
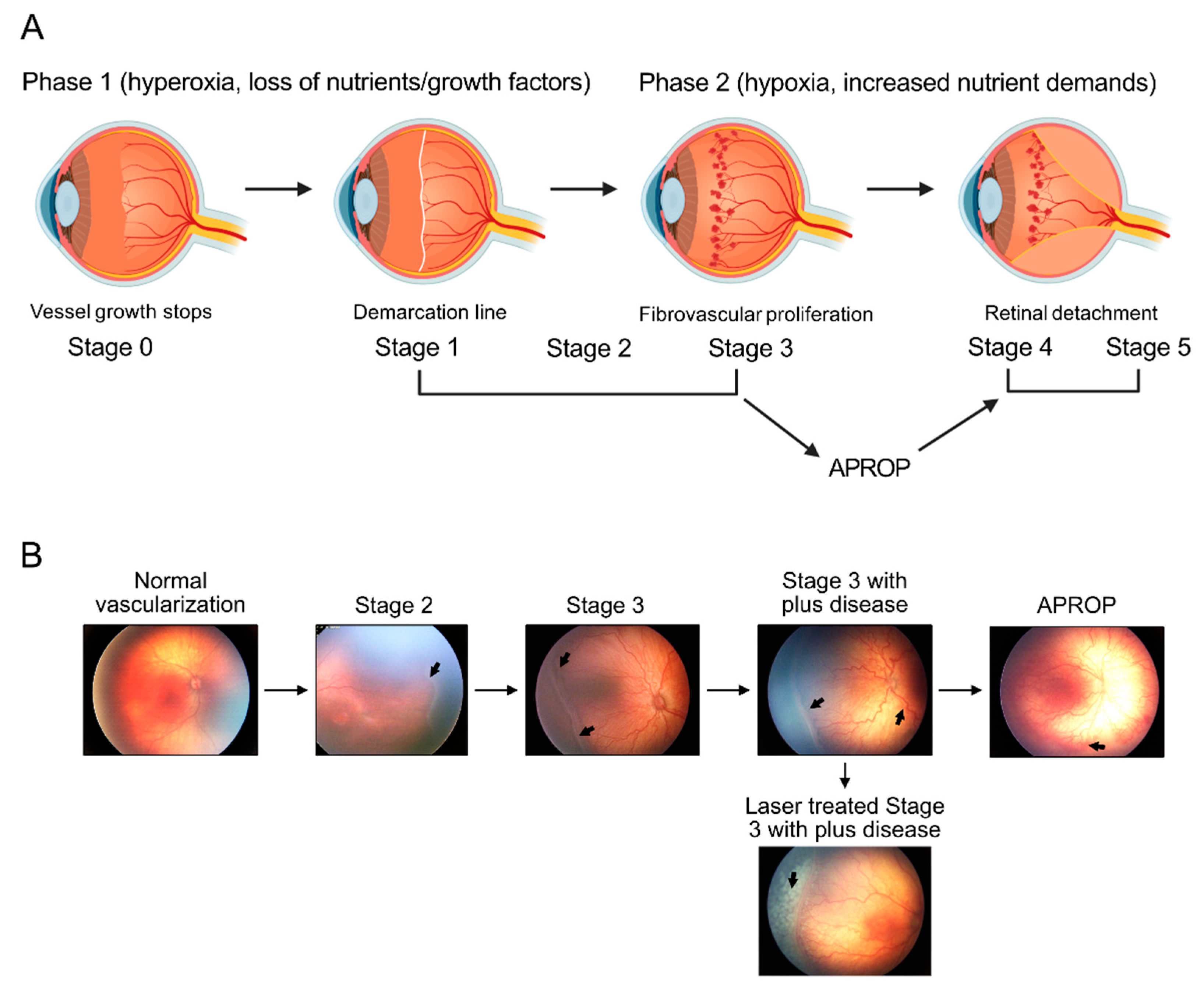
Metabolism in Retinopathy of Prematurity

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Investigations of Metabolic Changes in ROP
2.1. Lipidomics
2.2. Proteomics
2.3. Metabolomics
3. Experimental Investigations of Retinal Metabolism in ROP
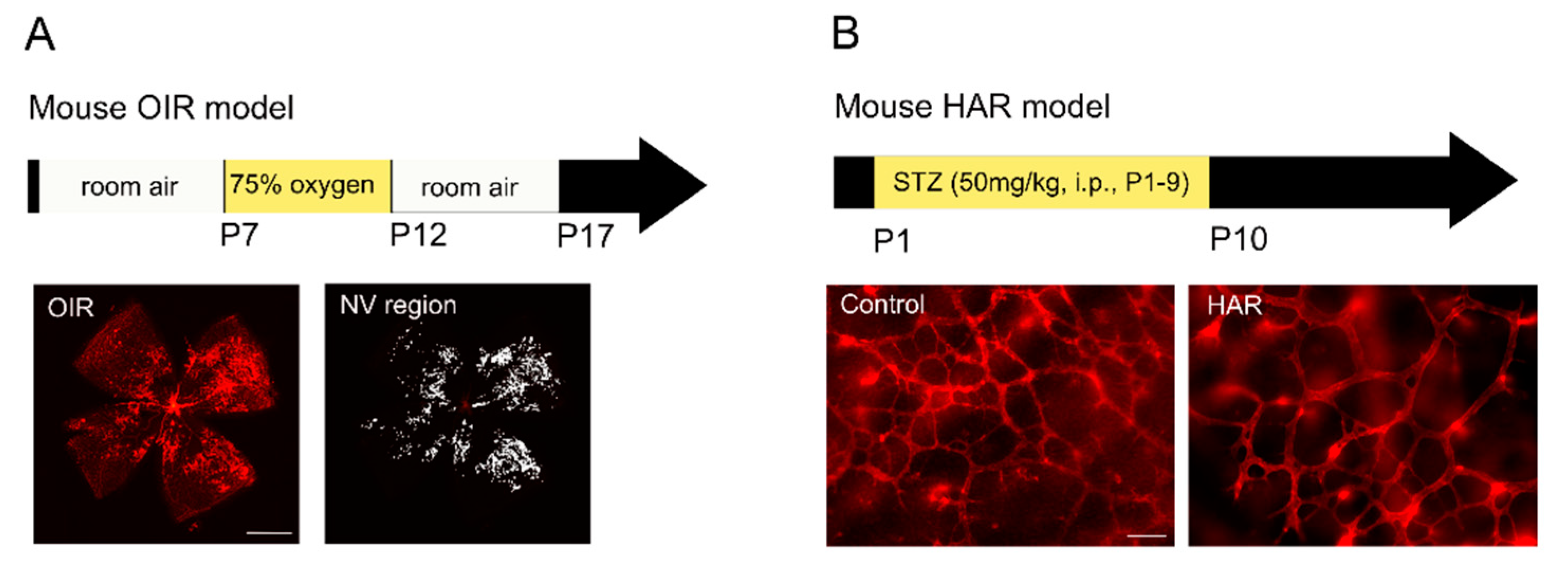
3.1. Oxygen-Induced Retinopathy (OIR)
3.2. Hyperglycemia-Associated Retinopathy (HAR)
4. Regulation of Retinal Metabolism
4.1. Nutrients
4.1.1. Glucose
4.1.2. Amino Acids
4.1.3. Fatty Acids
4.2. Hormones
4.2.1. Adiponectin (APN)
4.2.2. Insulin-Growth Factor 1 (IGF-1)
4.3. Other Related to Metabolism
4.3.1. Peroxisome Proliferator-Activated Receptor α (PPARα) Agonist
4.3.2. Rapamycin
4.3.3. Rho-Associated Kinase (ROCK) Inhibitor
4.3.4. Autophagy
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hellstrom, A.; Smith, L.E.; Dammann, O. Retinopathy of prematurity. Lancet 2013, 382, 1445–1457. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Gong, Y.; Lofqvist, C.; Hellstrom, A.; Smith, L.E. Review: Adiponectin in retinopathy. Biochim. Biophys. Acta 2016, 1862, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Hansen, R.M.; Moskowitz, A.; Akula, J.D.; Fulton, A.B. The neural retina in retinopathy of prematurity. Prog. Retin. Eye Res. 2017, 56, 32–57. [Google Scholar] [CrossRef] [Green Version]
- Fulton, A.B.; Dodge, J.; Hansen, R.M.; Williams, T.P. The rhodopsin content of human eyes. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1878–1883. [Google Scholar]
- Akula, J.D.; Hansen, R.M.; Tzekov, R.; Favazza, T.L.; Vyhovsky, T.C.; Benador, I.Y.; Mocko, J.A.; McGee, D.; Kubota, R.; Fulton, A.B. Visual cycle modulation in neurovascular retinopathy. Exp. Eye Res. 2010, 91, 153–161. [Google Scholar] [CrossRef]
- Lofqvist, C.A.; Najm, S.; Hellgren, G.; Engstrom, E.; Savman, K.; Nilsson, A.K.; Andersson, M.X.; Hard, A.L.; Smith, L.E.H.; Hellstrom, A. Association of Retinopathy of Prematurity With Low Levels of Arachidonic Acid: A Secondary Analysis of a Randomized Clinical Trial. JAMA Ophthalmol. 2018, 136, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Lofqvist, C.A.; Shao, Z.; Sun, Y.; Joyal, J.S.; Hurst, C.G.; Cui, R.Z.; Evans, L.P.; Tian, K.; SanGiovanni, J.P.; et al. Dietary omega-3 polyunsaturated fatty acids decrease retinal neovascularization by adipose-endoplasmic reticulum stress reduction to increase adiponectin. Am. J. Clin. Nutr. 2015, 101, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.E. IGF-1 and retinopathy of prematurity in the preterm infant. Biol. Neonate 2005, 88, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, P.; Stoltz Sjostrom, E.; Domellof, M.; Kallen, K.; Holmstrom, G.; Hard, A.L.; Smith, L.E.; Lofqvist, C.; Hellstrom, A. WINROP identifies severe retinopathy of prematurity at an early stage in a nation-based cohort of extremely preterm infants. PLoS ONE 2013, 8, e73256. [Google Scholar] [CrossRef]
- Wallace, D.K.; Kylstra, J.A.; Phillips, S.J.; Hall, J.G. Poor postnatal weight gain: A risk factor for severe retinopathy of prematurity. JAAPOS 2000, 4, 343–347. [Google Scholar] [CrossRef]
- Alexandre-Gouabau, M.C.; Moyon, T.; David-Sochard, A.; Fenaille, F.; Cholet, S.; Royer, A.L.; Guitton, Y.; Billard, H.; Darmaun, D.; Roze, J.C.; et al. Comprehensive Preterm Breast Milk Metabotype Associated with Optimal Infant Early Growth Pattern. Nutrients 2019, 11, 528. [Google Scholar] [CrossRef] [Green Version]
- Alexandre-Gouabau, M.C.; Moyon, T.; Cariou, V.; Antignac, J.P.; Qannari, E.M.; Croyal, M.; Soumah, M.; Guitton, Y.; David-Sochard, A.; Billard, H.; et al. Breast Milk Lipidome Is Associated with Early Growth Trajectory in Preterm Infants. Nutrients 2018, 10, 164. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, A.K.; Lofqvist, C.; Najm, S.; Hellgren, G.; Savman, K.; Andersson, M.X.; Smith, L.E.H.; Hellstrom, A. Long-chain polyunsaturated fatty acids decline rapidly in milk from mothers delivering extremely preterm indicating the need for supplementation. Acta Paediatr. 2018, 107, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Crawford, M.A.; Costeloe, K.; Ghebremeskel, K.; Phylactos, A.; Skirvin, L.; Stacey, F. Are deficits of arachidonic and docosahexaenoic acids responsible for the neural and vascular complications of preterm babies? Am. J. Clin. Nutr. 1997, 66, 1032S–1041S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanderVeen, D.K.; Martin, C.R.; Mehendale, R.; Allred, E.N.; Dammann, O.; Leviton, A.; Investigators, E.S. Early nutrition and weight gain in preterm newborns and the risk of retinopathy of prematurity. PLoS ONE 2013, 8, e64325. [Google Scholar] [CrossRef] [PubMed]
- Lapillonne, A.; Eleni dit Trolli, S.; Kermorvant-Duchemin, E. Postnatal docosahexaenoic acid deficiency is an inevitable consequence of current recommendations and practice in preterm infants. Neonatology 2010, 98, 397–403. [Google Scholar] [CrossRef]
- Pawlik, D.; Lauterbach, R.; Walczak, M.; Hurkala, J.; Sherman, M.P. Fish-oil fat emulsion supplementation reduces the risk of retinopathy in very low birth weight infants: A prospective, randomized study. JPEN J. Parenter. Enter. Nutr. 2014, 38, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, D.; Lauterbach, R.; Turyk, E. Fish-oil fat emulsion supplementation may reduce the risk of severe retinopathy in VLBW infants. Pediatrics 2011, 127, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Beken, S.; Dilli, D.; Fettah, N.D.; Kabatas, E.U.; Zenciroglu, A.; Okumus, N. The influence of fish-oil lipid emulsions on retinopathy of prematurity in very low birth weight infants: A randomized controlled trial. Early Hum. Dev. 2014, 90, 27–31. [Google Scholar] [CrossRef]
