
Cloning, Expression and Characterization of UDP-Glucose Dehydrogenases




Abstract
:1. Introduction
2. Materials and Methods
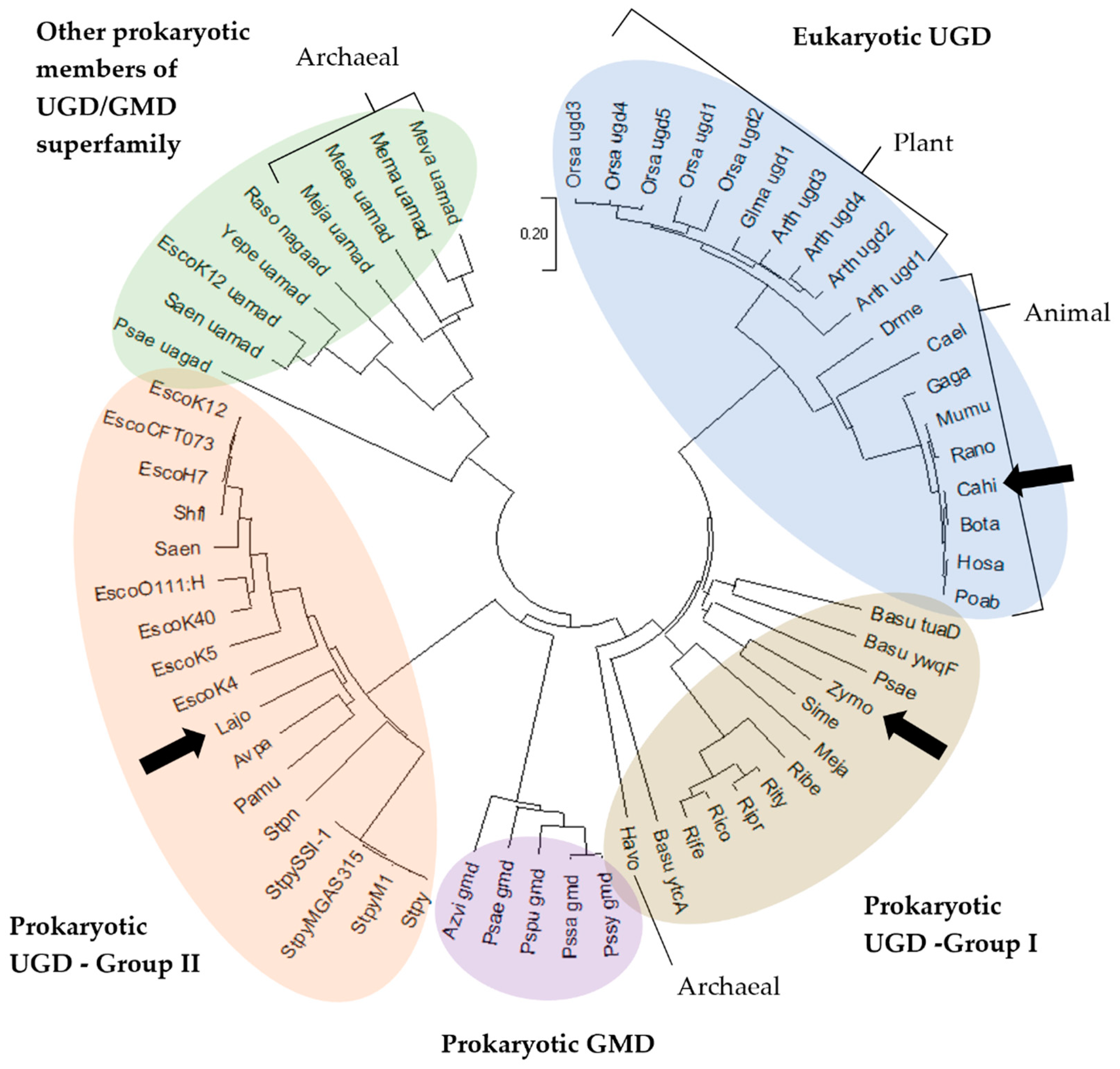
2.1. Multiple Sequence Alignment and Evolutionary Relationships
2.2. Strains and Plasmids
2.3. Gene Sources and Cloning Strategy
2.4. Culture Conditions
2.5. Preparation of E. coli and S. cerevisiae Protein Extracts
2.6. Purification of UGD
2.7. UGD In Vitro Assays
2.8. Analysis and Quantification of UDP-Sugars
2.9. Statistical Analysis
3. Results
3.1. Sequences and Phylogenetic Analysis
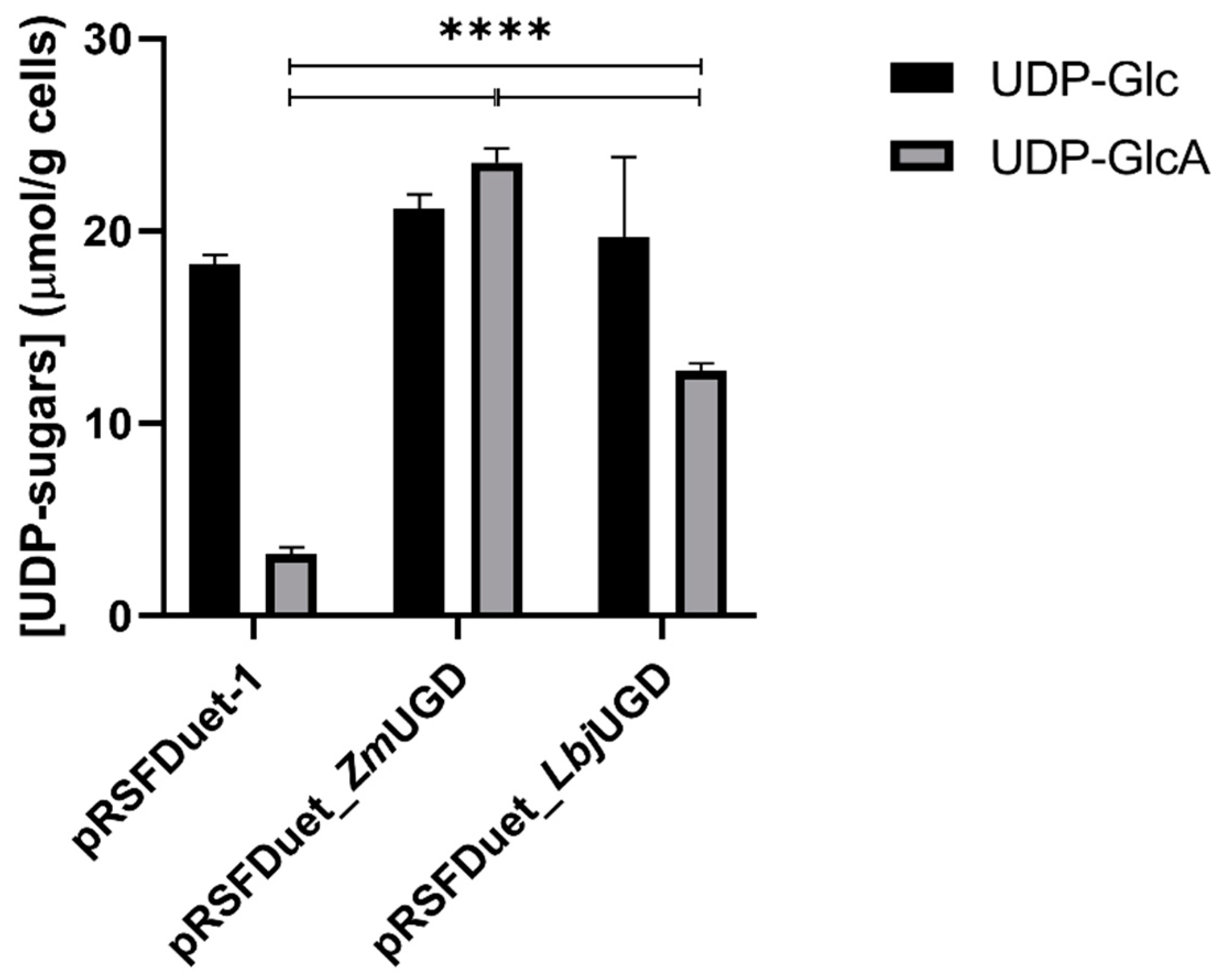
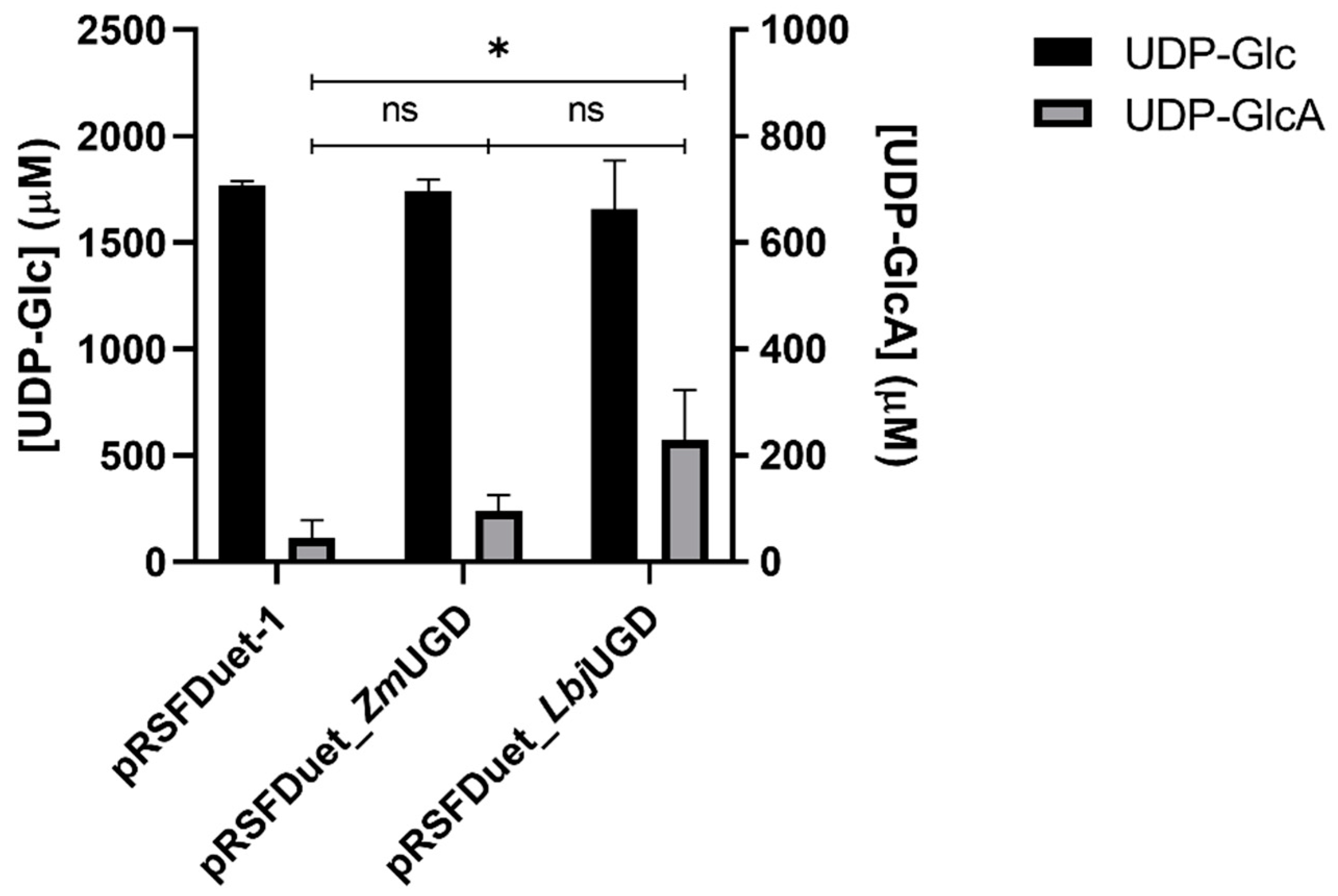
3.2. E. coli as UGD Expression Host
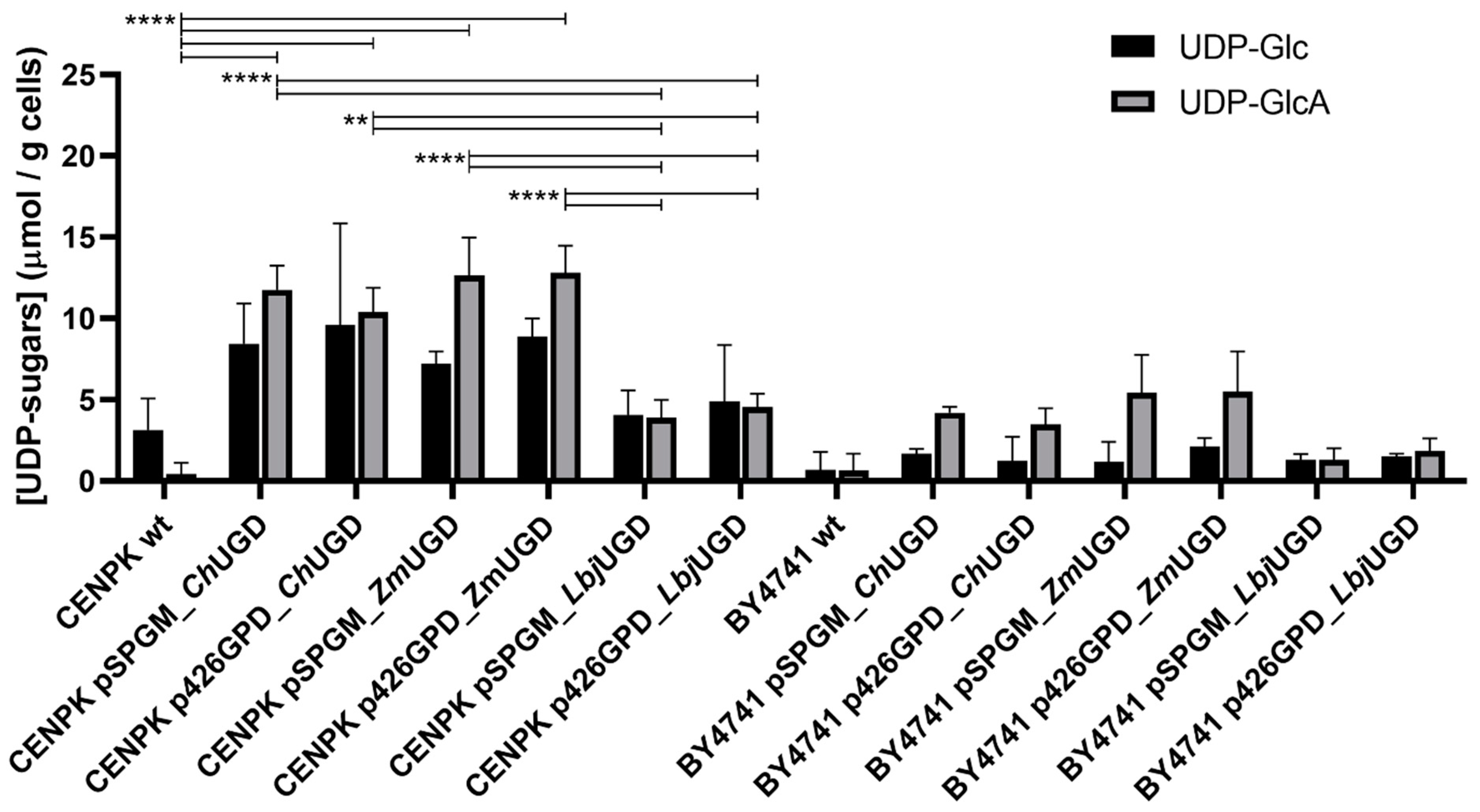
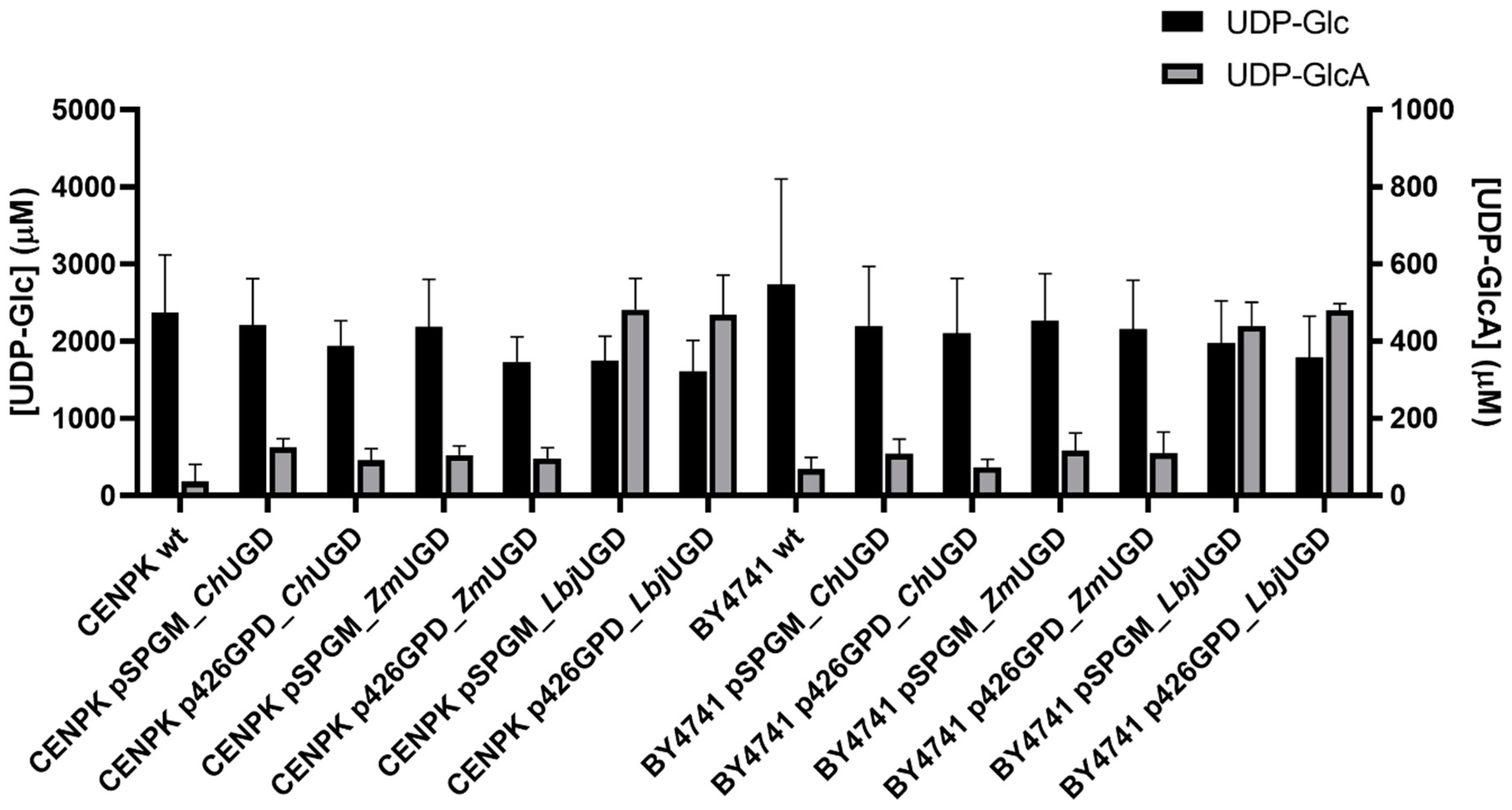
3.3. Three Novel UGDs Expressed in Yeasts
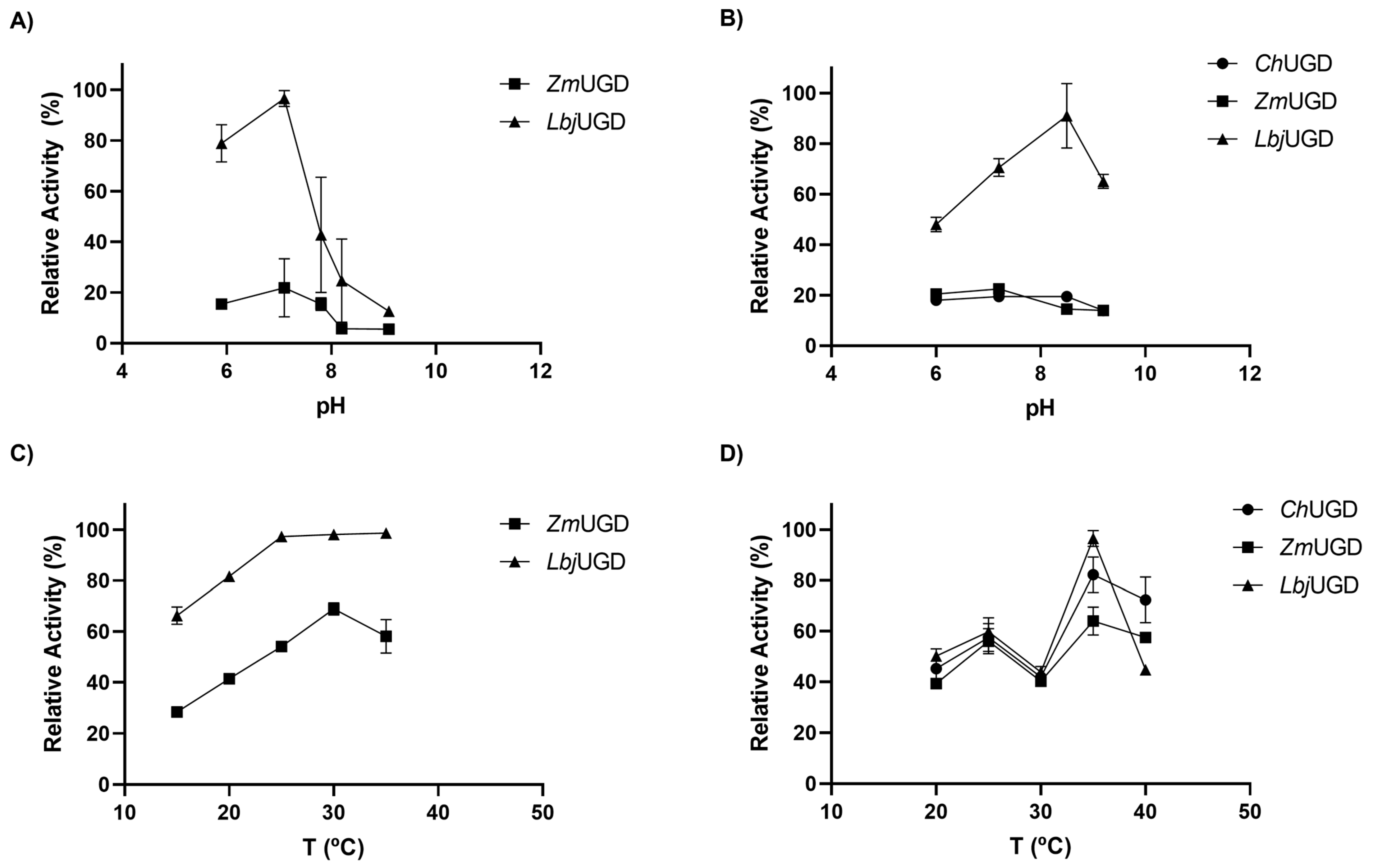
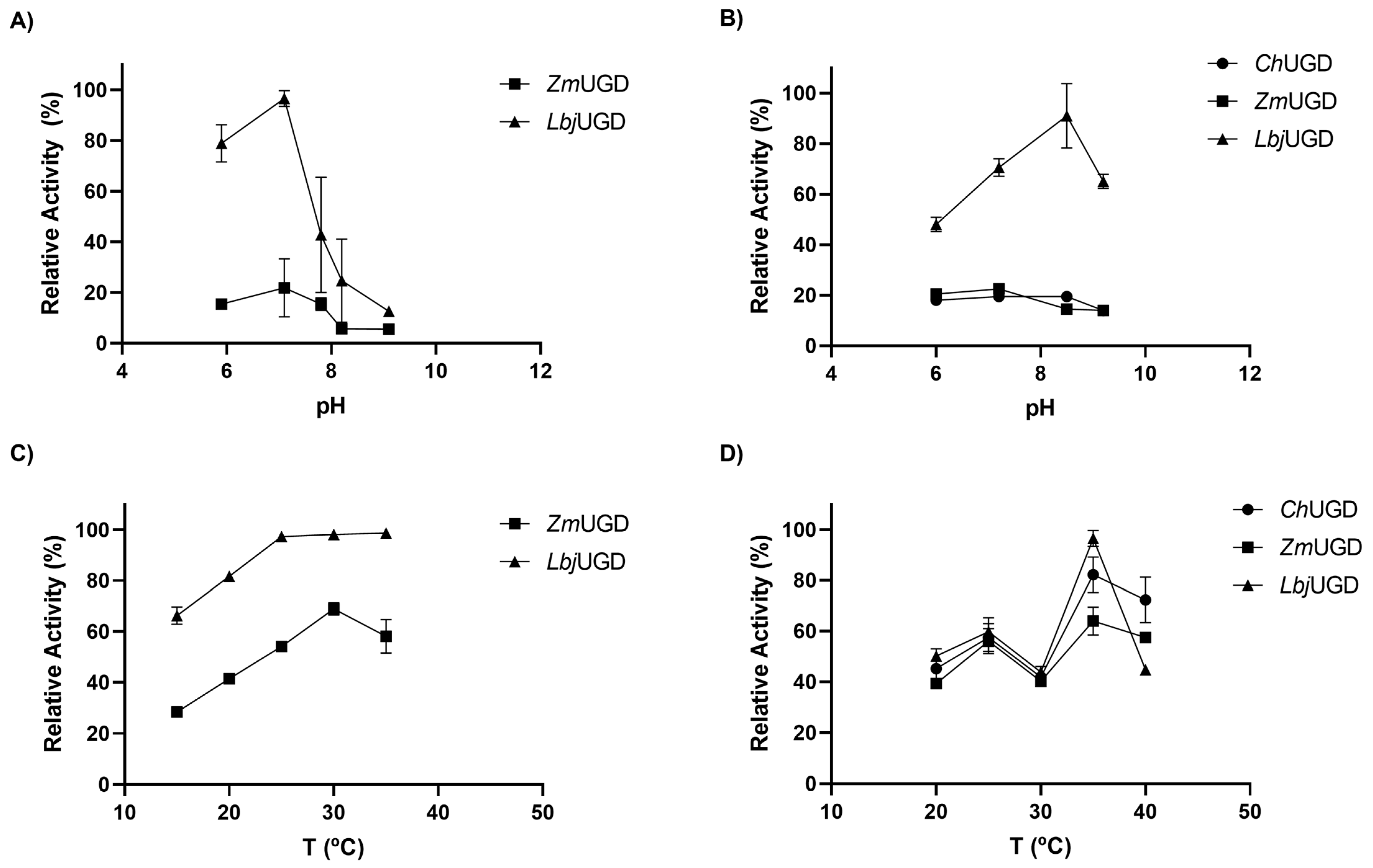
3.4. Biochemical Characterization of Cloned UGDs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Q.; Yao, R.; Chen, X.; Liu, L.; Xu, S.; Chen, J.; Wu, J. Enhancing fructosylated chondroitin production in Escherichia coli K4 by balancing the UDP-precursors. Metab. Eng. 2018, 47, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, L.; Huang, H.; Linhardt, R.J. Chemoenzymatic Synthesis of Glycosaminoglycans. Acc. Chem. Res. 2020, 53, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Badri, A.; Williams, A.; Linhardt, R.J.; Koffas, M.A. The road to animal-free glycosaminoglycan production: Current efforts and bottlenecks. Curr. Opin. Biotechnol. 2018, 53, 85–92. [Google Scholar] [CrossRef]
- Kang, Z.; Zhou, Z.; Wang, Y.; Huang, H.; Du, G.; Chen, J. Bio-Based Strategies for Producing Glycosaminoglycans and Their Oligosaccharides. Trends Biotechnol. 2018, 36, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Restaino, O.F.; Schiraldi, C. Microbial production and metabolic engineering of chondroitin and chondroitin sulfate. Emerg. Top. Life Sci. 2018, 2, 349–361. [Google Scholar] [CrossRef]
- Zakeri, A.; Rasaee, M.J.; Pourzardosht, N. Enhanced hyluronic acid production in Streptococcus zooepidemicus by over expressing HasA and molecular weight control with Niscin and glucose. Biotechnol. Rep. 2017, 16, 65–70. [Google Scholar] [CrossRef]
