
A Global Mutational Profile of SARS-CoV-2: A Systematic Review and Meta-Analysis of 368,316 COVID-19 Patients


 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Protocol
2.2. Literature Review
2.3. Inclusion and Exclusion Criteria for Studies
2.4. Quality Assessment
2.5. Data Extraction
2.6. Data Synthesis and Analysis
3. Results
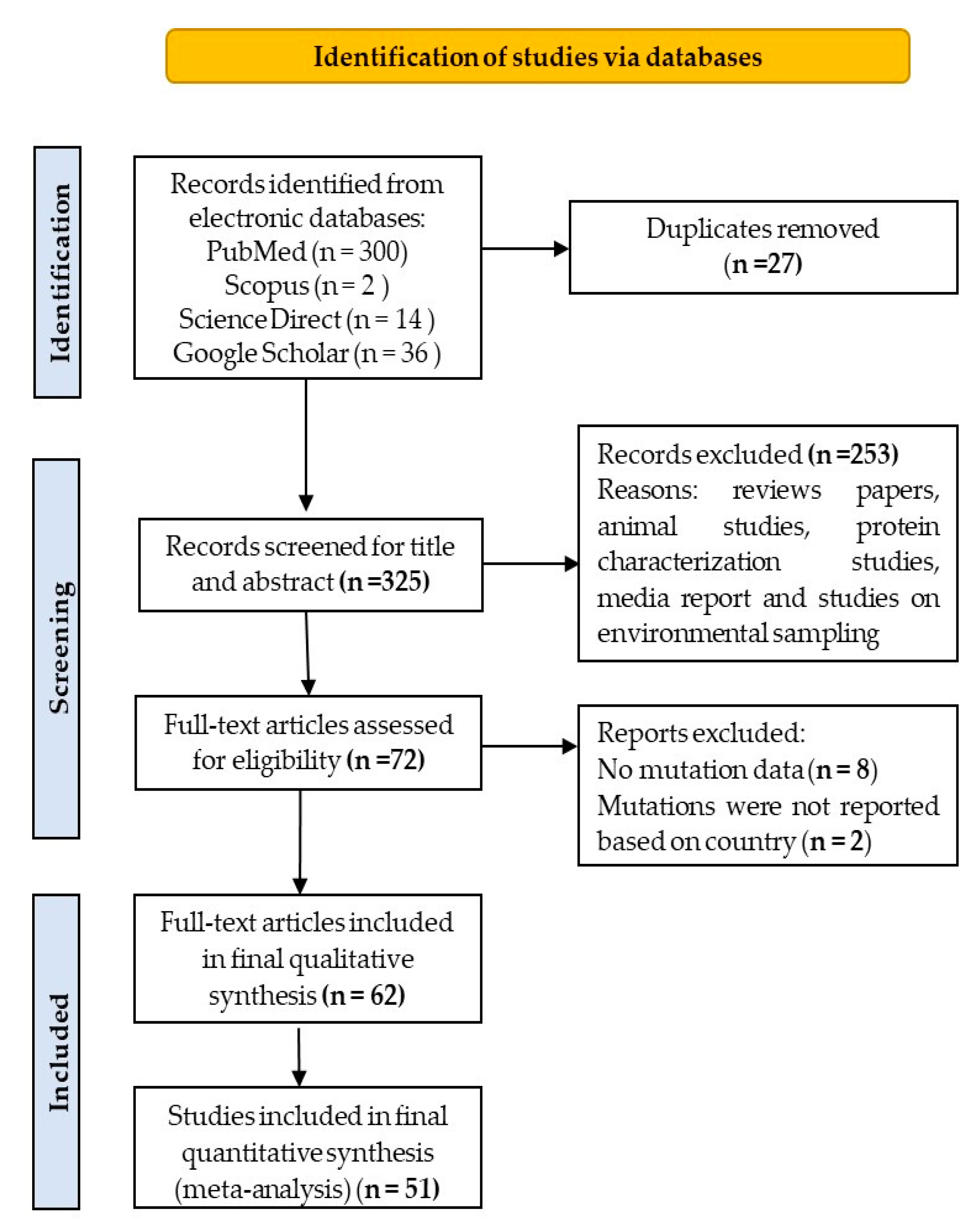
3.1. Search Result and Eligible Studies
3.2. Characteristics of the Eligible Studies
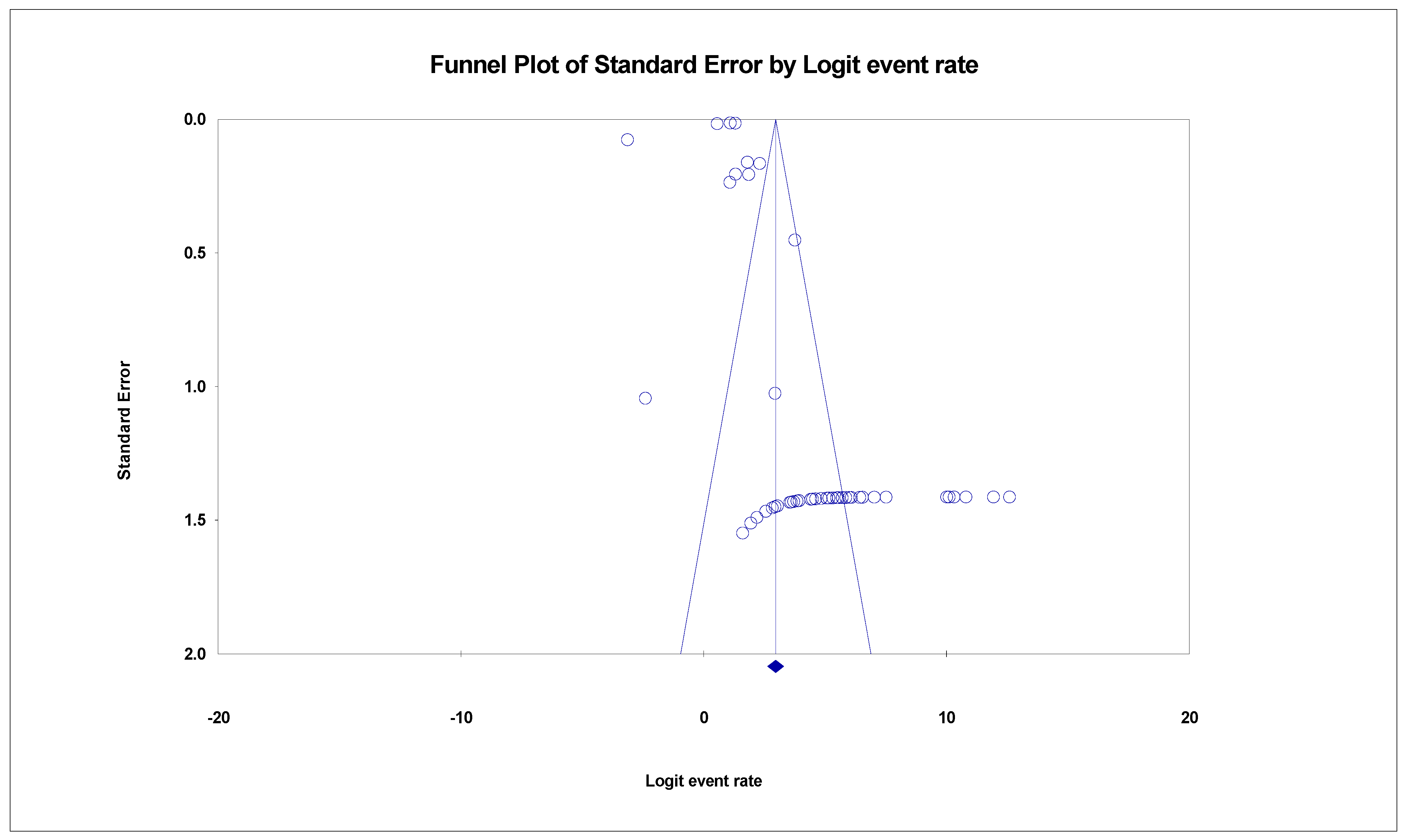
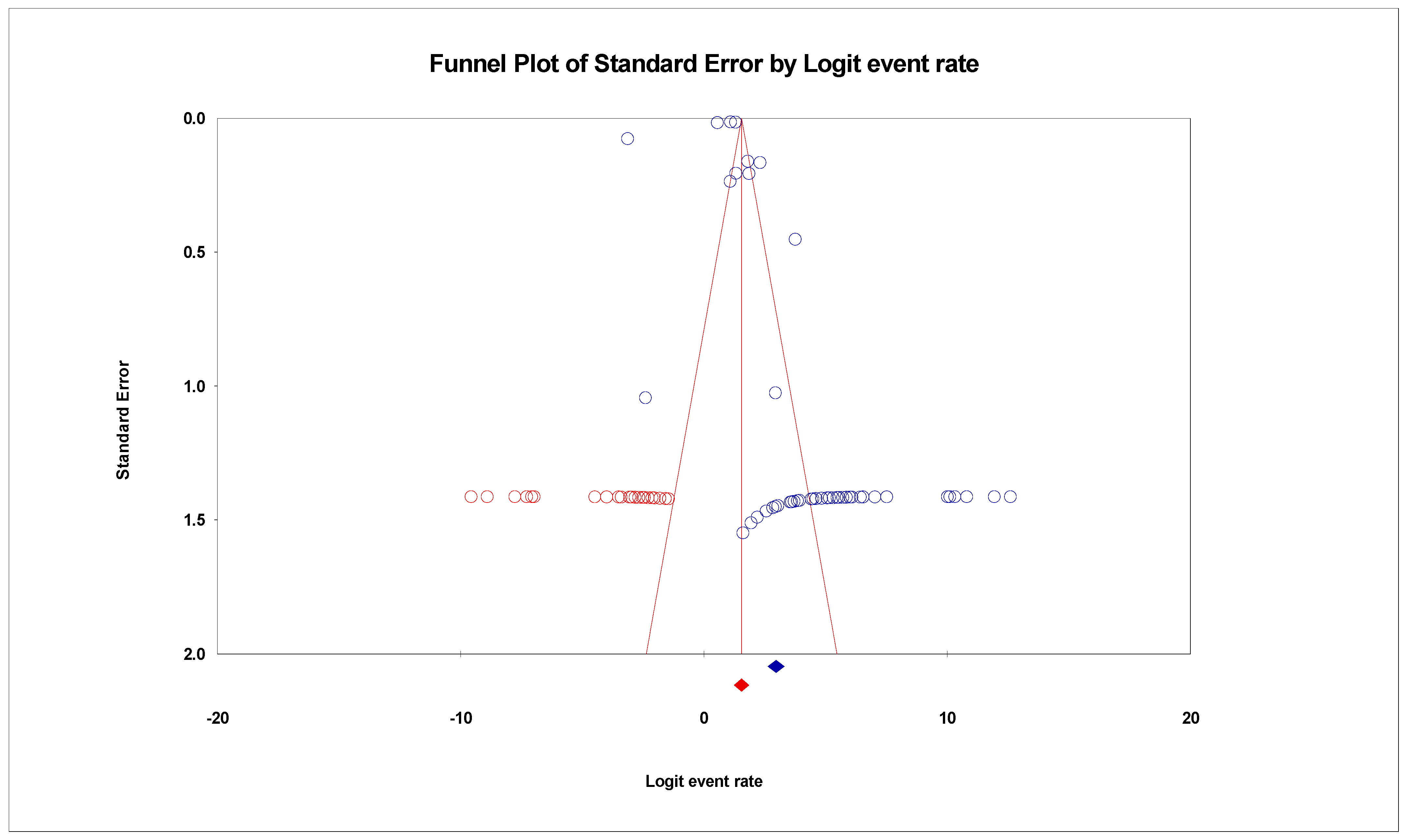
3.3. The Pooled Prevalence of SARS-CoV-2 Variants
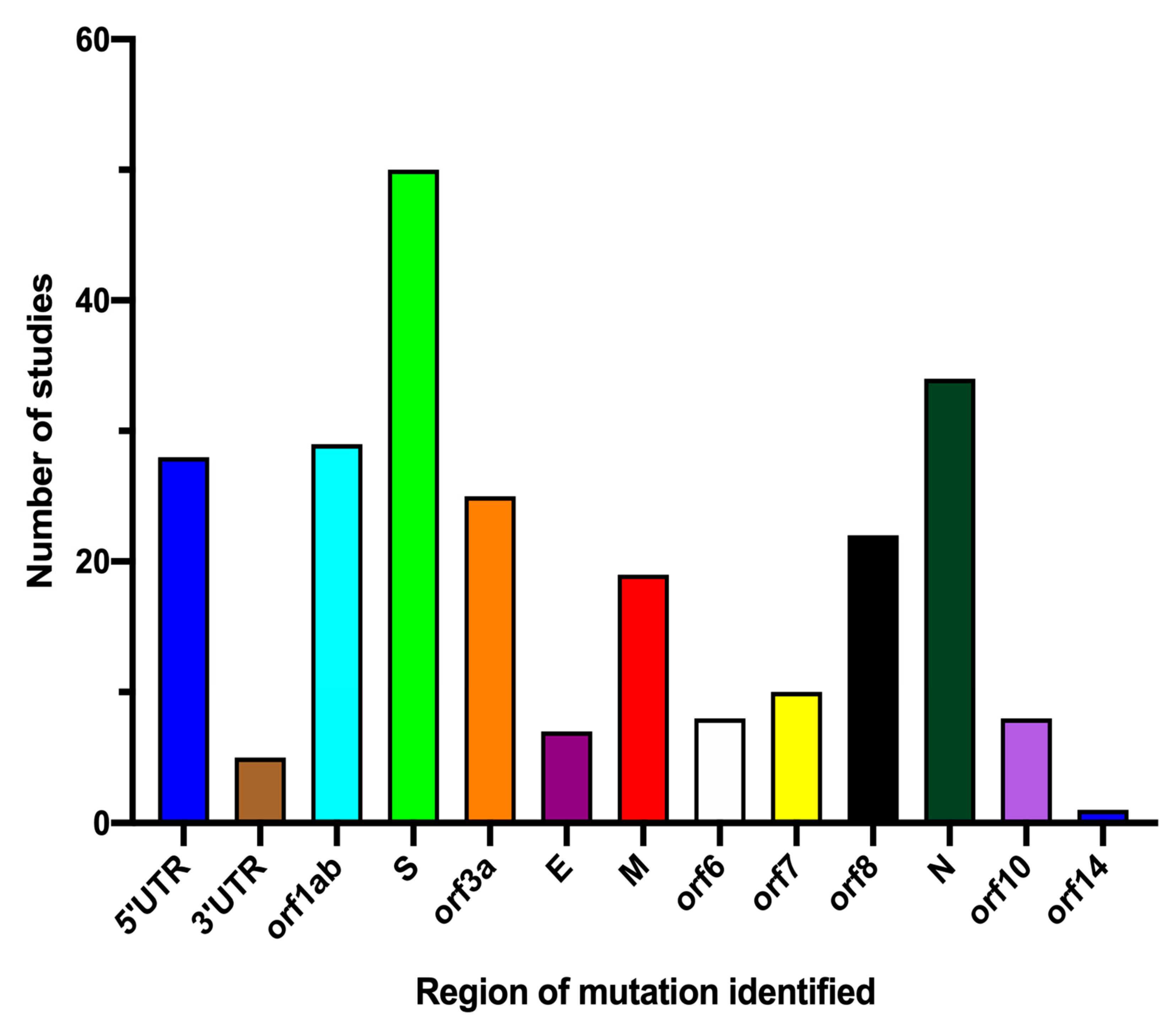
3.4. Subgroup Meta-Analysis
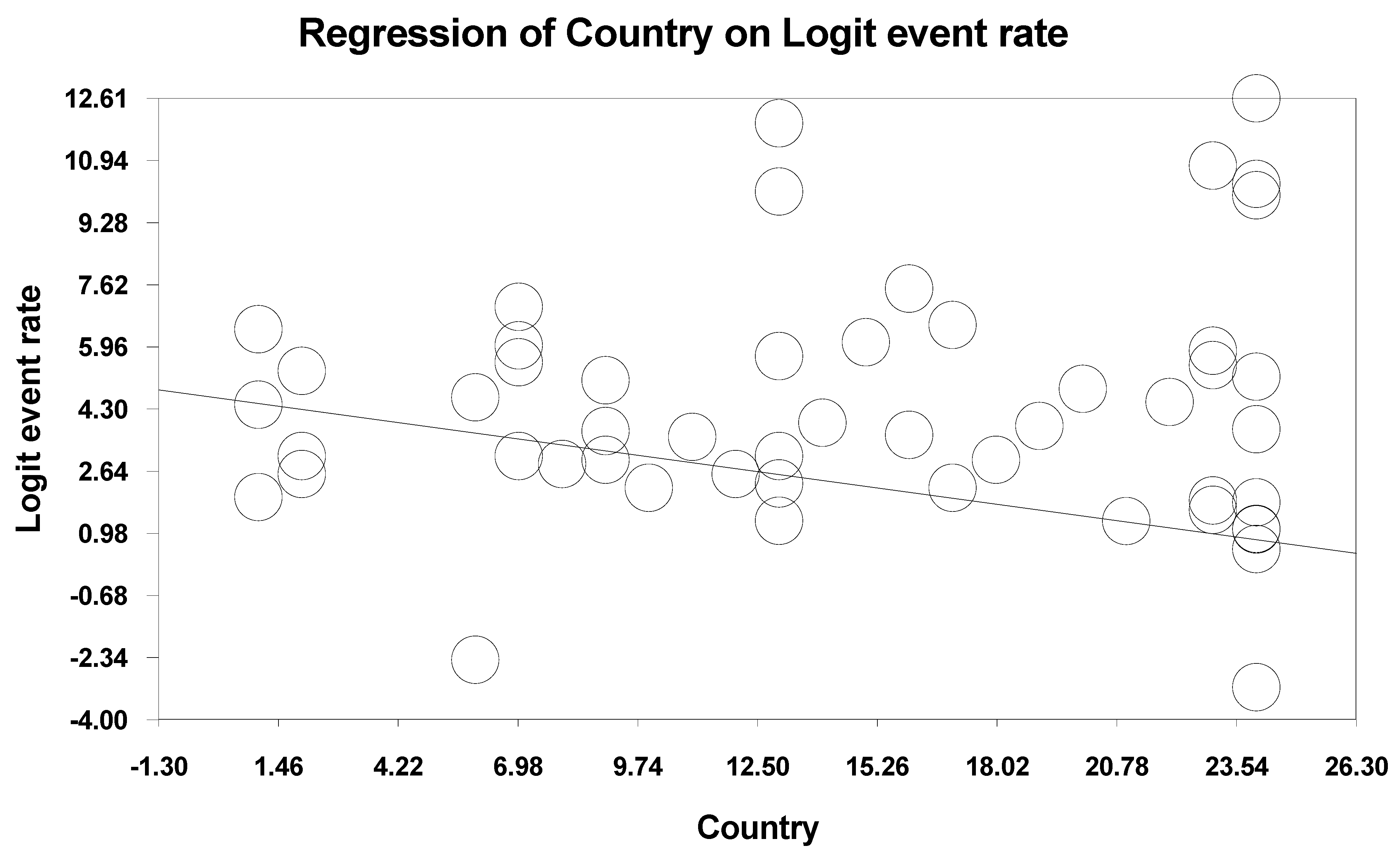
3.5. Meta-Regression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sahlan, M.; Irdiani, R.; Flamandita, D.; Aditama, R.; Alfarraj, S.; Ansari, M.J.; Khayrani, A.C.; Pratami, D.K.; Lischer, L. Molecular Interaction Analysis of Sulawesi Propolis Compounds with SARS-CoV-2 Main Protease as Preliminary Study for COVID-19 Drug Discovery. J. King Saud. Univ. Sci. 2021, 33, 101234. [Google Scholar] [CrossRef]
- Soratto, T.A.T.; Darban, H.; Bjerkner, A.; Coorens, M.; Albert, J.; Allander, T.; Andersson, B. Four SARS-CoV-2 Genome Sequences from Late April in Stockholm, Sweden, Reveal a Rare Mutation in the Spike Protein. Microbiol. Resour. Announc. 2020, 9, e00934-20. [Google Scholar] [CrossRef]
- Dinnes, J.; Deeks, J.J.; Berhane, S.; Taylor, M.; Adriano, A.; Davenport, C.; Dittrich, S.; Emperador, D.; Takwoingi, Y.; Cunningham, J.; et al. Rapid, Point-of-Care Antigen and Molecular-Based Tests for Diagnosis of SARS-CoV-2 Infection. Cochrane Database Syst. Rev. 2021, 3, CD013705. [Google Scholar] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The Biogrid Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Shakoor, H.; Feehan, J.; al Dhaheri, A.S.; Ali, H.I.; Platat, C.; Ismail, L.C.; Apostolopoulos, V.; Stojanovska, L. Immune-Boosting Role of Vitamins D, C, E, Zinc, Selenium and Omega-3 Fatty Acids: Could They Help against COVID-19? Maturitas 2021, 143, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Jatoi, M.A.; Al-Wraikat, M.; Ahmed, N.; Li, J. Time to Enhance Immunity Via Functional Foods and Supplements: Hope for SARS-CoV-2 Outbreak. Altern. Ther. Health Med. 2021, 27 (Suppl. S1), 30–44. [Google Scholar] [PubMed]
- Gelardi, M.; Notargiacomo, M.; Trecca, E.M.C.; Cassano, M.; Ciprandi, G. COVID-19 and Nasal Cytobrush Cytology. Acta Cytol. 2020, 64, 397–398. [Google Scholar] [CrossRef]
- Samudrala, P.K.; Kumar, P.; Choudhary, K.; Thakur, N.; Wadekar, G.S.; Dayaramani, R.; Agrawal, M.; Alexander, A. Virology, Pathogenesis, Diagnosis and in-Line Treatment of COVID-19. Eur. J. Pharmacol. 2020, 883, 173375. [Google Scholar] [CrossRef]
- Qiang, X.L.; Xu, P.; Fang, G.; Liu, W.B.; Kou, Z. Using the Spike Protein Feature to Predict Infection Risk and Monitor the Evolutionary Dynamic of Coronavirus. Infect. Dis. Poverty 2020, 9, 33. [Google Scholar] [CrossRef]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A. Preferred Reporting Items for Systematic Review and Meta-Analysis Protocols (Prisma-P) 2015: Elaboration and Explanation. BMJ Br. Med. J. 2015, 349, g7647. [Google Scholar] [CrossRef] [Green Version]