- Najm, S.; Lofqvist, C.; Hellgren, G.; Engstrom, E.; Lundgren, P.; Hard, A.L.; Lapillonne, A.; Savman, K.; Nilsson, A.K.; Andersson, M.X.; et al. Effects of a lipid emulsion containing fish oil on polyunsaturated fatty acid profiles, growth and morbidities in extremely premature infants: A randomized controlled trial. Clin. Nutr. ESPEN 2017, 20, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Bernabe-Garcia, M.; Villegas-Silva, R.; Villavicencio-Torres, A.; Calder, P.C.; Rodriguez-Cruz, M.; Maldonado-Hernandez, J.; Macias-Loaiza, D.; Lopez-Alarcon, M.; Inda-Icaza, P.; Cruz-Reynoso, L. Enteral Docosahexaenoic Acid and Retinopathy of Prematurity: A Randomized Clinical Trial. JPEN J. Parenter. Enter. Nutr. 2019, 43, 874–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, C.T.; Makrides, M.; McPhee, A.J.; Sullivan, T.R.; Davis, P.G.; Thio, M.; Simmer, K.; Rajadurai, V.S.; Travadi, J.; Berry, M.J.; et al. Docosahexaenoic Acid and Bronchopulmonary Dysplasia in Preterm Infants. N. Engl. J. Med. 2017, 376, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, A.; Nilsson, A.K.; Wackernagel, D.; Pivodic, A.; Vanpee, M.; Sjobom, U.; Hellgren, G.; Hallberg, B.; Domellof, M.; Klevebro, S.; et al. Effect of Enteral Lipid Supplement on Severe Retinopathy of Prematurity: A Randomized Clinical Trial. JAMA Pediatr. 2021, 175, 359–367. [Google Scholar] [CrossRef]
- Kapoor, V.; Malviya, M.N.; Soll, R. Lipid emulsions for parenterally fed preterm infants. Cochrane Database Syst. Rev. 2019, 6, CD013163. [Google Scholar] [CrossRef] [PubMed]
- Vayalthrikkovil, S.; Bashir, R.A.; Rabi, Y.; Amin, H.; Spence, J.M.; Robertson, H.L.; Lodha, A. Parenteral Fish-Oil Lipid Emulsions in the Prevention of Severe Retinopathy of Prematurity: A Systematic Review and Meta-Analysis. Am. J. Perinatol. 2017, 34, 705–715. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, Y.; Pei, J.; Chen, Z.; Wang, Q.; Xiang, B. Safety and efficacy of parenteral fish oil-containing lipid emulsions in premature neonates. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 708–716. [Google Scholar] [CrossRef]
- D’Ascenzo, R.; Savini, S.; Biagetti, C.; Bellagamba, M.P.; Marchionni, P.; Pompilio, A.; Cogo, P.E.; Carnielli, V.P. Higher docosahexaenoic acid, lower arachidonic acid and reduced lipid tolerance with high doses of a lipid emulsion containing 15% fish oil: A randomized clinical trial. Clin. Nutr. 2014, 33, 1002–1009. [Google Scholar] [CrossRef]
- Hellstrom, A.; Pivodic, A.; Granse, L.; Lundgren, P.; Sjobom, U.; Nilsson, A.K.; Soderling, H.; Hard, A.L.; Smith, L.E.H.; Lofqvist, C.A. Association of Docosahexaenoic Acid and Arachidonic Acid Serum Levels With Retinopathy of Prematurity in Preterm Infants. JAMA Netw. Open 2021, 4, e2128771. [Google Scholar] [CrossRef]
- Birch, E.E.; Carlson, S.E.; Hoffman, D.R.; Fitzgerald-Gustafson, K.M.; Fu, V.L.; Drover, J.R.; Castaneda, Y.S.; Minns, L.; Wheaton, D.K.; Mundy, D.; et al. The DIAMOND (DHA Intake And Measurement Of Neural Development) Study: A double-masked, randomized controlled clinical trial of the maturation of infant visual acuity as a function of the dietary level of docosahexaenoic acid. Am. J. Clin. Nutr. 2010, 91, 848–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SanGiovanni, J.P.; Parra-Cabrera, S.; Colditz, G.A.; Berkey, C.S.; Dwyer, J.T. Meta-analysis of dietary essential fatty acids and long-chain polyunsaturated fatty acids as they relate to visual resolution acuity in healthy preterm infants. Pediatrics 2000, 105, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Molloy, C.S.; Stokes, S.; Makrides, M.; Collins, C.T.; Anderson, P.J.; Doyle, L.W. Long-term effect of high-dose supplementation with DHA on visual function at school age in children born at <33 wk gestational age: Results from a follow-up of a randomized controlled trial. Am. J. Clin. Nutr. 2016, 103, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; SanGiovanni, J.P.; Lofqvist, C.; Aderman, C.M.; Chen, J.; Higuchi, A.; Hong, S.; Pravda, E.A.; Majchrzak, S.; Carper, D.; et al. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat. Med. 2007, 13, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Stahl, A.; Sapieha, P.; Connor, K.M.; Sangiovanni, J.P.; Chen, J.; Aderman, C.M.; Willett, K.L.; Krah, N.M.; Dennison, R.J.; Seaward, M.R.; et al. Short communication: PPAR gamma mediates a direct antiangiogenic effect of omega 3-PUFAs in proliferative retinopathy. Circ. Res. 2010, 107, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapieha, P.; Stahl, A.; Chen, J.; Seaward, M.R.; Willett, K.L.; Krah, N.M.; Dennison, R.J.; Connor, K.M.; Aderman, C.M.; Liclican, E.; et al. 5-Lipoxygenase metabolite 4-HDHA is a mediator of the antiangiogenic effect of omega-3 polyunsaturated fatty acids. Sci. Transl. Med. 2011, 3, 69ra12. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Fu, Z.; Edin, M.L.; Liu, C.H.; Wang, Z.; Shao, Z.; Fredrick, T.W.; Saba, N.J.; Morss, P.C.; Burnim, S.B.; et al. Cytochrome P450 Oxidase 2C Inhibition Adds to omega-3 Long-Chain Polyunsaturated Fatty Acids Protection Against Retinal and Choroidal Neovascularization. Arter. Thromb. Vasc. Biol. 2016, 36, 1919–1927. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Shao, Z.; Fu, Z.; Edin, M.L.; Sun, Y.; Liegl, R.G.; Wang, Z.; Liu, C.H.; Burnim, S.B.; Meng, S.S.; et al. Fenofibrate Inhibits Cytochrome P450 Epoxygenase 2C Activity to Suppress Pathological Ocular Angiogenesis. EBioMedicine 2016, 13, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Bibli, S.I.; Wittig, J.; Zukunft, S.; Lin, J.; Hammes, H.P.; Popp, R.; Fleming, I. Soluble epoxide hydrolase promotes astrocyte survival in retinopathy of prematurity. J. Clin. Investig. 2019, 129, 5204–5218. [Google Scholar] [CrossRef]
- Khairallah, R.J.; Kim, J.; O’Shea, K.M.; O’Connell, K.A.; Brown, B.H.; Galvao, T.; Daneault, C.; Des Rosiers, C.; Polster, B.M.; Hoppel, C.L.; et al. Improved mitochondrial function with diet-induced increase in either docosahexaenoic acid or arachidonic acid in membrane phospholipids. PLoS ONE 2012, 7, e34402. [Google Scholar] [CrossRef] [Green Version]
- Khairallah, R.J.; Sparagna, G.C.; Khanna, N.; O’Shea, K.M.; Hecker, P.A.; Kristian, T.; Fiskum, G.; Des Rosiers, C.; Polster, B.M.; Stanley, W.C. Dietary supplementation with docosahexaenoic acid, but not eicosapentaenoic acid, dramatically alters cardiac mitochondrial phospholipid fatty acid composition and prevents permeability transition. Biochim. Biophys. Acta 2010, 1797, 1555–1562. [Google Scholar] [CrossRef] [Green Version]
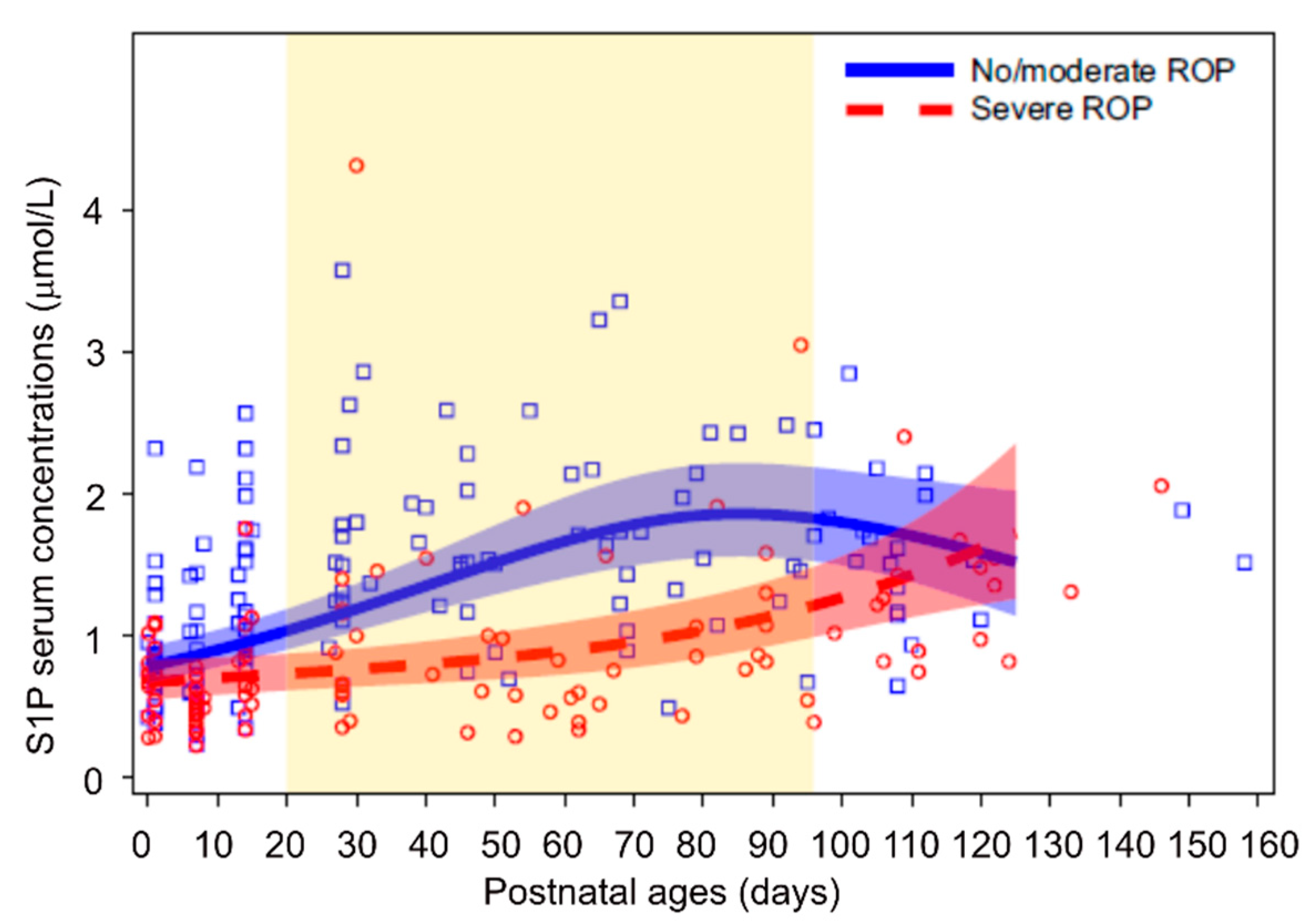