- Chu, X.; Han, J.; Guo, D.; Fu, Z.; Liu, W.; Tao, Y. Characterization of UDP-glucose dehydrogenase from Pasteurella multocida CVCC 408 and its application in hyaluronic acid biosynthesis. Enzyme Microb. Technol. 2016, 85, 64–70. [Google Scholar] [CrossRef]
- Restaino, O.F.; di Lauro, I.; Di Nuzzo, R.; De Rosa, M.; Schiraldi, C. New insight into chondroitin and heparosan-like capsular polysaccharide synthesis by profiling of the nucleotide sugar precursors. Biosci. Rep. 2017, 37, BSR20160548. [Google Scholar] [CrossRef] [Green Version]
- DeAngelis, P.L.; Gunay, N.S.; Toida, T.; Mao, W.; Linhardt, R.J. Identification of the capsular polysaccharides of Type D and F Pasteurella multocida as unmodified heparin and chondroitin, respectively. Carbohydr. Res. 2002, 337, 1547–1552. [Google Scholar] [CrossRef]
- Wu, J.-R.; Chen, P.-Y.; Shien, J.-H.; Shyu, C.-L.; Shieh, H.K.; Chang, F.; Chang, P.-C. Analysis of the biosynthesis genes and chemical components of the capsule of Avibacterium paragallinarum. Vet. Microbiol. 2010, 145, 90–99. [Google Scholar] [CrossRef]
- Schiraldi, C.; Cimini, D.; De Rosa, M. Production of chondroitin sulfate and chondroitin. Appl. Microbiol. Biotechnol. 2010, 87, 1209–1220. [Google Scholar] [CrossRef]
- He, W.; Fu, L.; Li, G.; Jones, J.A.; Linhardt, R.J.; Koffas, M. Production of chondroitin in metabolically engineered E. coli. Metab. Eng. 2015, 27, 92–100. [Google Scholar] [CrossRef]
- Jin, P.; Zhang, L.; Yuan, P.; Kang, Z.; Du, G.; Chen, J. Efficient biosynthesis of polysaccharides chondroitin and heparosan by metabolically engineered Bacillus subtilis. Carbohydr. Polym. 2016, 140, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, L.; Teng, L.; Chen, J.; Liu, J.; Li, J.; Du, G.; Chen, J. Metabolic engineering of Escherichia coli BL21 for biosynthesis of heparosan, a bioengineered heparin precursor. Metab. Eng. 2012, 14, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Gong, Q.; Yu, H.; Stephanopoulos, G. High-titer biosynthesis of hyaluronic acid by recombinant Corynebacterium glutamicum. Biotechnol. J. 2016, 11, 574–584. [Google Scholar] [CrossRef]
- Yu, H.; Stephanopoulos, G. Metabolic engineering of Escherichia coli for biosynthesis of hyaluronic acid. Metab. Eng. 2008, 10, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.Z.; Ling, P.X.; Zhu, X.Q.; Guo, X.P.; Zhang, T.M.; He, Y.L.; Wang, F.S. Use of induction promoters to regulate hyaluronan synthase and UDP-glucose-6-dehydrogenase of Streptococcus zooepidemicus expression in Lactococcus lactis: A case study of the regulation mechanism of hyaluronic acid polymer. J. Appl. Microbiol. 2009, 107, 136–144. [Google Scholar] [CrossRef]