- Munn, Z.; Moola, S.; Lisy, K.; Riitano, D.; Tufanaru, C. Methodological Guidance for Systematic Reviews of Observational Epidemiological Studies Reporting Prevalence and Cumulative Incidence Data. JBI Evid. Implement. 2015, 13, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.P.; Thompson, S.G. Quantifying Heterogeneity in a Meta-Analysis. Stat. Med. 2002, 21, 1539–1558. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Banu, T.A.; Goswami, B.; Osman, E.; Uzzaman, M.S.; Habib, M.A.; Jahan, I.; Mahmud, A.S.M.; Sarker, M.M.H.; Hossain, M.S.; et al. Coding-Complete Genome Sequences of Three SARS-CoV-2 Strains from Bangladesh. Microbiol. Resour. Announc. 2020, 9, e00764-20. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.; Garcia-Cehic, D.; Gregori, J.; Piñana, M.; Rodriguez-Frias, F.; Guerrero-Murillo, M.; Esperalba, J.; Rando, A.; Goterris, L.; Codina, M.G.; et al. Naturally Occurring SARS-CoV-2 Gene Deletions Close to the Spike S1/S2 Cleavage Site in the Viral Quasispecies of Covid19 Patients. Emerg. Microbes Infect. 2020, 9, 1900–1911. [Google Scholar] [CrossRef]
- Badua, C.L.D.; Baldo, K.A.T.; Medina, P.M.B. Genomic and Proteomic Mutation Landscapes of SARS-CoV-2. J. Med. Virol. 2020, 93, 1702–1721. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.; Bura, A.C.; He, Q.; Huang, F.W.; Li, T.J.X.; Waterman, M.S.; Reidys, C.M. Multiscale Feedback Loops in SARS-CoV-2 Viral Evolution. J. Comput. Biol. 2020, 28, 248–256. [Google Scholar] [CrossRef]
- Bartolini, B.; Rueca, M.; Gruber, C.E.M.; Messina, F.; Carletti, F.; Giombini, E.; Lalle, E.; Bordi, L.; Matusali, G.; Colavita, F.; et al. SARS-CoV-2 Phylogenetic Analysis, Lazio Region, Italy, February–March 2020. Emerg. Infect. Dis. 2020, 26, 1842–1845. [Google Scholar] [CrossRef]
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 Viral Spike G614 Mutation Exhibits Higher Case Fatality Rate. Int. J. Clin. Pract. 2020, 74, e13525. [Google Scholar] [CrossRef]
- Benvenuto, D.; Demir, A.B.; Giovanetti, M.; Bianchi, M.; Angeletti, S.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evidence for Mutations in SARS-CoV-2 Italian Isolates Potentially Affecting Virus Transmission. J. Med. Virol. 2020, 92, 2232–2237. [Google Scholar] [CrossRef]
- Chang, T.J.; Yang, D.M.; Wang, M.L.; Liang, K.H.; Tsai, P.H.; Chiou, S.H.; Lin, T.H.; Wang, C.T. Genomic Analysis and Comparative Multiple Sequences of SARS-CoV-2. J. Chin. Med. Assoc. 2020, 83, 537–543. [Google Scholar] [CrossRef]
- Chen, J.; Hilt, E.E.; Li, F.; Wu, H.; Jiang, Z.; Zhang, Q.; Wang, J.; Wang, Y.; Li, Z.; Tang, J.; et al. Epidemiological and Genomic Analysis of SARS-CoV-2 in 10 Patients from a Mid-Sized City Outside of Hubei, China in the Early Phase of the COVID-19 Outbreak. Front. Public Health 2020, 8, 567621. [Google Scholar] [CrossRef]
- Cusi, M.G.; Pinzauti, D.; Gandolfo, C.; Anichini, G.; Pozzi, G.; Santoro, F. Whole-Genome Sequence of SARS-CoV-2 Isolate Siena-1/2020. Microbiol. Resour. Announc. 2020, 9, e00944-20. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.B.; Benvenuto, D.; Abacioğlu, H.; Angeletti, S.; Ciccozzi, M. Identification of the Nucleotide Substitutions in 62 SARS-CoV-2 Sequences from Turkey. Turk. J. Biol. 2020, 44, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Devendran, R.; Kumar, M.; Chakraborty, S. Genome Analysis of SARS-CoV-2 Isolates Occurring in India: Present Scenario. Indian J. Public Health 2020, 64, S147–S55. [Google Scholar] [PubMed]
- Du, P.; Ding, N.; Li, J.; Zhang, F.; Wang, Q.; Chen, Z.; Song, C.; Han, K.; Xie, W.; Liu, J.; et al. Genomic Surveillance of COVID-19 Cases in Beijing. Nat. Commun. 2020, 11, 5503. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, V.; Harkins, G.W.; Mabvakure, B.; Smidt, S.; Zappile, P.; Marier, C.; Maurano, M.; Perez, V.; Mazza, N.; Beloso, C.; et al. SARS-CoV-2 Genomic Characterization and Clinical Manifestation of the COVID-19 Outbreak in Uruguay. Emerg. Microbes Infect. 2020, 10, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Eskier, D.; Suner, A.; Karakülah, G.; Oktay, Y. Mutation Density Changes in SARS-CoV-2 Are Related to the Pandemic Stage but to a Lesser Extent in the Dominant Strain with Mutations in Spike and Rdrp. PeerJ 2020, 8, e9703. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; del Molino, M.L.P.; Martinón-Torres, F.; Salas, A. Phylogeography of SARS-CoV-2 Pandemic in Spain: A Story of Multiple Introductions, Micro-Geographic Stratification, Founder Effects, and Super-Spreaders. Zool. Res. 2020, 41, 605–620. [Google Scholar] [CrossRef]