- Nilsson, A.K.; Andersson, M.X.; Sjobom, U.; Hellgren, G.; Lundgren, P.; Pivodic, A.; Smith, L.E.H.; Hellstrom, A. Sphingolipidomics of serum in extremely preterm infants: Association between low sphingosine-1-phosphate levels and severe retinopathy of prematurity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158939. [Google Scholar] [CrossRef]
- Victoria, S.M.; Basu, S.K.; Bano, Q.; Richard, G.; Rotstein, N.P.; Nawajes, M. Sphingolipids as critical players in retinal physiology and pathology. J. Lipid Res. 2021, 62, 100037. [Google Scholar] [CrossRef]
- Miranda, G.E.; Abrahan, C.E.; Politi, L.E.; Rotstein, N.P. Sphingosine-1-phosphate is a key regulator of proliferation and differentiation in retina photoreceptors. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4416–4428. [Google Scholar] [CrossRef]
- Yanagida, K.; Engelbrecht, E.; Niaudet, C.; Jung, B.; Gaengel, K.; Holton, K.; Swendeman, S.; Liu, C.H.; Levesque, M.V.; Kuo, A.; et al. Sphingosine 1-Phosphate Receptor Signaling Establishes AP-1 Gradients to Allow for Retinal Endothelial Cell Specialization. Dev. Cell 2020, 52, 779–793.e7. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Shen, J.; Dong, A.; Rashid, A.; Stoller, G.; Campochiaro, P.A. Blockade of sphingosine-1-phosphate reduces macrophage influx and retinal and choroidal neovascularization. J. Cell. Physiol. 2009, 218, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, A.M.; Wagner, B.D.; Mandava, N.; Palestine, A.G.; Mourani, P.M.; McCourt, E.A.; Oliver, S.C.; Abman, S.H. The Relationship of Novel Plasma Proteins in the Early Neonatal Period With Retinopathy of Prematurity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5076–5082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spierer, A.; Rabinowitz, R.; Pri-Chen, S.; Rosner, M. An increase in superoxide dismutase ameliorates oxygen-induced retinopathy in transgenic mice. Eye 2005, 19, 86–91. [Google Scholar] [CrossRef]
- Boskabadi, H.; Marefat, M.; Maamouri, G.; Abrishami, M.; Abrishami, M.; Shoeibi, N.; Sanjari, M.S.; Mobarhan, M.G.; Shojaei, S.R.H.; Tavallaei, S.; et al. Evaluation of pro-oxidant antioxidant balance in retinopathy of prematurity. Eye 2021. [Google Scholar] [CrossRef]
- Banjac, L.; Banjac, G.; Kotur-Stevuljevic, J.; Spasojevic-Kalimanovska, V.; Gojkovic, T.; Bogavac-Stanojevic, N.; Jelic-Ivanovic, Z.; Banjac, G. Pro-Oxidants and Antioxidants in Retinopathy of Prematurity. Acta Clin. Croat. 2018, 57, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Ozieblo-Kupczyk, M.; Bakunowicz-Lazarczyk, A.; Dzienis, K.; Skrzydlewska, E.; Szczepanski, M.; Waszkiewiczz, E. The estimation of selected parameters in antioxidant system in red blood cells in ROP screening of premature infants. Klin. Ocz. 2006, 108, 413–415. [Google Scholar]
- Kumar, A.; Ranjan, R.; Basu, S.; Khanna, H.D.; Bhargava, V. Antioxidant levels in cord blood of low birth weight newborns. Indian Pediatr. 2008, 45, 583–585. [Google Scholar] [PubMed]
- Ramiro-Cortijo, D.; Lopez de Pablo, A.L.; Lopez-Gimenez, M.R.; Martin, C.R.; Brown, J.; de Pipaon, M.S.; Arribas, S.M. Plasma Oxidative Status in Preterm Infants Receiving LCPUFA Supplementation: A Pilot Study. Nutrients 2020, 12, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsson, H.; Tebani, A.; Zhong, W.; Fagerberg, L.; Brusselaers, N.; Hard, A.L.; Uhlen, M.; Hellstrom, A. Blood protein profiles related to preterm birth and retinopathy of prematurity. Pediatr. Res. 2021. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Z.; Li, S.; Yang, M.; Xiao, X.; Lian, C.; Wen, W.; He, H.; Zeng, J.; Wang, J.; et al. Targeted Blood Metabolomic Study on Retinopathy of Prematurity. Investig. Ophthalmol. Vis. Sci. 2020, 61, 12. [Google Scholar] [CrossRef] [Green Version]
- Hozyasz, K.K.; Oltarzewski, M.; Dudkiewicz, Z. Malonylcarnitine in newborns with non-syndromic cleft lip with or without cleft palate. Int. J. Oral Sci. 2010, 2, 136–141. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Ko, J.M.; Song, M.K.; Song, J.; Park, K.S. A Korean child diagnosed with malonic aciduria harboring a novel start codon mutation following presentation with dilated cardiomyopathy. Mol. Genet. Genom. Med. 2020, 8, e1379. [Google Scholar] [CrossRef]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Xu, Y.; Zhang, X.; Zhao, P.; Gong, X.; He, M.; Cao, J.; Jiang, B.; Yoshida, S.; Li, Y. Plasma metabolites in treatment-requiring retinopathy of prematurity: Potential biomarkers identified by metabolomics. Exp. Eye Res. 2020, 199, 108198. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Cagnone, G.; Fu, Z.; Cakir, B.; Kotoda, Y.; Asakage, M.; Wakabayashi, Y.; Hellstrom, A.; Joyal, J.S.; Talukdar, S.; et al. Vitreous metabolomics profiling of proliferative diabetic retinopathy. Diabetologia 2021, 64, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Paris, L.P.; Johnson, C.H.; Aguilar, E.; Usui, Y.; Cho, K.; Hoang, L.T.; Feitelberg, D.; Benton, H.P.; Westenskow, P.D.; Kurihara, T.; et al. Global metabolomics reveals metabolic dysregulation in ischemic retinopathy. Metabolomics 2016, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouda, A.Y.; Eldahshan, W.; Narayanan, S.P.; Caldwell, R.W.; Caldwell, R.B. Arginase Pathway in Acute Retina and Brain Injury: Therapeutic Opportunities and Unexplored Avenues. Front. Pharmacol. 2020, 11, 277. [Google Scholar] [CrossRef] [PubMed]
- Neu, J.; Afzal, A.; Pan, H.; Gallego, E.; Li, N.; Li Calzi, S.; Caballero, S.; Spoerri, P.E.; Shaw, L.C.; Grant, M.B. The dipeptide Arg-Gln inhibits retinal neovascularization in the mouse model of oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3151–3155. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- McLeod, D.S.; D’Anna, S.A.; Lutty, G.A. Clinical and histopathologic features of canine oxygen-induced proliferative retinopathy. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1918–1932. [Google Scholar]
- Kremer, I.; Kissun, R.; Nissenkorn, I.; Ben-Sira, I.; Garner, A. Oxygen-induced retinopathy in newborn kittens. A model for ischemic vasoproliferative retinopathy. Investig. Ophthalmol. Vis. Sci. 1987, 28, 126–130. [Google Scholar]