- Jin, P.; Kang, Z.; Yuan, P.; Du, G.; Chen, J. Production of specific-molecular-weight hyaluronan by metabolically engineered Bacillus subtilis 168. Metab. Eng. 2016, 35, 21–30. [Google Scholar] [CrossRef]
- Badri, A.; Williams, A.; Awofiranye, A.; Datta, P.; Xia, K.; He, W.; Fraser, K.; Dordick, J.S.; Linhardt, R.J.; Koffas, M.A.G. Complete biosynthesis of a sulfated chondroitin in Escherichia coli. Nat. Commun. 2021, 12, 1389. [Google Scholar] [CrossRef]
- Cimini, D.; De Rosa, M.; Viggiani, A.; Restaino, O.F.; Carlino, E.; Schiraldi, C. Improved fructosylated chondroitin production by kfoC overexpression in E. coli K4. J. Biotechnol. 2010, 150, 324–331. [Google Scholar] [CrossRef]
- Strominger, J.L.; Kalckar, H.M.; Axelrod, J.; Maxwell, E.S. Enzymatic oxidation of uridine diphosphate glucose to uridine diphosphate glucuronic acid. J. Am. Chem. Soc. 1954, 76, 6411–6412. [Google Scholar] [CrossRef]
- Rodionova, I.A.; Zhang, Z.; Aboulwafa, M.; Saier, J.M.H. UDP-glucose dehydrogenase Ugd in E. coli is activated by Gmd and RffD, is inhibited by CheY, and regulates swarming. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Zhang, J. Evolutionary Distance: Estimation. In Encyclopedia of Life Sciences (eLS); John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; pp. 1–4. [Google Scholar]
- Entian, K.-D.; Kötter, P. 25 Yeast Genetic Strain and Plasmid Collections. In Methods in Microbiology; Stansfield, I., Stark, M.J.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 36, pp. 629–666. ISBN 0123694787. [Google Scholar]
- Brachmann, C.B.; Davies, A.; Cost, G.J.; Caputo, E.; Li, J.; Hieter, P.; Boeke, J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 1998, 14, 115–132. [Google Scholar] [CrossRef]
- Chen, Y.; Partow, S.; Scalcinati, G.; Siewers, V.; Nielsen, J. Enhancing the copy number of episomal plasmids in Saccharomyces cerevisiae for improved protein production. FEMS Yeast Res. 2012, 12, 598–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gietz, R.D.; Woods, R.A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. In Methods in Enzymology; Guthrie, C., Fink, G.R., Eds.; Academic Press: Cambridge, MA, USA, 2002; Volume 350, pp. 87–96. ISBN 9780121822538. [Google Scholar]