- Gong, Y.N.; Tsao, K.C.; Hsiao, M.J.; Huang, C.G.; Huang, P.N.; Huang, P.W.; Lee, K.M.; Liu, Y.C.; Yang, S.L.; Kuo, R.L.; et al. SARS-CoV-2 Genomic Surveillance in Taiwan Revealed Novel Orf8-Deletion Mutant and Clade Possibly Associated with Infections in Middle East. Emerg. Microbes Infect. 2020, 9, 1457–1466. [Google Scholar] [CrossRef]
- Gupta, A.M.; Chakrabarti, J.; Mandal, S. Non-Synonymous Mutations of SARS-CoV-2 Leads Epitope Loss and Segregates Its Variants. Microbes Infect. 2020, 22, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Hartley, P.; Tillett, R.L.; Xu, Y.; AuCoin, D.P.; Sevinsky, J.R.; Gorzalski, A.; Pandori, M.C.; Buttery, E.; Hansen, H.; Picker, M.; et al. Genomic Surveillance Revealed Prevalence of Unique SARS-CoV-2 Variants Bearing Mutation in the Rdrp Gene among Nevada Patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Hassan, S.S.; Choudhury, P.P.; Roy, B.; Jana, S.S. Missense Mutations in SARS-CoV2 Genomes from Indian Patients. Genomics 2020, 112, 4622–4627. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Chen, C.H.; Wang, J.H.; Liao, H.C.; Yang, C.T.; Chen, C.W.; Lin, Y.C.; Kao, C.H.; Lu, M.J.; Liao, J.C. Analysis of Genomic Distributions of SARS-CoV-2 Reveals a Dominant Strain Type with Strong Allelic Associations. Proc. Natl. Acad. Sci. USA 2020, 117, 30679–30686. [Google Scholar] [CrossRef]
- Ip, J.D.; Kok, K.H.; Chan, W.M.; Chu, A.W.; Wu, W.L.; Yip, C.C.; To, W.K.; Tsang, O.T.; Leung, W.S.; Chik, T.S.; et al. Intra-Host Non-Synonymous Diversity at a Neutralizing Antibody Epitope of SARS-CoV-2 Spike Protein N-Terminal Domain. Clin. Microbiol. Infect. 2020, 27, 1350-e1. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-Wide Analysis of SARS-CoV-2 Virus Strains Circulating Worldwide Implicates heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.J.; Vasudevan, K.; Veeraraghavan, B.; Iyadurai, R.; Gunasekaran, K. Genomic Evolution of Severe Acute Respiratory Syndrome Coronavirus 2 in India and Vaccine Impact. Indian J. Med. Microbiol. 2020, 38, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Jary, A.; Leducq, V.; Malet, I.; Marot, S.; Klement-Frutos, E.; Teyssou, E.; Soulié, C.; Abdi, B.; Wirden, M.; Pourcher, V.; et al. Evolution of Viral Quasispecies During SARS-CoV-2 Infection. Clin. Microbiol. Infect. 2020, 26, 1560-e1. [Google Scholar] [CrossRef] [PubMed]
- Jenjaroenpun, P.; Wanchai, V.; Ono-Moore, K.D.; Laudadio, J.; James, L.P.; Adams, S.H.; Prior, F.; Nookaew, I.; Ussery, D.W.; Wongsurawat, T. Two SARS-CoV-2 Genome Sequences of Isolates from Rural U.S. Patients Harboring the D614g Mutation, Obtained Using Nanopore Sequencing. Microbiol. Resour. Announc. 2020, 10, e01109-20. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic Characterization of a Novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Kim, J.S.; Jang, J.H.; Kim, J.M.; Chung, Y.S.; Yoo, C.K.; Han, M.G. Genome-Wide Identification and Characterization of Point Mutations in the SARS-CoV-2 Genome. Osong Public Health Res. Perspect. 2020, 11, 101–111. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.H.; Lee, S.; Shim, S.; Nguyen, T.T.; Hwang, J.; Kim, H.; Choi, Y.O.; Hong, J.; Bae, S.; et al. The Progression of Sars Coronavirus 2 (SARS-CoV2): Mutation in the Receptor Binding Domain of Spike Gene. Immune Netw. 2020, 20, e41. [Google Scholar] [CrossRef]