- Ricci, B. Oxygen-induced retinopathy in the rat model. Doc. Ophthalmol. Proc. Ser. 1990, 74, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Pierce, E.A.; Foley, E.D.; Smith, L.E. Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch. Ophthalmol. 1996, 114, 1219–1228. [Google Scholar] [CrossRef]
- Rabinowitz, R.; Priel, A.; Rosner, M.; Pri-Chen, S.; Spierer, A. Avastin treatment reduces retinal neovascularization in a mouse model of retinopathy of prematurity. Curr. Eye Res 2012, 37, 624–629. [Google Scholar] [CrossRef]
- Jiang, C.; Ruan, L.; Zhang, J.; Huang, X. Inhibitory Effects On Retinal Neovascularization by Ranibizumab and sTie2-Fc in An Oxygen-Induced Retinopathy Mouse Model. Curr. Eye Res 2018, 43, 1190–1198. [Google Scholar] [CrossRef]
- Sone, H.; Kawakami, Y.; Segawa, T.; Okuda, Y.; Sekine, Y.; Honmura, S.; Segawa, T.; Suzuki, H.; Yamashita, K.; Yamada, N. Effects of intraocular or systemic administration of neutralizing antibody against vascular endothelial growth factor on the murine experimental model of retinopathy. Life Sci. 1999, 65, 2573–2580. [Google Scholar] [CrossRef]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Miwa, Y.; Hoshino, Y.; Shoda, C.; Jiang, X.; Tsubota, K.; Kurihara, T. Pharmacological HIF inhibition prevents retinal neovascularization with improved visual function in a murine oxygen-induced retinopathy model. Neurochem. Int. 2019, 128, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Usui-Ouchi, A.; Aguilar, E.; Murinello, S.; Prins, M.; Gantner, M.L.; Wright, P.E.; Berlow, R.B.; Friedlander, M. An allosteric peptide inhibitor of HIF-1alpha regulates hypoxia-induced retinal neovascularization. Proc. Natl. Acad. Sci. USA 2020, 117, 28297–28306. [Google Scholar] [CrossRef]
- Hoppe, G.; Bolok, Y.; McCollum, L.; Zhang, J.; Sears, J.E. Rank Order of Small Molecule Induced Hypoxiamimesis to Prevent Retinopathy of Prematurity. Front. Cell Dev. Biol. 2020, 8, 488. [Google Scholar] [CrossRef]
- Singh, C.; Hoppe, G.; Tran, V.; McCollum, L.; Bolok, Y.; Song, W.; Sharma, A.; Brunengraber, H.; Sears, J.E. Serine and 1-carbon metabolism are required for HIF-mediated protection against retinopathy of prematurity. JCI Insight 2019, 4, e129398. [Google Scholar] [CrossRef]
- Hoppe, G.; Yoon, S.; Gopalan, B.; Savage, A.R.; Brown, R.; Case, K.; Vasanji, A.; Chan, E.R.; Silver, R.B.; Sears, J.E. Comparative systems pharmacology of HIF stabilization in the prevention of retinopathy of prematurity. Proc. Natl. Acad. Sci. USA 2016, 113, E2516–E2525. [Google Scholar] [CrossRef] [Green Version]
- Gantner, M.L.; Eade, K.; Wallace, M.; Handzlik, M.K.; Fallon, R.; Trombley, J.; Bonelli, R.; Giles, S.; Harkins-Perry, S.; Heeren, T.F.C.; et al. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. N. Engl. J. Med. 2019, 381, 1422–1433. [Google Scholar] [CrossRef]
- Shen, W.; Lee, S.R.; Mathai, A.E.; Zhang, R.; Du, J.; Yam, M.X.; Pye, V.; Barnett, N.L.; Rayner, C.L.; Zhu, L.; et al. Effect of selectively knocking down key metabolic genes in Muller glia on photoreceptor health. Glia 2021, 69, 1966–1986. [Google Scholar] [CrossRef]
- Guo, D.; Murdoch, C.E.; Xu, H.; Shi, H.; Duan, D.D.; Ahmed, A.; Gu, Y. Vascular endothelial growth factor signaling requires glycine to promote angiogenesis. Sci. Rep. 2017, 7, 14749. [Google Scholar] [CrossRef]
- Lu, F.; Liu, Y.; Guo, Y.; Gao, Y.; Piao, Y.; Tan, S.; Tang, Y. Metabolomic changes of blood plasma associated with two phases of rat OIR. Exp. Eye Res. 2020, 190, 107855. [Google Scholar] [CrossRef] [PubMed]
- Dungan, K.M.; Braithwaite, S.S.; Preiser, J.C. Stress hyperglycaemia. Lancet 2009, 373, 1798–1807. [Google Scholar] [CrossRef]
- Pelikanova, T. Diabetic retinopathy: Pathogenesis and therapeutic implications. Vnitr. Lek. 2016, 62, 620–628. [Google Scholar]
- Au, S.C.; Tang, S.M.; Rong, S.S.; Chen, L.J.; Yam, J.C. Association between hyperglycemia and retinopathy of prematurity: A systemic review and meta-analysis. Sci. Rep. 2015, 5, 9091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadpour-Kacho, M.; Motlagh, A.J.; Rasoulinejad, S.A.; Jahangir, T.; Bijani, A.; Pasha, Y.Z. Correlation between hyperglycemia and retinopathy of prematurity. Pediatr. Int. 2014, 56, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Agthe, A.G.; Donohue, P.K.; Lehmann, C.U. Hyperglycemia and retinopathy of prematurity in very low birth weight infants. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2003, 23, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, S.; Murray, J.C.; Dagle, J.M.; Colaizy, T. Hyperglycemia as a risk factor for the development of retinopathy of prematurity. BMC Pediatr. 2013, 13, 78. [Google Scholar] [CrossRef] [Green Version]
- Kaempf, J.W.; Kaempf, A.J.; Wu, Y.; Stawarz, M.; Niemeyer, J.; Grunkemeier, G. Hyperglycemia, insulin and slower growth velocity may increase the risk of retinopathy of prematurity. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2011, 31, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Mohsen, L.; Abou-Alam, M.; El-Dib, M.; Labib, M.; Elsada, M.; Aly, H. A prospective study on hyperglycemia and retinopathy of prematurity. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2014, 34, 453–457. [Google Scholar] [CrossRef]
- Ertl, T.; Gyarmati, J.; Gaal, V.; Szabo, I. Relationship between hyperglycemia and retinopathy of prematurity in very low birth weight infants. Biol. Neonate 2006, 89, 56–59. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; McGowan, J.; Cannon, E.; Lehmann, C.U. Contribution of early glycemic status in the development of severe retinopathy of prematurity in a cohort of ELBW infants. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2011, 31, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Cakir, B.; Hellstrom, W.; Tomita, Y.; Fu, Z.; Liegl, R.; Winberg, A.; Hansen-Pupp, I.; Ley, D.; Hellstrom, A.; Lofqvist, C.; et al. IGF1, serum glucose, and retinopathy of prematurity in extremely preterm infants. JCI Insight 2020, 5, e140363. [Google Scholar] [CrossRef]
- Lei, C.; Duan, J.; Ge, G.; Zhang, M. Association between neonatal hyperglycemia and retinopathy of prematurity: A meta-analysis. Eur. J. Pediatr. 2021. [Google Scholar] [CrossRef] [PubMed]
