sequencing | V3–V4 319 F 806 R+ | QIIME software package (v. 1.8) SPSS software ver. 21.0 | The microbial abundance and diversity in the PPROM was higher than in PTL women. |
| 3 | [90] | To explore the profiling of the vaginal microbiota associated with HPV16 infection (control) | N = 52 27 HPV16 + ve and 25 HPV − ve | Vaginal fornix and cervix | Illumina Hiseq X-ten platform shotgun metagenomic sequencing | SOAPdenovo (Version 1.05) Software MetaGeneMark | The abundance of Lactobacillus (Firmicutes) was lower in HPV16-positive women than in controls. | |
| 4 | [91] | Identification of vaginal microbial signatures in women with PTL | N = 1572 | Vaginal and rectal samples | Illumina MiSeq | V1–V3 | ASGARD, HUMAnN2 and ShortBRED | Coupled with genetic factors, microbiome-associated taxonomic, metabolic and immunologic biomarkers may be useful in defining the risk of PTL. |
| 5 | [92] | Vaginal microbial gene catalogue differs in pregnancy | N = 68 | Vaginal introitus, posterior fornix, and mid- vagina | 454FLX Titanium platform | V3–V5 354F 926R | QIIME | Characterization of healthy, gravid vaginal microbiome. |
| 6 | [93] | Characterization of changes in the composition of the vaginal microbiota of pregnant women | N = 54 | Posterior fornix | Pyrosequencing | V1–V2 27F 338R | UCHIME UCLUST | Vaginal microbiota in normal pregnancy is different and stable from that of non-pregnant women. |
| 7 | [94] | To examine composition of the vaginal microbiome of women of African and non-African ancestry differently during pregnancy | N = 2582 | Vagina (Self-sampling) | Roche 454 titanium Illumina MiSeq | V1–V3 | CLARK-S MetaPhlAn2 | Women of European, African and Hispanic ancestry exhibit different vaginal microbiome compositions and dynamics during pregnancy |
| 8 | [95] | To determine the vaginal bacterial composition in healthy Nigerian women and BV women | N = 28 | Vagina | 16S rRNA sequencing | V4 region | QIIME-UCLUST | Characterization of vaginal bacteriome compositions of healthy and often times annoying condition known as BV in Nigerian women. |
| 8 | [4] | Understanding of the composition and ecology of the vagina microbial ecosystem in asymptomatic women | N = 396 | Vagina | 454 Life Sciences FLX sequencing | V1–V2 27F 338R | Differences in vaginal bacterial community composition in different ethnic group of North American women. | |
| 9 | [96] | Comparison of vaginal microbiomes of African American women with women of European ancestry with and without a diagnosis of BV | N = 1268 AAW N = 416 EA | Mid-vagina | Roche 454 GS FLX Titanium | V1–V3 | African women have high rates of BV. | |
| 10 | [97] | To examine the composition of the vaginal microbiome throughout pregnancy and in the postpartum period | N = 42 | Posterior fornix | MiSeq sequencing | V1–V2 28F 388R | Mothur | Biogeographical and ethnic differences exist between microbial communities in the vaginal microbiome during pregnancy and in the postpartum period. |
| 11 | [98] | Pregnant women at high and low risk of PTB were studied. | N = 88 | Posterior fornix | Sanger sequencing | 8F 1492R | QIIME | PTB is related to the variety of the vaginal microbiome during human pregnancy, and race/ethnicity and sampling site are relevant determinants. |
| 12 | [99] | To assess the vaginal microbiome throughout full-term uncomplicated pregnancy | N = 12 | Posterior fornix and cervix | Illumina MiSeq | V3–V5 357F 926R | IM-TORNADO QIIME | Normal pregnancy has a less diverse and highly stable microbiome. |
| 13 | [100] | Characterization of the vaginal microbial community in African-American, pregnant women associated with the risk for preterm birth. | N = 149 | Mid-vagina | Roche 454 | V1–V3 27F 534R V3–V5 357F 926R | Preterm birth is linked to decreases in the richness and variety of the vaginal microbial community in the African-American population. | |
| 14 | [101] | Comparison of changes in the vaginal microbiota and metabolome of females as a result of frequent genital illnesses. | N = 79 | Vagina | NGS | V3–V4 | PANDAseq (v. 2.5.0) QIIME pipeline (release 1.8.0) | Women with vulvovaginal candidiasis (VVC) and Chlamydia trachomatis infection (CT) had a vaginal microbiome that was positioned between eubiosis (healthy women) and dysbiosis (BV-positive subjects), with lactobacilli depletion and an increase in several anaerobe genera (e.g., Gardnerella, Megasphaera, Roseburia, and Atopobium |