- Koyama, T.; Platt, D.; Parida, L. Variant Analysis of SARS-CoV-2 Genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Kozlovskaya, L.; Piniaeva, A.; Ignatyev, G.; Selivanov, A.; Shishova, A.; Kovpak, A.; Gordeychuk, I.; Ivin, Y.; Berestovskaya, A.; Prokhortchouk, E.; et al. Isolation and Phylogenetic Analysis of SARS-CoV-2 Variants Collected in Russia During the COVID-19 Outbreak. Int. J. Infect. Dis. 2020, 99, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Verma, H.; Singhvi, N.; Sood, U.; Gupta, V.; Singh, M.; Kumari, R.; Hira, P.; Nagar, S.; Talwar, C.; et al. Comparative Genomic Analysis of Rapidly Evolving SARS-CoV-2 Reveals Mosaic Pattern of Phylogeographical Distribution. mSystems 2020, 5, e00505-20. [Google Scholar] [CrossRef] [PubMed]
- Laamarti, M.; Chemao-Elfihri, M.W.; Kartti, S.; Laamarti, R.; Allam, L.; Ouadghiri, M.; Smyej, I.; Rahoui, J.; Benrahma, H.; Diawara, I.; et al. Genome Sequences of Six SARS-CoV-2 Strains Isolated in Morocco, Obtained Using Oxford Nanopore Minion Technology. Microbiol. Resour. Announc. 2020, 9, e00767-20. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.S.; Ng, T.T.; Wu, A.K.; Yau, M.C.; Lao, H.Y.; Choi, M.P.; Tam, K.K.; Lee, L.K.; Wong, B.K.; Ho, A.Y.M.; et al. Territorywide Study of Early Coronavirus Disease Outbreak, Hong Kong, China. Emerg. Infect. Dis. 2021, 27, 196–204. [Google Scholar] [CrossRef]
- Ling, J.; Hickman, R.A.; Li, J.; Lu, X.; Lindahl, J.F.; Lundkvist, Å.; Järhult, J.D. Spatio-Temporal Mutational Profile Appearances of Swedish SARS-CoV-2 During the Early Pandemic. Viruses 2020, 12, 1026. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.P.; Caro-Vegas, C.; Landis, J.T.; Moorad, R.; Pluta, L.J.; Eason, A.B.; Thompson, C.; Bailey, A.; Villamor, F.C.S.; Lange, P.T.; et al. High-Density Amplicon Sequencing Identifies Community Spread and Ongoing Evolution of SARS-CoV-2 in the Southern United States. Cell Rep. 2020, 33, 108352. [Google Scholar] [CrossRef] [PubMed]
- Micheli, V.; Rimoldi, S.G.; Romeri, F.; Comandatore, F.; Mancon, A.; Gigantiello, A.; Perini, M.; Mileto, D.; Pagani, C.; Lombardi, A.; et al. Geographical Reconstruction of the SARS-CoV-2 Outbreak in Lombardy (Italy) During the Early Phase. J. Med. Virol. 2020, 93, 1752–1757. [Google Scholar] [CrossRef]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different Mutations in SARS-CoV-2 Associate with Severe and Mild Outcome. Int. J. Antimicrob. Agents 2020, 57, 106272. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 Mutation Hot Spots Include a Novel Rna-Dependent-Rna Polymerase Variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Parvez, M.S.A.; Rahman, M.M.; Morshed, M.N.; Rahman, D.; Anwar, S.; Hosen, M.J. Genetic Analysis of SARS-CoV-2 Isolates Collected from Bangladesh: Insights into the Origin, Mutational Spectrum and Possible Pathomechanism. Comput. Biol. Chem. 2020, 90, 107413. [Google Scholar] [CrossRef] [PubMed]
- Raghav, S.; Ghosh, A.; Turuk, J.; Kumar, S.; Jha, A.; Madhulika, S.; Priyadarshini, M.; Biswas, V.K.; Shyamli, P.S.; Singh, B.; et al. Analysis of Indian SARS-CoV-2 Genomes Reveals Prevalence of D614g Mutation in Spike Protein Predicting an Increase in Interaction with Tmprss2 and Virus Infectivity. Front. Microbiol. 2020, 11, 594928. [Google Scholar] [CrossRef]
- Rito, T.; Richards, M.B.; Pala, M.; Correia-Neves, M.; Soares, P.A. Phylogeography of 27,000 SARS-CoV-2 Genomes: Europe as the Major Source of the COVID-19 Pandemic. Microorganisms 2020, 8, 1678. [Google Scholar] [CrossRef] [PubMed]