- Waśko, A.; Kieliszek, M.; Targoński, Z. Purification and characterization of a proteinase from the probiotic Lactobacillus rhamnosus OXY. Prep. Biochem. Biotechnol. 2012, 42, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.; Bhattacharyya, D. UDP-glucose dehydrogenase from Capra hircus liver: Purification, partial characterization and evaluation as a coupling enzyme in UDP-galactose 4-epimerase assay. J. Mol. Catal. B Enzym. 2011, 68, 37–43. [Google Scholar] [CrossRef]
- Silbir, S.; Dagbagli, S.; Yegin, S.; Baysal, T.; Goksungur, Y. Levan production by Zymomonas mobilis in batch and continuous fermentation systems. Carbohydr. Polym. 2014, 99, 454–461. [Google Scholar] [CrossRef]
- Jofré, E.; Becker, A. Production of Succinoglycan Polymer in Sinorhizobium meliloti Is Affected by SMb21506 and Requires the N-terminal Domain of ExoP. Mol. Plant-Microbe Interact. 2009, 22, 1656–1668. [Google Scholar] [CrossRef] [Green Version]
- Marvasi, M.; Visscher, P.T.; Casillas Martinez, L. Exopolymeric substances (EPS) from Bacillus subtilis: Polymers and genes encoding their synthesis. FEMS Microbiol. Lett. 2010, 313, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bhavsar, A.P.; Erdman, L.K.; Schertzer, J.W.; Brown, E.D. Teichoic Acid Is an Essential Polymer in Bacillus subtilis. J. Bacteriol. 2004, 186, 7865–7873. [Google Scholar] [CrossRef] [Green Version]
- Valentine, M.E.; Kirby, B.D.; Withers, T.R.; Johnson, S.L.; Long, T.E.; Hao, Y.; Lam, J.S.; Niles, R.M.; Yu, H.D. Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate. Microb. Biotechnol. 2020, 13, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Górska-Frączek, S.; Sandström, C.; Kenne, L.; Paściak, M.; Brzozowska, E.; Strus, M.; Heczko, P.; Gamian, A. The structure and immunoreactivity of exopolysaccharide isolated from Lactobacillus johnsonii strain 151. Carbohydr. Res. 2013, 378, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Kralj, S.; van der Maarel, M.J.E.C.; Dijkhuizen, L. The Probiotic Lactobacillus johnsonii NCC 533 Produces High-Molecular-Mass Inulin from Sucrose by Using an Inulosucrase Enzyme. Appl. Environ. Microbiol. 2008, 74, 3426–3433. [Google Scholar] [CrossRef] [Green Version]