- Vannadil, H.; Moulick, P.S.; Khan, M.A.; Shankar, S.; Kaushik, J.; Sati, A. Hyperglycaemia as a risk factor for the development of retinopathy of prematurity: A cohort study. Med. J. Armed Forces India 2020, 76, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. 2015, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Fu, Z.; Lofqvist, C.A.; Liegl, R.; Wang, Z.; Sun, Y.; Gong, Y.; Liu, C.H.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Photoreceptor glucose metabolism determines normal retinal vascular growth. EMBO Mol. Med. 2018, 10, 76–90. [Google Scholar] [CrossRef]
- Fu, Z.; Sun, Y.; Cakir, B.; Tomita, Y.; Huang, S.; Wang, Z.; Liu, C.H.; Cho, S.C.; Britton, W.; Kern, T.S.; et al. Targeting Neurovascular Interaction in Retinal Disorders. Int. J. Mol. Sci. 2020, 21, 1503. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Sapieha, P. Neurons and guidance cues in retinal vascular diseases. Oncotarget 2016, 7, 9618–9619. [Google Scholar] [CrossRef] [Green Version]
- Hoang, Q.V.; Linsenmeier, R.A.; Chung, C.K.; Curcio, C.A. Photoreceptor inner segments in monkey and human retina: Mitochondrial density, optics, and regional variation. Vis. Neurosci. 2002, 19, 395–407. [Google Scholar]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef]
- Narayan, D.S.; Chidlow, G.; Wood, J.P.; Casson, R.J. Glucose metabolism in mammalian photoreceptor inner and outer segments. Clin. Exp. Ophthalmol. 2017, 45, 730–741. [Google Scholar] [CrossRef] [Green Version]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; An, X.; Guo, X.; Habtetsion, T.G.; Wang, Y.; Xu, X.; Kandala, S.; Li, Q.; Li, H.; Zhang, C.; et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1231–1239. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell. Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Kong, J.; Hartnett, M.E.; Wang, H. Enhancing Retinal Endothelial Glycolysis by Inhibiting UCP2 Promotes Physiologic Retinal Vascular Development in a Model of Retinopathy of Prematurity. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1604–1613. [Google Scholar] [CrossRef]
- Liu, Z.; Yan, S.; Wang, J.; Xu, Y.; Wang, Y.; Zhang, S.; Xu, X.; Yang, Q.; Zeng, X.; Zhou, Y.; et al. Endothelial adenosine A2a receptor-mediated glycolysis is essential for pathological retinal angiogenesis. Nat. Commun. 2017, 8, 584. [Google Scholar] [CrossRef] [Green Version]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef]
- Fu, Z.J.; Li, S.Y.; Kociok, N.; Wong, D.; Chung, S.K.; Lo, A.C. Aldose reductase deficiency reduced vascular changes in neonatal mouse retina in oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5698–5712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Nian, S.; Li, S.Y.; Wong, D.; Chung, S.K.; Lo, A.C. Deficiency of aldose reductase attenuates inner retinal neuronal changes in a mouse model of retinopathy of prematurity. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Goveia, J.; Garcia-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e14. [Google Scholar] [CrossRef] [PubMed]
- Neu, J. Glutamine supplements in premature infants: Why and how. J. Pediatr. Gastroenterol. Nutr. 2003, 37, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Jaeger, L.A.; Bazer, F.W.; Rhoads, J.M. Arginine deficiency in preterm infants: Biochemical mechanisms and nutritional implications. J. Nutr. Biochem. 2004, 15, 442–451. [Google Scholar] [CrossRef]
- Wu, G.; Haynes, T.E.; Li, H.; Meininger, C.J. Glutamine metabolism in endothelial cells: Ornithine synthesis from glutamine via pyrroline-5-carboxylate synthase. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2000, 126, 115–123. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Eade, K.; Gantner, M.L.; Hostyk, J.A.; Nagasaki, T.; Giles, S.; Fallon, R.; Harkins-Perry, S.; Baldini, M.; Lim, E.W.; Scheppke, L.; et al. Serine biosynthesis defect due to haploinsufficiency of PHGDH causes retinal disease. Nat. Metab. 2021, 3, 366–377. [Google Scholar] [CrossRef]
- Vandekeere, S.; Dubois, C.; Kalucka, J.; Sullivan, M.R.; Garcia-Caballero, M.; Goveia, J.; Chen, R.; Diehl, F.F.; Bar-Lev, L.; Souffreau, J.; et al. Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab. 2018, 28, 573–587.e13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Gillies, M.C.; Madigan, M.C.; Shen, W.; Du, J.; Grunert, U.; Zhou, F.; Yam, M.; Zhu, L. Disruption of De Novo Serine Synthesis in Muller Cells Induced Mitochondrial Dysfunction and Aggravated Oxidative Damage. Mol. Neurobiol. 2018, 55, 7025–7037. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, L.; Madigan, M.C.; Liu, W.; Shen, W.; Cherepanoff, S.; Zhou, F.; Zeng, S.; Du, J.; Gillies, M.C. Human macular Muller cells rely more on serine biosynthesis to combat oxidative stress than those from the periphery. eLife 2019, 8, e43598. [Google Scholar] [CrossRef]
- Becker, S.; Wang, H.; Simmons, A.B.; Suwanmanee, T.; Stoddard, G.J.; Kafri, T.; Hartnett, M.E. Targeted Knockdown of Overexpressed VEGFA or VEGF164 in Muller cells maintains retinal function by triggering different signaling mechanisms. Sci. Rep. 2018, 8, 2003. [Google Scholar] [CrossRef] [Green Version]
- Le, Y.Z. VEGF production and signaling in Muller glia are critical to modulating vascular function and neuronal integrity in diabetic retinopathy and hypoxic retinal vascular diseases. Vis. Res. 2017, 139, 108–114. [Google Scholar] [CrossRef]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Schneider, J.G.; Shenouda, S.M.; Lee, A.; Towler, D.A.; Chakravarthy, M.V.; Vita, J.A.; Semenkovich, C.F. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (eNOS) palmitoylation. J. Biol. Chem. 2011, 286, 2933–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmasri, H.; Karaaslan, C.; Teper, Y.; Ghelfi, E.; Weng, M.; Ince, T.A.; Kozakewich, H.; Bischoff, J.; Cataltepe, S. Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. 2009, 23, 3865–3873. [Google Scholar] [CrossRef] [Green Version]