- Saha, I.; Ghosh, N.; Maity, D.; Sharma, N.; Sarkar, J.P.; Mitra, K. Genome-Wide Analysis of Indian SARS-CoV-2 Genomes for the Identification of Genetic Mutation and Snp. Infect. Genet. Evol. 2020, 85, 104457. [Google Scholar] [CrossRef]
- Saha, O.; Hossain, M.S.; Rahaman, M.M. Genomic Exploration Light on Multiple Origin with Potential Parsimony-Informative Sites of the Severe Acute Respiratory Syndrome Coronavirus 2 in Bangladesh. Gene Rep. 2020, 21, 100951. [Google Scholar] [CrossRef] [PubMed]
- San, J.E.; Ngcapu, S.; Kanzi, A.M.; Tegally, H.; Fonseca, V.; Giandhari, J.; Wilkinson, E.; Nelson, C.W.; Smidt, W.; Kiran, A.M.; et al. Transmission Dynamics of SARS-CoV-2 within-Host Diversity in Two Major Hospital Outbreaks in South Africa. Virus Evol. 2021, 7, veab041. [Google Scholar] [CrossRef] [PubMed]
- Skums, P.; Kirpich, A.; Baykal, P.I.; Zelikovsky, A.; Chowell, G. Global Transmission Network of SARS-CoV-2: From Outbreak to Pandemic. medRxiv 2020. [Google Scholar] [CrossRef]
- Soliman, M.S.; AbdelFattah, M.; Aman, S.M.N.; Ibrahim, L.M.; Aziz, R.K. A Gapless, Unambiguous Rna Metagenome-Assembled Genome Sequence of a Unique SARS-CoV-2 Variant Encoding Spike S813i and Orf1a A859v Substitutions. Omics 2020, 25, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.S.; Xu, F.; An, Q.; Chen, C.; Yang, Z.N.; Lu, H.J.; Chen, J.C.; Yao, P.P.; Jiang, J.M.; Zhu, H.P. A SARS-CoV-2 Variant with the 12-Bp Deletion at E Gene. Emerg. Microbes Infect. 2020, 9, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Surleac, M.; Banica, L.; Casangiu, C.; Cotic, M.; Florea, D.; Sandulescu, O.; Milu, P.; Streinu-Cercel, A.; Vlaicu, O.; Paraskevis, D.; et al. Molecular Epidemiology Analysis of SARS-CoV-2 Strains Circulating in Romania During the First Months of the Pandemic. Life 2020, 10, 152. [Google Scholar] [CrossRef]
- Taboada, B.; Vazquez-Perez, J.A.; Muñoz-Medina, J.E.; Ramos-Cervantes, P.; Escalera-Zamudio, M.; Boukadida, C.; Sanchez-Flores, A.; Isa, P.; Mendieta-Condado, E.; Martínez-Orozco, J.A.; et al. Genomic Analysis of Early SARS-CoV-2 Variants Introduced in Mexico. J. Virol. 2020, 94, e01056-20. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 Genomic Variations Associated with Mortality Rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef]
- Velasco, J.M.; Chinnawirotpisan, P.; Joonlasak, K.; Manasatienkij, W.; Huang, A.; Valderama, M.T.; Diones, P.C.; Leonardia, S.; Timbol, M.L.; Navarro, F.C.; et al. Coding-Complete Genome Sequences of 23 SARS-CoV-2 Samples from the Philippines. Microbiol. Resour. Announc. 2020, 9, e01031-20. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614g on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G. Characterizing SARS-CoV-2 Mutations in the United States. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Hozumi, Y.; Yin, C.; Wei, G.W. Decoding Asymptomatic COVID-19 Infection and Transmission. J. Phys. Chem. Lett. 2020, 11, 10007–10015. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.W. Decoding SARS-CoV-2 Transmission and Evolution and Ramifications for COVID-19 Diagnosis, Vaccine, and Medicine. J. Chem. Inf. Model 2020, 60, 5853–5865. [Google Scholar] [CrossRef]
- Yap, P.S.X.; Tan, T.S.; Chan, Y.F.; Tee, K.K.; Kamarulzaman, A.; Teh, C.S.J. An Overview of the Genetic Variations of the SARS-CoV-2 Genomes Isolated in Southeast Asian Countries. J. Microbiol. Biotechnol. 2020, 30, 962–966. [Google Scholar] [CrossRef]