- Wessels, M.R. Capsular Polysaccharide of Group A Streptococcus. Microbiol. Spectr. 2019, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, Q.; Luo, H.; Liang, K.; Han, Y.; Roland, K.L.; Curtiss, R.; Kong, Q. Bi-valent polysaccharides of Vi capsular and O9 O-antigen in attenuated Salmonella Typhimurium induce strong immune responses against these two antigens. NPJ Vaccines 2018, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, C. Biosynthesis and Assembly of Capsular Polysaccharides in Escherichia coli. Annu. Rev. Biochem. 2006, 75, 39–68. [Google Scholar] [CrossRef]
- Stenutz, R.; Weintraub, A.; Widmalm, G. The structures of Escherichia coli O-polysaccharide antigens. FEMS Microbiol. Rev. 2006, 30, 382–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granja, A.T.; Popescu, A.; Marques, A.R.; Sá-Correia, I.; Fialho, A.M. Biochemical characterization and phylogenetic analysis of UDP-glucose dehydrogenase from the gellan gum producer Sphingomonas elodea ATCC 31461. Appl. Microbiol. Biotechnol. 2007, 76, 1319–1327. [Google Scholar] [CrossRef]
- Klinghammer, M.; Tenhaken, R. Genome-wide analysis of the UDP-glucose dehydrogenase gene family in Arabidopsis, a key enzyme for matrix polysaccharides in cell walls. J. Exp. Bot. 2007, 58, 3609–3621. [Google Scholar] [CrossRef] [PubMed]
- Tenhaken, R.; Thulke, O. Cloning of an Enzyme That Synthesizes a Key Nucleotide-Sugar Precursor of Hemicellulose Biosynthesis from Soybean: UDP-Glucose Dehydrogenase. Plant Physiol. 1996, 112, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Bindschedler, L.V.; Wheatley, E.; Gay, E.; Cole, J.; Cottage, A.; Bolwell, G.P. Characterisation and expression of the pathway from UDP-glucose to UDP-xylose in differentiating tobacco tissue. Plant Mol. Biol. 2005, 57, 285–301. [Google Scholar] [CrossRef]
- Zhou, T.; Song, Y.C.; Feng, M.Q.; Tan, R.X. Cloning, expression and biochemical characterization of UDP-glucose 6-dehydrogenase, a key enzyme in the biosynthesis of an anti-tumor polysaccharide from the marine fungus Phoma herbarum YS4108. Process Biochem. 2011, 46, 2263–2268. [Google Scholar] [CrossRef]