- Joyal, J.S.; Gantner, M.L.; Smith, L.E.H. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog. Retin. Eye Res. 2018, 64, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, A.; Ohashi, K.; Kihara, S.; Walsh, K.; Ouchi, N. Adiponectin suppresses pathological microvessel formation in retina through modulation of tumor necrosis factor-alpha expression. Circ. Res. 2009, 104, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.S.; Calandria, J.M.; Gordon, W.C.; Jun, B.; Zhou, Y.; Gelfman, C.M.; Li, S.; Jin, M.; Knott, E.J.; Chang, B.; et al. Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell survival. Nat Commun. 2015, 6, 6228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluch, V.M.; Banks, A.; Li, H.; Crowley, M.A.; Davis, V.; Xiang, C.; Yang, J.; Demirs, J.T.; Vrouvlianis, J.; Leehy, B.; et al. ADIPOR1 is essential for vision and its RPE expression is lost in the Mfrp(rd6) mouse. Sci. Rep. 2018, 8, 14339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Liegl, R.; Wang, Z.; Gong, Y.; Liu, C.H.; Sun, Y.; Cakir, B.; Burnim, S.B.; Meng, S.S.; Lofqvist, C.; et al. Adiponectin Mediates Dietary Omega-3 Long-Chain Polyunsaturated Fatty Acid Protection Against Choroidal Neovascularization in Mice. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3862–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, C.; Li, L.; Man, Q.; Meng, L.; Song, P.; Froyland, L.; Du, Z.Y. Dietary inclusion of salmon, herring and pompano as oily fish reduces CVD risk markers in dyslipidaemic middle-aged and elderly Chinese women. Br. J. Nutr. 2012, 108, 1455–1465. [Google Scholar] [CrossRef] [Green Version]
- Olza, J.; Mesa, M.D.; Aguilera, C.M.; Moreno-Torres, R.; Jimenez, A.; Perez de la Cruz, A.; Gil, A. Influence of an eicosapentaenoic and docosahexaenoic acid-enriched enteral nutrition formula on plasma fatty acid composition and biomarkers of insulin resistance in the elderly. Clin. Nutr. 2010, 29, 31–37. [Google Scholar] [CrossRef]
- Kuda, O.; Jelenik, T.; Jilkova, Z.; Flachs, P.; Rossmeisl, M.; Hensler, M.; Kazdova, L.; Ogston, N.; Baranowski, M.; Gorski, J.; et al. n-3 fatty acids and rosiglitazone improve insulin sensitivity through additive stimulatory effects on muscle glycogen synthesis in mice fed a high-fat diet. Diabetologia 2009, 52, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Prostek, A.; Gajewska, M.; Kamola, D.; Balasinska, B. The influence of EPA and DHA on markers of inflammation in 3T3-L1 cells at different stages of cellular maturation. Lipids Health Dis. 2014, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013, 17, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Owen, B.M.; Mangelsdorf, D.J.; Kliewer, S.A. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 2015, 26, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Kharitonenkov, A.; Larsen, P. FGF21 reloaded: Challenges of a rapidly growing field. Trends Endocrinol. Metab. 2011, 22, 81–86. [Google Scholar] [CrossRef]
- Lin, Z.; Gong, Q.; Wu, C.; Yu, J.; Lu, T.; Pan, X.; Lin, S.; Li, X. Dynamic change of serum FGF21 levels in response to glucose challenge in human. J. Clin. Endocrinol. Metab. 2012, 97, E1224–E1228. [Google Scholar] [CrossRef] [Green Version]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ramos, D.; Mehta, R.; Aguilar-Salinas, C.A. Fibroblast Growth Factor 21 and Browning of White Adipose Tissue. Front. Physiol. 2019, 10, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Gong, Y.; Liegl, R.; Wang, Z.; Liu, C.H.; Meng, S.S.; Burnim, S.B.; Saba, N.J.; Fredrick, T.W.; Morss, P.C.; et al. FGF21 Administration Suppresses Retinal and Choroidal Neovascularization in Mice. Cell Rep. 2017, 18, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Wang, Z.; Liu, C.H.; Gong, Y.; Cakir, B.; Liegl, R.; Sun, Y.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Fibroblast Growth Factor 21 Protects Photoreceptor Function in Type 1 Diabetic Mice. Diabetes 2018, 67, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Qiu, C.; Cagnone, G.; Tomita, Y.; Huang, S.; Cakir, B.; Kotoda, Y.; Allen, W.; Bull, E.; Akula, J.D.; et al. Retinal glial remodeling by FGF21 preserves retinal function during photoreceptor degeneration. iScience 2021, 24, 102376. [Google Scholar] [CrossRef]
- Sanchez-Infantes, D.; Gallego-Escuredo, J.M.; Diaz, M.; Aragones, G.; Sebastiani, G.; Lopez-Bermejo, A.; de Zegher, F.; Domingo, P.; Villarroya, F.; Ibanez, L. Circulating FGF19 and FGF21 surge in early infancy from infra- to supra-adult concentrations. Int. J. Obes. 2015, 39, 742–746. [Google Scholar] [CrossRef]
- Guasti, L.; Silvennoinen, S.; Bulstrode, N.W.; Ferretti, P.; Sankilampi, U.; Dunkel, L. Elevated FGF21 leads to attenuated postnatal linear growth in preterm infants through GH resistance in chondrocytes. J. Clin. Endocrinol. Metab. 2014, 99, E2198–E2206. [Google Scholar] [CrossRef] [Green Version]
- Mericq, V.; De Luca, F.; Hernandez, M.I.; Pena, V.; Rossel, K.; Garcia, M.; Avila, A.; Cavada, G.; Iniguez, G. Serum fibroblast growth factor 21 levels are inversely associated with growth rates in infancy. Horm. Res. Paediatr. 2014, 82, 324–331. [Google Scholar] [CrossRef]
- Daughaday, W.H.; Rotwein, P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 1989, 10, 68–91. [Google Scholar] [CrossRef]
- Liegl, R.; Lofqvist, C.; Hellstrom, A.; Smith, L.E. IGF-1 in retinopathy of prematurity, a CNS neurovascular disease. Early Hum. Dev. 2016, 102, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom, A.; Perruzzi, C.; Ju, M.; Engstrom, E.; Hard, A.L.; Liu, J.L.; Albertsson-Wikland, K.; Carlsson, B.; Niklasson, A.; Sjodell, L.; et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: Direct correlation with clinical retinopathy of prematurity. Proc. Natl. Acad. Sci. USA 2001, 98, 5804–5808. [Google Scholar] [CrossRef] [Green Version]