- Yuan, F.; Wang, L.; Fang, Y.; Wang, L. Global Snp Analysis of 11,183 SARS-CoV-2 Strains Reveals High Genetic Diversity. Transbound. Emerg. Dis. 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, Y.; Zhao, X.; Shi, W.; Chen, Z.; Zhang, S.; Liu, P.; Xiao, J.; Tan, W.; Wang, D.; et al. Genomic Characterization of SARS-CoV-2 Identified in a Reemerging COVID-19 Outbreak in Beijing’s Xinfadi Market in 2020. Biosaf. Health 2020, 2, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, K.; Steininger, P.; Ziegler, R.; Steinmann, J.; Korn, K.; Ensser, A. SARS-CoV-2 Samples May Escape Detection Because of a Single Point Mutation in the N Gene. Eurosurveillance 2020, 25, 2001650. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, N.S.; Pando, R.; Bucris, E.; Drori, Y.; Lustig, Y.; Erster, O.; Mor, O.; Mendelson, E.; Mandelboim, M. Comprehensive Analyses of SARS-CoV-2 Transmission in a Public Health Virology Laboratory. Viruses 2020, 12, 854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-Protein D614g Mutation Increases Virion Spike Density and Infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614g SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Study ID (Ref) | Country of Study | Period of Study | No. of Participant | No. of Mutated Cases | Mutation Detection Method | Regions of Mutation |
|---|---|---|---|---|---|---|---|
| 1 | Akter et al., 2020 [13] | Bangladesh | May–June 2020 | 3 | 3 | Whole-genome sequencing | ORF1ab, N and S gene |
| 2 | Andrés et al., 2020 [14] | Spain | March 2020 | 18 | 18 | Deep sequencing of S gene | S gene |
| 3 | Badua et al., 2020 [15] | Multiple countries | January–May 2020 | 151 | 151 | NGS | ORF1ab, ORF8, ORF3a, 5′UTR, 3′UTR, ORF6, ORF7a, ORF10, S, E, M and N gene. |
| 4 | Barret et al., 2020 [16] | USA | December 2019–May 2020 | 119 | 119 | NGS | 5′UTR, ORF1ab, S gene |
| 5 | Bartolini et al., 2020 [17] | Italy | February–March 2020 | 9 | 9 | NGS (SARS-CoV-2 panel) | ORF1ab, UTR, S, N and M gene, |
| 6 | Becerra-Flores 2020 [18] | Worldwide | March–April 2020 | NR | NR | NGS | S gene |
| 7 | Benvenuto et al., 2020 [19] | Italy | January–April 2020 | 79 | 79 | NGS | S and N gene |
| 8 | Chang et al., 2020 [20] | Multiple countries | NR | 10 | 10 | NGS | ORF1ab, ORF8, S and E gene |
| 9 | Chen et al., 2020 [21] | China | January–February 2020 | 10 | 10 | qRT-PCR on ORF1ab and N gene; RNA sequencing | ORF1ab, ORF3a, ORF8, ORF10, S and N gene |
| 10 | Cusi et al., 2020 [22] | Italy | March 2020 | 1 | 1 | Direct RNA and amplicon sequencing | S gene |
| 11 | Demİr et al., 2020 [23] | Turkey | March–May 2020 | 63 | 63 | NGS | ORF1ab, ORF3a, 3′UTR, 5′UTR, S, N and M gene |
| 12 | Devendran et al., 2021 [24] | India | as of April 2020 | 10 | 10 | NGS/WGS | ORF1ab, ORF8, S and N gene |
| 13 | Du et al., 2020 [25] | China | January–April 2020 | 102 | 102 | qRT-PCR, meta-transcriptomic sequencing | 5′UTR, ORF1ab, S, ORF3a, ORF8, N gene |
| 14 | Elizondo et al., 2020 [26] | Uruguay | March–May 2020 | 44 | 44 | qRT-PCR, NGS | ORF8, ORF3a, ORF1ab |
| 15 | Eskier et al., 2020 [27] | USA and UK | January–March 2020 | 11,701 | 11,701 | NGS | Whole genome |
| 16 | Gómez-Carballa et al., 2020 [28] | Spain | as of June 2020 | 922 | 922 | NGS | Whole genome |
| 17 | Gong et al., 2020 [29] | Taiwan | January–March 2020 | 20 | 19 | RT-PCR & WGS | ORF1ab, ORF8, ORF3a, S gene, N gene |
| 18 | Gupta 2020 [30] | Worldwide | January–April 2020 | 87 | 87 | NGS | ORF1ab, ORF3a, ORF7a, ORF8, N, S and M gene |
| 19 | Hartley et al., 2021 [31] | USA | March–June 2020 | 200 | 173 | NGS | ORF1ab, S gene |
| 20 | Hassan et al., 2020 [32] | India | as of May 2020 | 128 | 128 | NGS | ORF1, ORF3a, ORF8, ORF7a, S, M and N gene |
| 21 | Yang et al., 2020 [33] | Worldwide | December 2019–June 2020 | 46,414 | 46,414 | NGS | Whole genome |