- Robichon, C.; Luo, J.; Causey, T.B.; Benner, J.S.; Samuelson, J.C. Engineering Escherichia coli BL21(DE3) Derivative Strains To Minimize E. coli Protein Contamination after Purification by Immobilized Metal Affinity Chromatography. Appl. Environ. Microbiol. 2011, 77, 4634–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morán-Zorzano, M.T.; Viale, A.M.; Muñoz, F.J.; Alonso-Casajús, N.; Eydallín, G.G.; Zugasti, B.; Baroja-Fernández, E.; Pozueta-Romero, J. Escherichia coli AspP activity is enhanced by macromolecular crowding and by both glucose-1,6-bisphosphate and nucleotide-sugars. FEBS Lett. 2007, 581, 1035–1040. [Google Scholar] [CrossRef] [Green Version]
- Cimini, D.; Carlino, E.; Giovane, A.; Argenzio, O.; Dello Iacono, I.; De Rosa, M.; Schiraldi, C. Engineering a branch of the UDP-precursor biosynthesis pathway enhances the production of capsular polysaccharide in Escherichia coli O5:K4:H4. Biotechnol. J. 2015, 10, 1307–1315. [Google Scholar] [CrossRef]
- Chen, W.Y.; Marcellin, E.; Hung, J.; Nielsen, L.K. Hyaluronan Molecular Weight Is Controlled by UDP-N-acetylglucosamine Concentration in Streptococcus zooepidemicus. J. Biol. Chem. 2009, 284, 18007–18014. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Zhou, H.; Zhou, J.; Chen, J. Promoter-Library-Based Pathway Optimization for Efficient (2S)-Naringenin Production from p-Coumaric Acid in Saccharomyces cerevisiae. J. Agric. Food Chem. 2020, 68, 6884–6891. [Google Scholar] [CrossRef]
- Partow, S.; Siewers, V.; Bjørn, S.; Nielsen, J.; Maury, J. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast 2010, 27, 955–964. [Google Scholar] [CrossRef]
- Sun, J.; Shao, Z.; Zhao, H.; Nair, N.; Wen, F.; Xu, J.-H.; Zhao, H. Cloning and characterization of a panel of constitutive promoters for applications in pathway engineering in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2012, 109, 2082–2092. [Google Scholar] [CrossRef]
- Xiong, L.; Zeng, Y.; Tang, R.-Q.; Alper, H.S.; Bai, F.-W.; Zhao, X.-Q. Condition-specific promoter activities in Saccharomyces cerevisiae. Microb. Cell Fact. 2018, 17, 58. [Google Scholar] [CrossRef]
- Oka, T.; Jigami, Y. Reconstruction of de novo pathway for synthesis of UDP-glucuronic acid and UDP-xylose from intrinsic UDP-glucose in Saccharomyces cerevisiae. FEBS J. 2006, 273, 2645–2657. [Google Scholar] [CrossRef]
- Isogai, S.; Okahashi, N.; Asama, R.; Nakamura, T.; Hasunuma, T.; Matsuda, F.; Ishii, J.; Kondo, A. Synthetic production of prenylated naringenins in yeast using promiscuous microbial prenyltransferases. Metab. Eng. Commun. 2021, 12, e00169. [Google Scholar] [CrossRef] [PubMed]