| 15 | [102] | To study the relationship between the vaginal microbiota and CIN disease progression | N = 169 | Posterior vaginal fornix | Illumina MiSeq sequencing | V1–V2 | Mothur | Vaginal microbial diversity is associated not only with HPV infection, but also with advancing cervical intra-epithelial neoplasia (CIN) severity |
| 16 | [103] | To discover the vaginal microbiome of postmenopausal women who were either healthy or experienced vaginal dryness | N = 500 | Mid-vagina | Illumina sequencing | V6 | Uclust version 3.0.617 | In women with moderate to severe vaginal dryness, there is an inverse relationship between Lactobacillus ratio and dryness, as well as an increase in bacterial diversity. |
| Serial No. | In-Silico Tools | Functions | URL | References |
|---|---|---|---|---|
| 1 | QIIME | Used to perform demultiplexing, quality filtering, operational taxonomic unit picking, taxonomic assignment, phylogenetic reconstruction, diversity analyses and visualizations | http://qiime.org/ | [125] |
| 2 | Mothur | Used to analyze raw sequences to the generation of visualization tools to describe α and β diversity | http://mothur.org/ | [126] |
| 3 | VAMPS | VAMPS is the collection of tools used to visualize and analyze data for microbial population structures and distributions | https://vamps2.mbl.edu/ | [140] |
| 4 | FastTree | Command line tool used to generate phylogenetic trees by maximum-likelihood from nucleotide and protein sequences | http://www.microbesonline.org/fasttree/ | [141] |
| 5 | BLAST | Tool used to find similarity between nucleotide or protein sequences with reference database | https://blast.ncbi.nlm.nih.gov/Blast.cgi | [142] |
| 6 | MG-RAST | Open source application used for the phylogenetic and functional analysis of metagenomic data | https://www.mg-rast.org/ | [143] |
| 7 | IMG/M | Analysis and annotation of genome and metagenome datasets | https://img.jgi.doe.gov/cgi-bin/m/main.cgi | [144] |
| 8 | iMAP | This is a bioinformatic pipeline that performs metadata profiling, quality control of reads, sequence processing and classification, and diversity analysis of operational taxonomic units | https://github.com/tmbuza/iMAP | [145] |
| 9 | Phyloseq | Phyloseq is an R programming language package used to import, store, analyze, and graphically display complex phylogenetic sequencing data that has already been clustered into Operational Taxonomic Units (OTUs). | https://joey711.github.io/phyloseq/ | [146] |
| 10 | SILVA | Database of ribosomal RNA database with web based tools used for sequence alignment and many interactive analysis | https://www.arb-silva.de/ | [147] |
| 11 | HUMAN 3.0 | Used for profiling the microbial metabolic pathways and other molecular based functions from metagenomic or metatranscriptomic data | https://huttenhower.sph.harvard.edu/humann | [148] |
| 12 | PICRUSt | This software is used to predict metagenome functional content from marker gene and full genomes. | http://picrust.github.io/picrust/ | [149] |
| 13 | Meta Gene Mark | Tool is used to identify the protein coding regions from the metagenomic sequences. | http://exon.gatech.edu/Genemark/meta_gmhmmp.cgi | [150] |
| 14 | Glimmer-MG | Gene finding tool for microbial (bacteria, archaea and viruses) DNA | https://github.com/davek44/Glimmer-MG | [151] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, M.; Chopra, C.; Mehta, M.; Sharma, V.; Mallubhotla, S.; Sistla, S.; Sistla, J.C.; Bhushan, I. An Insight into Vaginal Microbiome Techniques. Life 2021, 11, 1229. https://doi.org/10.3390/life11111229
Sharma M, Chopra C, Mehta M, Sharma V, Mallubhotla S, Sistla S, Sistla JC, Bhushan I. An Insight into Vaginal Microbiome Techniques. Life. 2021; 11(11):1229. https://doi.org/10.3390/life11111229
Chicago/Turabian StyleSharma, Mahima, Chitrakshi Chopra, Malvika Mehta, Varun Sharma, Sharada Mallubhotla, Srinivas Sistla, Jyothi C. Sistla, and Indu Bhushan. 2021. "An Insight into Vaginal Microbiome Techniques" Life 11, no. 11: 1229. https://doi.org/10.3390/life11111229
APA StyleSharma, M., Chopra, C., Mehta, M., Sharma, V., Mallubhotla, S., Sistla, S., Sistla, J. C., & Bhushan, I. (2021). An Insight into Vaginal Microbiome Techniques. Life, 11(11), 1229. https://doi.org/10.3390/life11111229