| 22 | Ip et al., 2020 [34] | Hong Kong | January–March 2020 | 12 | 1 | Sanger sequencing, Nanopore and Illumina sequencing | S gene |
| 23 | Islam et al., 2020 [35] | Multiple countries | as of May 2020 | 444 | 404 | NGS | ORF1ab, N, E, M, S |
| 24 | Jacob et al., 2020 [36] | India | until June 2020 | >600 | NR | NGS/WGS | S gene |
| 25 | Jary et al., 2021 [37] | France | January–February 2020 | 1 | 1 | NGS | ORF3a, ORF7a, ORF6, ORF7b, ORF8, ORF10, N, M and E gene |
| 26 | Jenjaroenpun et al., 2021 [38] | USA | July 2020 | 2 | 2 | Oxford Nanopore Technologies (ONT) MinION sequencing technology | ORF1ab, ORF3a, ORF14 and S gene |
| 27 | Khailany et al., 2020 [39] | Worldwide | December 2019–April 2020 | 95 | 71 | NGS/WGS | ORF1ab, ORF8, ORF3a, ORF10, S, N and M gene |
| 28 | Kim et al., 2020 [40] | Worldwide | NR | 178 | 178 | NGS | ORF1ab, ORF3, ORF6, ORF7a, ORF7b, ORF8, ORF10 S, M, E and N gene |
| 29 | Kim et al., 2020 [41] | Korea | NR | 4 | 4 | qRT-PCR and Sanger sequencing | S gene |
| 30 | Koyama et al., 2020 [42] | Worldwide | February–May 2020 | 15,755 | 10,022 | NGS | Whole genome |
| 31 | Kozlovskaya et al., 2020 [43] | Russia | March–April 2020 | 220 | 220 | NGS | ORF1ab, S and N gene |
| 32 | Kumar et al., 2020 [44] | Multiple countries | December 2019–March 2020 | 95 | 95 | NGS/WGS | Whole genome |
| 33 | Laamarti et al., 2020 [45] | Morocco | NR | 6 | 6 | Oxford NanoporeTechnologies [ONT] | ORF1ab, S gene, 5′UTR |
| 34 | Leung et al., 2021 [46] | Hong Kong | as of February 2020 | 50 | 50 | Nanopore and NGS | ORF3a, ORF1ab, S gene |
| 35 | Ling et al., 2020 [47] | Sweden | February–May 2020 | 348 | 348 | NGS | 5′-UTR, ORF1ab, S, ORF3a, M and N gene |
| 36 | McNamara et al., 2020 [48] | USA | March–May 2020 | 175 | 175 | NGS | S and 3′UTR |
| 37 | Micheli et al., 2020 [49] | Italy | February–April 2020 | 20 | 20 | NGS | M and N gene |
| 38 | Nagy et al. 2021 [50] | Worldwide | December 2019–September 2020 | 149,061 | 149,061 | NGS | ORF1ab, ORF3a, ORF8, ORF6, N and S gene |
| 39 | Pachetti et al., 2020 [51] | Worldwide | December 2019–March 2020 | 220 | 215 | NGS/WGS | Whole genome |
| 40 | Parvez et al., 2021 [52] | Bangladesh | as of August 2020 | 311 | 311 | NGS/WGS | ORF1a, S and N gene |
| 41 | Raghav et al., 2020 [53] | India | March–June 2020 | 202 | 202 | NGS | ORF1ab, 5′-UTR, ORF3a, ORF6, ORF7b, ORF84, ORF10, M, N and S |
| 42 | Rito et al., 2020 [54] | Worldwide | May–20 | 26,869 | 20,163 | NGS/WGS | Whole genome |
| 43 | Saha et al., 2020 [55] | India | NR | 566 | 566 | NGS | 5′UTR, ORF1ab, ORF3a, S, M and N |
| 44 | Saha et al., 2020 [56] | Bangladesh | April–July 2020 | 41 | 41 | NGS | ORF1ab, ORF3a, ORF6, ORF7a, ORF8, Matrix (M gene), S and N gene |
| 45 | San et al., 2021 [57] | South Africa | March–June 2020 | 109 | 109 | NGS | ORF1ab, ORF3a, ORF6, ORF7a, ORF7b, ORF8, ORF10, S, E, M and N gene |
| 46 | Skums et al., 2020 [58] | Worldwide | NR | 319 | 274 | NGS/WGS | Whole genome |
| 47 | Soliman et al., 2021 [59] | Egypt | June 2020 | 1 | 1 | NGS | ORF1ab and S gene |
| 48 | Soratto et al., 2020 [2] | Sweden | April 2020 | 4 | 4 | NGS | ORF1ab, ORF3a, ORF7a, S and N gene |
| 49 | Sun et al., 2020 [60] | China | NR | 1 | 1 | RT-PCR | E gene |
| 50 | Surleac et al., 2020 [61] | Romania | January–February 2020 | 25 | 25 | NGS | ORF1ab, S and N gene |
| 51 | Taboada et al., 2020 [62] | Mexico | February–March 2020 | 17 | 17 | NGS | ORF1ab, ORF8 and S gene |
| 52 | Toyoshima et al., 2020 [63] | Multiple countries | As of May 2020 | 12,343 | 12,343 | NGS | ORF1ab, ORF3a, ORF8, S, N and M gene |