- Ankel, H.; Ankel, E.; Feingold, D.S. Biosynthesis of Uridine Diphosphate D-Xylose. III. Uridine Diphosphate D-Glucose Dehydrogenase of Cryptococcus laurentii. Biochemistry 1966, 5, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Hinterberg, B.; Klos, C.; Tenhaken, R. Recombinant UDP-glucose dehydrogenase from soybean. Plant Physiol. Biochem. 2002, 40, 1011–1017. [Google Scholar] [CrossRef]
- Turner, W.; Botha, F.C. Purification and kinetic properties of UDP-glucose dehydrogenase from sugarcane. Arch. Biochem. Biophys. 2002, 407, 209–216. [Google Scholar] [CrossRef]
- Wei, S.; Kulinich, A.; Duan, X.; Liu, L.; Voglmeir, J. Discovery and Biochemical Characterization of UDP-Glucose Dehydrogenase from Granulibacter bethesdensis. Protein Pept. Lett. 2015, 22, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Zhang, X.-Y.; Sun, Y.; Conway, L.P.; Liu, L. Discovery and Biochemical Characterization of UDP-Glucose Dehydrogenase from Akkermansia muciniphila. Protein Pept. Lett. 2017, 24, 735–741. [Google Scholar] [CrossRef]
- Rocha, J.; Popescu, A.O.; Borges, P.; Mil-Homens, D.; Moreira, L.M.; Sá-Correia, I.; Fialho, A.M.; Frazão, C. Structure of Burkholderia cepacia UDP-Glucose Dehydrogenase (UGD) BceC and Role of Tyr10 in Final Hydrolysis of UGD Thioester Intermediate. J. Bacteriol. 2011, 193, 3978–3987. [Google Scholar] [CrossRef] [Green Version]
- Broach, B.; Gu, X.; Bar-Peled, M. Biosynthesis of UDP-glucuronic acid and UDP-galacturonic acid in Bacillus cereus subsp. cytotoxis NVH 391-98. FEBS J. 2012, 279, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, A.S.; Thelen, A.M.; Barycki, J.J.; Simpson, M.A. UDP-glucose dehydrogenase activity and optimal downstream cellular function require dynamic reorganization at the dimer-dimer subunit interfaces. J. Biol. Chem. 2013, 288, 35049–35057. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Relevant Genotype | Source |
|---|---|---|
| Zymomonas mobilis subsp. mobilis ATCC 29191 | Wild type | ATCC 29191 |
| Lactobacillus johnsonii ATCC 11506 | Wild type | ATCC 11506 |
| Escherichia coli NZY5α | fhuA2 Δ(argF−lacZ)U169 phoA glnV44Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | NZYTech (MB00401) |