- Hard, A.L.; Smith, L.E.; Hellstrom, A. Nutrition, insulin-like growth factor-1 and retinopathy of prematurity. Semin. Fetal Neonatal. Med. 2013, 18, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellstrom, A.; Engstrom, E.; Hard, A.L.; Albertsson-Wikland, K.; Carlsson, B.; Niklasson, A.; Lofqvist, C.; Svensson, E.; Holm, S.; Ewald, U.; et al. Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics 2003, 112, 1016–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, A.K.; Ying, G.S.; Huang, J.; Quinn, G.E.; Binenbaum, G. Postnatal Serum Insulin-Like Growth Factor I and Retinopathy of Prematurity. Retina 2017, 37, 867–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellgren, G.; Lundgren, P.; Pivodic, A.; Lofqvist, C.; Nilsson, A.K.; Ley, D.; Savman, K.; Smith, L.E.; Hellstrom, A. Decreased Platelet Counts and Serum Levels of VEGF-A, PDGF-BB, and BDNF in Extremely Preterm Infants Developing Severe ROP. Neonatology 2021, 118, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.E.; Shen, W.; Perruzzi, C.; Soker, S.; Kinose, F.; Xu, X.; Robinson, G.; Driver, S.; Bischoff, J.; Zhang, B.; et al. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat. Med. 1999, 5, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebrouck, S.; Daniels, H.; Moons, L.; Vanhole, C.; Carmeliet, P.; De Zegher, F. Oxygen-induced retinopathy in mice: Amplification by neonatal IGF-I deficit and attenuation by IGF-I administration. Pediatr. Res. 2009, 65, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Ley, D.; Hallberg, B.; Hansen-Pupp, I.; Dani, C.; Ramenghi, L.A.; Marlow, N.; Beardsall, K.; Bhatti, F.; Dunger, D.; Higginson, J.D.; et al. rhIGF-1/rhIGFBP-3 in Preterm Infants: A Phase 2 Randomized Controlled Trial. J. Pediatr. 2019, 206, 56–65.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakir, B.; Liegl, R.; Hellgren, G.; Lundgren, P.; Sun, Y.; Klevebro, S.; Lofqvist, C.; Mannheimer, C.; Cho, S.; Poblete, A.; et al. Thrombocytopenia is associated with severe retinopathy of prematurity. JCI Insight 2018, 3, e99448. [Google Scholar] [CrossRef]
- Jensen, A.K.; Ying, G.S.; Huang, J.; Quinn, G.E.; Binenbaum, G. Longitudinal study of the association between thrombocytopenia and retinopathy of prematurity. J. AAPOS Off. Publ. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2018, 22, 119–123. [Google Scholar] [CrossRef]
- Keech, A.C.; Mitchell, P.; Summanen, P.A.; O’Day, J.; Davis, T.M.; Moffitt, M.S.; Taskinen, M.R.; Simes, R.J.; Tse, D.; Williamson, E.; et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): A randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- Group, A.S.; Group, A.E.S.; Chew, E.Y.; Ambrosius, W.T.; Davis, M.D.; Danis, R.P.; Gangaputra, S.; Greven, C.M.; Hubbard, L.; Esser, B.A.; et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010, 363, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Najib, J. Fenofibrate in the treatment of dyslipidemia: A review of the data as they relate to the new suprabioavailable tablet formulation. Clin. Ther. 2002, 24, 2022–2050. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Lin, M.; Jenkins, A.J.; Keech, A.C.; Mott, R.; Lyons, T.J.; Ma, J.X. Therapeutic effects of PPARalpha agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 2013, 62, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csaicsich, D.; Russo-Schlaff, N.; Messerschmidt, A.; Weninger, M.; Pollak, A.; Aufricht, C. Renal failure, comorbidity and mortality in preterm infants. Wien Klin. Wochenschr. 2008, 120, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and Safety of Pemafibrate Versus Fenofibrate in Patients with High Triglyceride and Low HDL Cholesterol Levels: A Multicenter, Placebo-Controlled, Double-Blind, Randomized Trial. J. Atheroscler. Thromb. 2018, 25, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Abe, K.; Toma, T.; Nishikawa, M.; Ozawa, H.; Okuda, A.; Araki, T.; Oda, S.; Inoue, K.; Shibuya, K.; et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor alpha agonists. Bioorgan. Med. Chem. Lett. 2007, 17, 4689–4693. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Lee, D.; Tsubota, K.; Kurihara, T. PPARalpha Agonist Oral Therapy in Diabetic Retinopathy. Biomedicines 2020, 8, 433. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Ozawa, N.; Miwa, Y.; Ishida, A.; Ohta, M.; Tsubota, K.; Kurihara, T. Pemafibrate Prevents Retinal Pathological Neovascularization by Increasing FGF21 Level in a Murine Oxygen-Induced Retinopathy Model. Int. J. Mol. Sci. 2019, 20, 5878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, Y.; Lee, D.; Miwa, Y.; Jiang, X.; Ohta, M.; Tsubota, K.; Kurihara, T. Pemafibrate Protects Against Retinal Dysfunction in a Murine Model of Diabetic Retinopathy. Int. J. Mol. Sci. 2020, 21, 6243. [Google Scholar] [CrossRef]
- Swarbrick, A.W.; Frederiks, A.J.; Foster, R.S. Systematic review of sirolimus in dermatological conditions. Australas. J. Derm. 2021. [Google Scholar] [CrossRef]
- Yu, J.; Parkhitko, A.A.; Henske, E.P. Mammalian target of rapamycin signaling and autophagy: Roles in lymphangioleiomyomatosis therapy. Proc. Am. Thorac. Soc. 2010, 7, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Saunders, R.N.; Metcalfe, M.S.; Nicholson, M.L. Rapamycin in transplantation: A review of the evidence. Kidney Int. 2001, 59, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Cho, K. Lipid metabolism, inflammation, and foam cell formation in health and metabolic disorders: Targeting mTORC1. J. Mol. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Dejneka, N.S.; Kuroki, A.M.; Fosnot, J.; Tang, W.; Tolentino, M.J.; Bennett, J. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Mol. Vis. 2004, 10, 964–972. [Google Scholar]
- Yagasaki, R.; Nakahara, T.; Ushikubo, H.; Mori, A.; Sakamoto, K.; Ishii, K. Anti-angiogenic effects of mammalian target of rapamycin inhibitors in a mouse model of oxygen-induced retinopathy. Biol. Pharm. Bull. 2014, 37, 1838–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhu, M.; Ruan, L.; Jiang, C.; Yang, Q.; Chang, Q.; Huang, X. Protective effects of rapamycin on the retinal vascular bed during the vaso-obliteration phase in mouse oxygen-induced retinopathy model. FASEB J. 2020, 34, 15822–15836. [Google Scholar] [CrossRef] [PubMed]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Arita, R.; Hata, Y.; Nakao, S.; Kita, T.; Miura, M.; Kawahara, S.; Zandi, S.; Almulki, L.; Tayyari, F.; Shimokawa, H.; et al. Rho kinase inhibition by fasudil ameliorates diabetes-induced microvascular damage. Diabetes 2009, 58, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Kan, L.; Smith, A.; Chen, M.; Ledford, B.T.; Fan, H.; Liu, Z.; He, J.Q. Rho-Associated Kinase Inhibitor (Y-27632) Attenuates Doxorubicin-Induced Apoptosis of Human Cardiac Stem Cells. PLoS ONE 2015, 10, e0144513. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; Saito, Y.; Nakao, K.; Narumiya, S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996, 392, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Noda, K.; Nakajima, S.; Godo, S.; Saito, H.; Ikeda, S.; Shimizu, T.; Enkhjargal, B.; Fukumoto, Y.; Tsukita, S.; Yamada, T.; et al. Rho-kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway in mice. PLoS ONE 2014, 9, e110446. [Google Scholar] [CrossRef]