| 53 | Velasco et al., 2020 [64] | The Phillipines | April–July 2020 | 23 | 23 | NGS | ORF1ab, ORF6, ORF7a, OORF7b, ORF8, ORF10, S, N and M gene |
| 54 | Volz et al., 2021 [65] | UK | January–June 2020 | 26,986 | 21,231 | NGS | S gene |
| 55 | Wang et al., 2020 [66] | USA | July 2020 | 24,715 | 24,715 | NGS | ORF1ab, ORF3a, ORF8 and S gene |
| 56 | Wang et al., 2020 [67] | Multiple countries | as of October 2020 | 75,775 | 75,775 | NGS | ORF1ab |
| 57 | Wang et al., 2020 [68] | Worldwide | as of June 2020 | 15,140 | 15,140 | NGS | Whole genome |
| 58 | Yap et al., 2020 [69] | Multiple countries | January–April 2020 | 142 | 112 | NGS | ORF1ab, ORF8, S and N gene |
| 59 | Yuan et al., 2020 [70] | Worldwide | January–May 2020 | 11,183 | 11,183 | NGS | Whole genome |
| 60 | Zhang et al., 2020 [71] | China | June–July 2020 | 6 | 6 | NGS | ORF1ab gene, S and N gene |
| 61 | Ziegler et al., 2020 [72] | Germany | July 2020 | 1 | 1 | qRT-PCR, PCR & Sanger sequencing | N gene |
| 62 | Zuckerman et al., 2020 [73] | Isreal | March 2020 | 8 | 8 | qRT-PCR, NGS | 5-UTR, ORF1ab, S, ORF3a and N gene |
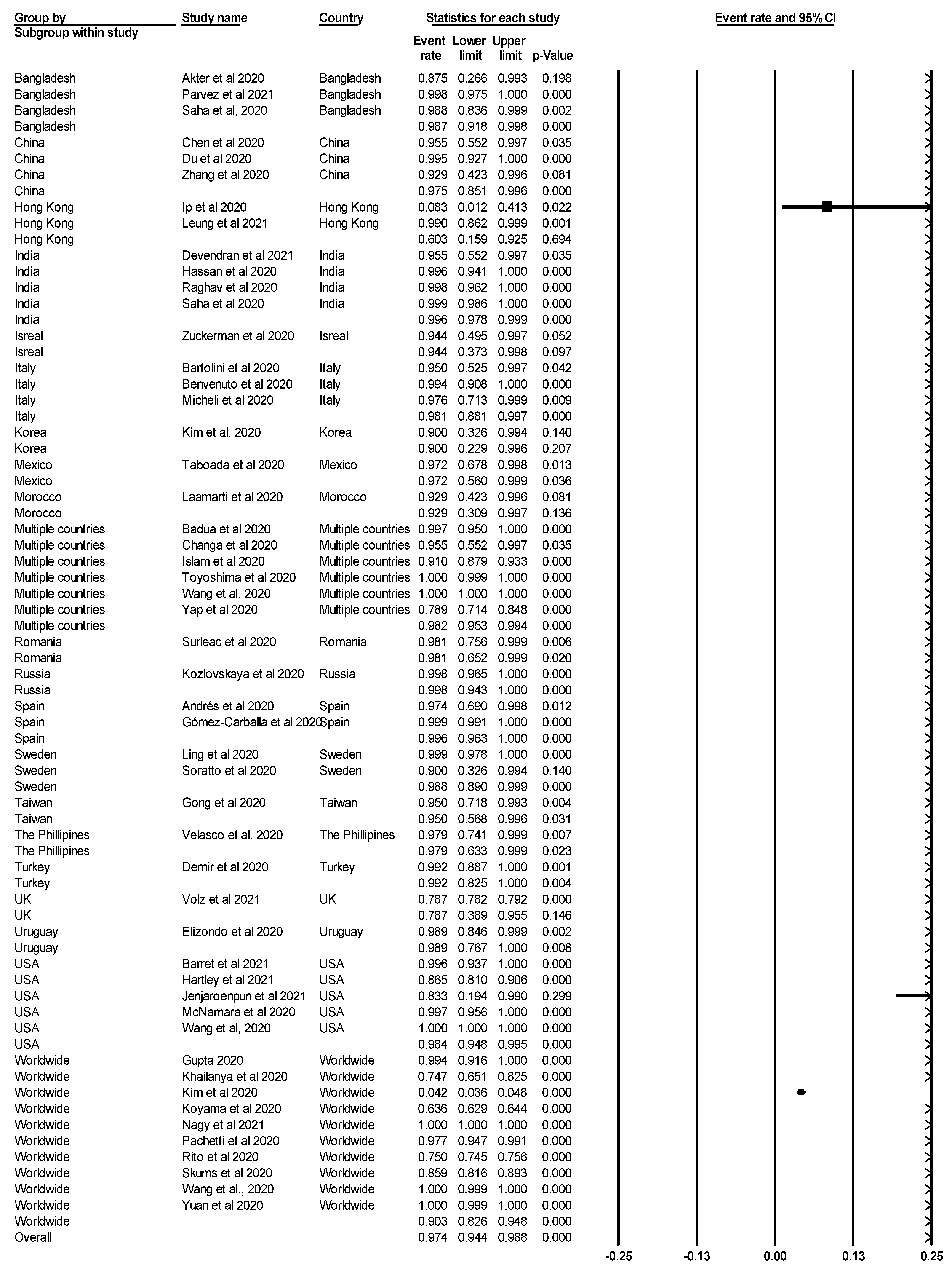
| Country of Study | Number of Studies | Prevalence (%) | 95% CI | I2 (%) | Q | Heterogeneity Test | |
|---|---|---|---|---|---|---|---|
| DF | p | ||||||
| Bangladesh | 3 | 98.7 | 91.9–99.8 | 57.445 | 4.700 | 2 | 0.095 |
| China | 3 | 97.5 | 85.1–99.6 | 5.356 | 2.113 | 2 | 0.348 |
| Hong Kong | 2 | 60.3 | 15.9–92.5 | 93.675 | 15.811 | 1 | 0.000 |
| India | 4 | 99.6 | 97.8–99.9 | 28.063 | 4.170 | 3 | 0.244 |
| Israel | 1 | 94.4 | 37.4–99.8 | - | - | - | 1.000 |
| Italy | 3 | 98.1 | 88.2–99.7 | 0.000 | 1.129 | 2 | 0.569 |
| Korea | 1 | 90.0 | 22.9–99.6 | - | - | - | 1.000 |
| Mexico | 1 | 97.2 | 56.0–99.9 | - | - | - | 1.000 |
| Morocco | 1 | 92.9 | 30.9–99.7 | - | - | - | 1.000 |
| Multiple countries | 6 | 98.2 | 95.3–99.3 | 95.168 | 103.483 | 5 | 0.000 |
| Romania | 1 | 98.1 | 65.2–99.9 | - | - | - | 1.000 |
| Russia | 1 | 99.8 | 94.3–100.0 | - | - | - | 1.000 |
| Spain | 2 | 99.6 | 96.3–100.0 | 73.466 | 3.769 | 1 | 0.052 |
| Sweden | 2 | 98.8 | 89.0–99.9 | 77.667 | 4.478 | 1 | 0.034 |
| Taiwan | 1 | 95.0 | 56.8–99.6 | - | - | - | 1.000 |
| The Philippines | 1 | 97.9 | 63.3–99.9 | - | - | - | 1.000 |
| Turkey | 1 | 99.2 | 82.5–100.0 | - | - | - | 1.000 |
| UK | 1 | 78.7 | 78.2–79.2 | - | - | - | 1.000 |
| Uruguay | 1 | 98.9 | 76.7–100.0 | - | - | - | 1.000 |
| USA | 5 | 98.4 | 94.8–99.5 | 92.301 | 51.954 | 4 | 0.000 |
| Worldwide | 10 | 90.3 | 82.6–94.8 | 99.747 | 3553.894 | 9 | 0.000 |
| Total | 51 | 97.4 | 94.4–98.8 | 98.952 | 4772.621 | 50 | 0.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yusof, W.; Irekeola, A.A.; Wada, Y.; Engku Abd Rahman, E.N.S.; Ahmed, N.; Musa, N.; Khalid, M.F.; Rahman, Z.A.; Hassan, R.; Yusof, N.Y.; et al. A Global Mutational Profile of SARS-CoV-2: A Systematic Review and Meta-Analysis of 368,316 COVID-19 Patients. Life 2021, 11, 1224. https://doi.org/10.3390/life11111224
Yusof W, Irekeola AA, Wada Y, Engku Abd Rahman ENS, Ahmed N, Musa N, Khalid MF, Rahman ZA, Hassan R, Yusof NY, et al. A Global Mutational Profile of SARS-CoV-2: A Systematic Review and Meta-Analysis of 368,316 COVID-19 Patients. Life. 2021; 11(11):1224. https://doi.org/10.3390/life11111224
Chicago/Turabian StyleYusof, Wardah, Ahmad Adebayo Irekeola, Yusuf Wada, Engku Nur Syafirah Engku Abd Rahman, Naveed Ahmed, Nurfadhlina Musa, Muhammad Fazli Khalid, Zaidah Abdul Rahman, Rosline Hassan, Nik Yusnoraini Yusof, and et al. 2021. "A Global Mutational Profile of SARS-CoV-2: A Systematic Review and Meta-Analysis of 368,316 COVID-19 Patients" Life 11, no. 11: 1224. https://doi.org/10.3390/life11111224