| E. coli BL21 (DE3) | F− ompT gal dcm lon hsdSB(rB- mB-) λ(DE3 *lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | NZYTech (MB006) |
| Saccharomyces cerevisiae CEN.PK2-1C | MATa ura3-52 his3Δ1 leu2-3, 112 trp1-289 MAL2-8c SUC2 | Euroscarf 30000A [26] |
| S. cerevisiae BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Euroscarf Y00000 [27] |
| Plasmids | Description | Source |
| pGEM®-T Easy | ColE1/pMB1/pBR322/pUC ori, lacZ, AmpR | Promega |
| pRSFDuet-1 | RSF1030 ori, lacI, double PT7lac, KanR | Novagen |
| pGEM_LbjUGD | pGEM®-T Easy harboring L. johnsonii UGD (LbjUGD) gene | This study |
| pRSFDuet_ZmUGD | pRSFDuet-1 carrying Z. mobilis UGD (ZmUGD) gene | This study |
| pRSFDuet_LbjUGD | pRSFDuet-1 carrying LbjUGD gene | This study |
| pUC57_ChUGD | pMB1 ori, AmpR; pUC57 carrying Capra hircus UGD (ChUGD) gene codon-optimized for S. cerevisiae | NZYTech |
| pSP-GM1 | pUC ori, AmpR, 2 µ ori, URA3 PTEF1 PPGK1 | Addgene #64739 [28] |
| p426GPD | pUC ori, AmpR, 2 µ ori, URA3 PGPD | ATCC 87361 |
| pSP-GM1_ZmUGD | pSP-GM1 carrying ZmUGD gene | This study |
| p426GPD_ZmUGD | p426GPD carrying ZmUGD gene | This study |
| pSP-GM1_LbjUGD | pSP-GM1 carrying LbjUGD gene | This study |
| p426GPD_LbjUGD | p426GPD carrying LbjUGD gene | This study |
| pSP-GM1_ChUGD | pSP-GM1 carrying ChUGD gene | This study |
| p426GPD_ChUGD | p426GPD carrying ChUGD gene | This study |
| Expression System | Enzyme Expressed | Vmax (μmol·min−1·gprotein−1) | Km (µM) | Vmax/Km (L·min−1·gprotein−1) |
|---|---|---|---|---|
| Escherichia coli | ZmUGD | 81 | 2586 | 3.1 × 10−2 |
| LbjUGD | 66 | 1939 | 3.4 × 10−2 | |
| Saccharomyces cerevisiae | ChUGD | 2.2 | 841 | 2.6 × 10−3 |
| ZmUGD | 3.7 | 1234 | 3.0 × 10−3 | |
| LbjUGD | 1.4 | 314 | 4.5 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couto, M.R.; Rodrigues, J.L.; Rodrigues, L.R. Cloning, Expression and Characterization of UDP-Glucose Dehydrogenases. Life 2021, 11, 1201. https://doi.org/10.3390/life11111201
Couto MR, Rodrigues JL, Rodrigues LR. Cloning, Expression and Characterization of UDP-Glucose Dehydrogenases. Life. 2021; 11(11):1201. https://doi.org/10.3390/life11111201
Chicago/Turabian StyleCouto, Márcia R., Joana L. Rodrigues, and Lígia R. Rodrigues. 2021. "Cloning, Expression and Characterization of UDP-Glucose Dehydrogenases" Life 11, no. 11: 1201. https://doi.org/10.3390/life11111201