- Fang, X.; Ueno, M.; Yamashita, T.; Ikuno, Y. RhoA activation and effect of Rho-kinase inhibitor in the development of retinal neovascularization in a mouse model of oxygen-induced retinopathy. Curr. Eye Res 2011, 36, 1028–1036. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Nakao, S.; Arita, R.; Kaizu, Y.; Arima, M.; Zhou, Y.; Kita, T.; Yoshida, S.; Kimura, K.; Isobe, T.; et al. Vascular Normalization by ROCK Inhibitor: Therapeutic Potential of Ripasudil (K-115) Eye Drop in Retinal Angiogenesis and Hypoxia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2264–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollanders, K.; Hove, I.V.; Sergeys, J.; Bergen, T.V.; Lefevere, E.; Kindt, N.; Castermans, K.; Vandewalle, E.; van Pelt, J.; Moons, L.; et al. AMA0428, A Potent Rock Inhibitor, Attenuates Early and Late Experimental Diabetic Retinopathy. Curr. Eye Res 2017, 42, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Petrovski, G.; Vereb, Z.; Facsko, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed. Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef] [PubMed]
- Pesce, N.A.; Canovai, A.; Lardner, E.; Cammalleri, M.; Kvanta, A.; Andre, H.; Dal Monte, M. Autophagy Involvement in the Postnatal Development of the Rat Retina. Cells 2021, 10, 177. [Google Scholar] [CrossRef]
- Wang, S.; Ji, L.Y.; Li, L.; Li, J.M. Oxidative stress, autophagy and pyroptosis in the neovascularization of oxygeninduced retinopathy in mice. Mol. Med. Rep. 2019, 19, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Li, L.; Zhao, Y.; Liu, S.; Li, J.; Zhang, J.; Zhao, Q.; Wang, S. Immunosubunit beta5i Knockout Suppresses Neovascularization and Restores Autophagy in Retinal Neovascularization by Targeting ATG5 for Degradation. Investig. Ophthalmol. Vis. Sci. 2020, 61, 30. [Google Scholar] [CrossRef]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Fader, C.M.; Chiabrando, G.A.; Sanchez, M.C. Effect of Autophagy Modulators on Vascular, Glial, and Neuronal Alterations in the Oxygen-Induced Retinopathy Mouse Model. Front. Cell. Neurosci. 2019, 13, 279. [Google Scholar] [CrossRef] [Green Version]
- Sprott, D.; Poitz, D.M.; Korovina, I.; Ziogas, A.; Phieler, J.; Chatzigeorgiou, A.; Mitroulis, I.; Deussen, A.; Chavakis, T.; Klotzsche-von Ameln, A. Endothelial-Specific Deficiency of ATG5 (Autophagy Protein 5) Attenuates Ischemia-Related Angiogenesis. Arter. Thromb. Vasc. Biol. 2019, 39, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Smithers, L.G.; Gibson, R.A.; McPhee, A.; Makrides, M. Higher dose of docosahexaenoic acid in the neonatal period improves visual acuity of preterm infants: Results of a randomized controlled trial. Am. J. Clin. Nutr. 2008, 88, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Dyall, S.C. Interplay Between n-3 and n-6 Long-Chain Polyunsaturated Fatty Acids and the Endocannabinoid System in Brain Protection and Repair. Lipids 2017, 52, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Fruttiger, M.; Zhu, L.; Chung, S.H.; Barnett, N.L.; Kirk, J.K.; Lee, S.; Coorey, N.J.; Killingsworth, M.; Sherman, L.S.; et al. Conditional Mullercell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J. Neurosci. 2012, 32, 15715–15727. [Google Scholar] [CrossRef] [PubMed]
- Poitry-Yamate, C.L.; Poitry, S.; Tsacopoulos, M. Lactate released by Muller glial cells is metabolized by photoreceptors from mammalian retina. J. Neurosci. 1995, 15, 5179–5191. [Google Scholar] [CrossRef]
- Toft-Kehler, A.K.; Skytt, D.M.; Poulsen, K.A.; Braendstrup, C.T.; Gegelashvili, G.; Waagepetersen, H.; Kolko, M. Limited energy supply in Muller cells alters glutamate uptake. Neurochem. Res. 2014, 39, 941–949. [Google Scholar] [CrossRef]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017, 6. [Google Scholar] [CrossRef]
- Bibb, C.; Young, R.W. Renewal of fatty acids in the membranes of visual cell outer segments. J. Cell Biol. 1974, 61, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K. Retinoids for treatment of retinal diseases. Trends Pharm. Sci. 2010, 31, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Orban, T.; Palczewska, G.; Palczewski, K. Retinyl ester storage particles (retinosomes) from the retinal pigmented epithelium resemble lipid droplets in other tissues. J. Biol. Chem. 2011, 286, 17248–17258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Bazan, N.G.; Gordon, W.C.; Rodriguez de Turco, E.B. Docosahexaenoic acid uptake and metabolism in photoreceptors: Retinal conservation by an efficient retinal pigment epithelial cell-mediated recycling process. Adv. Exp. Med. Biol. 1992, 318, 295–306. [Google Scholar] [CrossRef]
- Gordon, W.C.; Rodriguez de Turco, E.B.; Bazan, N.G. Retinal pigment epithelial cells play a central role in the conservation of docosahexaenoic acid by photoreceptor cells after shedding and phagocytosis. Curr. Eye Res 1992, 11, 73–83. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomita, Y.; Usui-Ouchi, A.; Nilsson, A.K.; Yang, J.; Ko, M.; Hellström, A.; Fu, Z. Metabolism in Retinopathy of Prematurity. Life 2021, 11, 1119. https://doi.org/10.3390/life11111119
Tomita Y, Usui-Ouchi A, Nilsson AK, Yang J, Ko M, Hellström A, Fu Z. Metabolism in Retinopathy of Prematurity. Life. 2021; 11(11):1119. https://doi.org/10.3390/life11111119
Chicago/Turabian StyleTomita, Yohei, Ayumi Usui-Ouchi, Anders K. Nilsson, Jay Yang, Minji Ko, Ann Hellström, and Zhongjie Fu. 2021. "Metabolism in Retinopathy of Prematurity" Life 11, no. 11: 1119. https://doi.org/10.3390/life11111119
APA StyleTomita, Y., Usui-Ouchi, A., Nilsson, A. K., Yang, J., Ko, M., Hellström, A., & Fu, Z. (2021). Metabolism in Retinopathy of Prematurity. Life, 11(11), 1119. https://doi.org/10.3390/life11111119