
CXCR4-CCR7 Heterodimerization Is a Driver of Breast Cancer Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Human Tissues
2.3. Human Gene Expression Analysis
2.4. Cell Lines
2.5. Isolation of Mouse Mammary Epithelial Cells
2.6. Isolation of Human Mammary Epithelial Cells
2.7. In Vivo Metastasis Assay
2.8. Ligand Cooperation Assay
2.9. Flow Cytometry
2.10. Immunofluorescence Analysis
2.11. Proximity Ligation Assay (PLA)
2.12. Forced Heterodimerization
2.13. Transient Transfection of Human Cell Lines
2.14. AlphaScreen cAMP Assay
2.15. Matrigel Invasion Assay
2.16. Statistical Analysis
3. Results
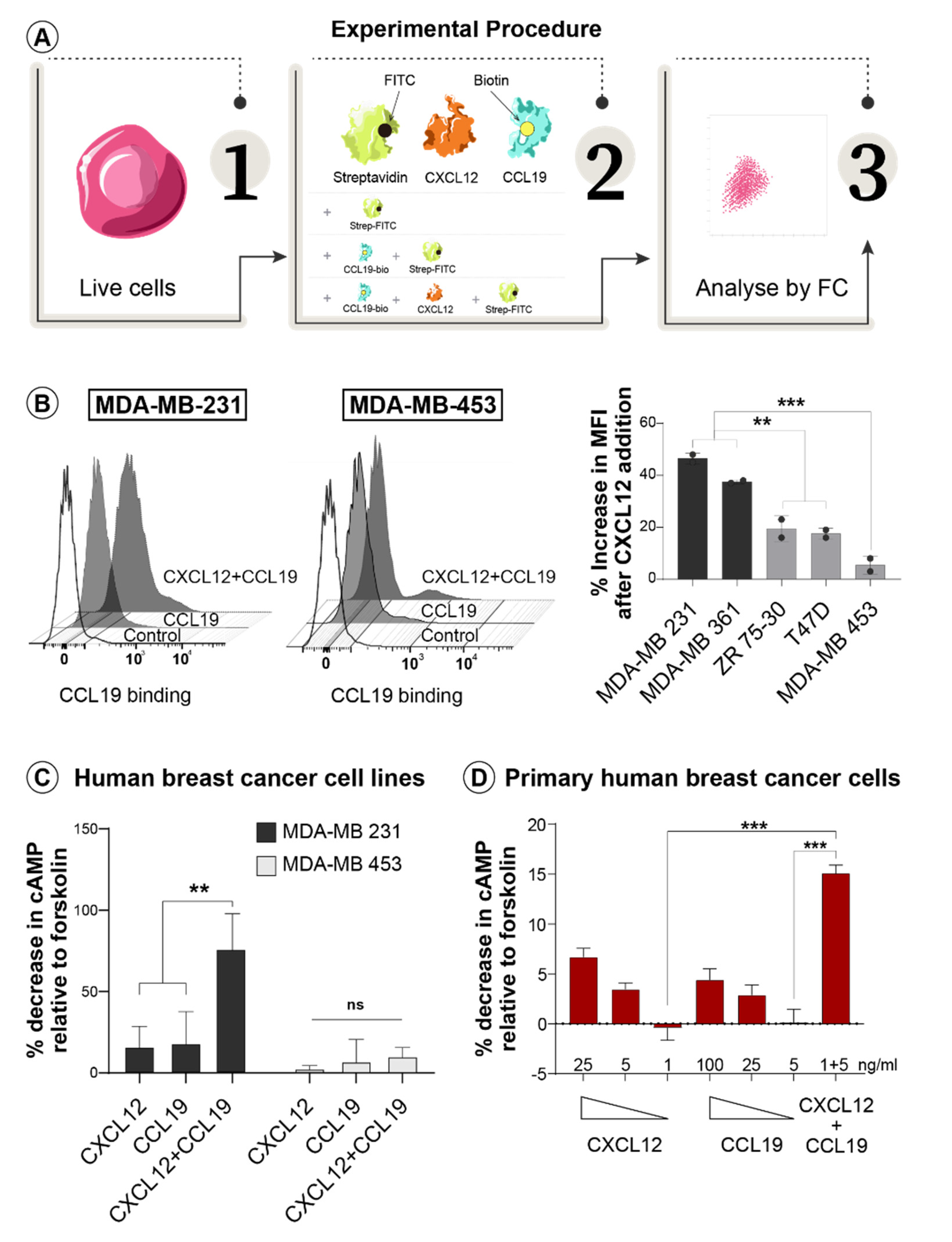
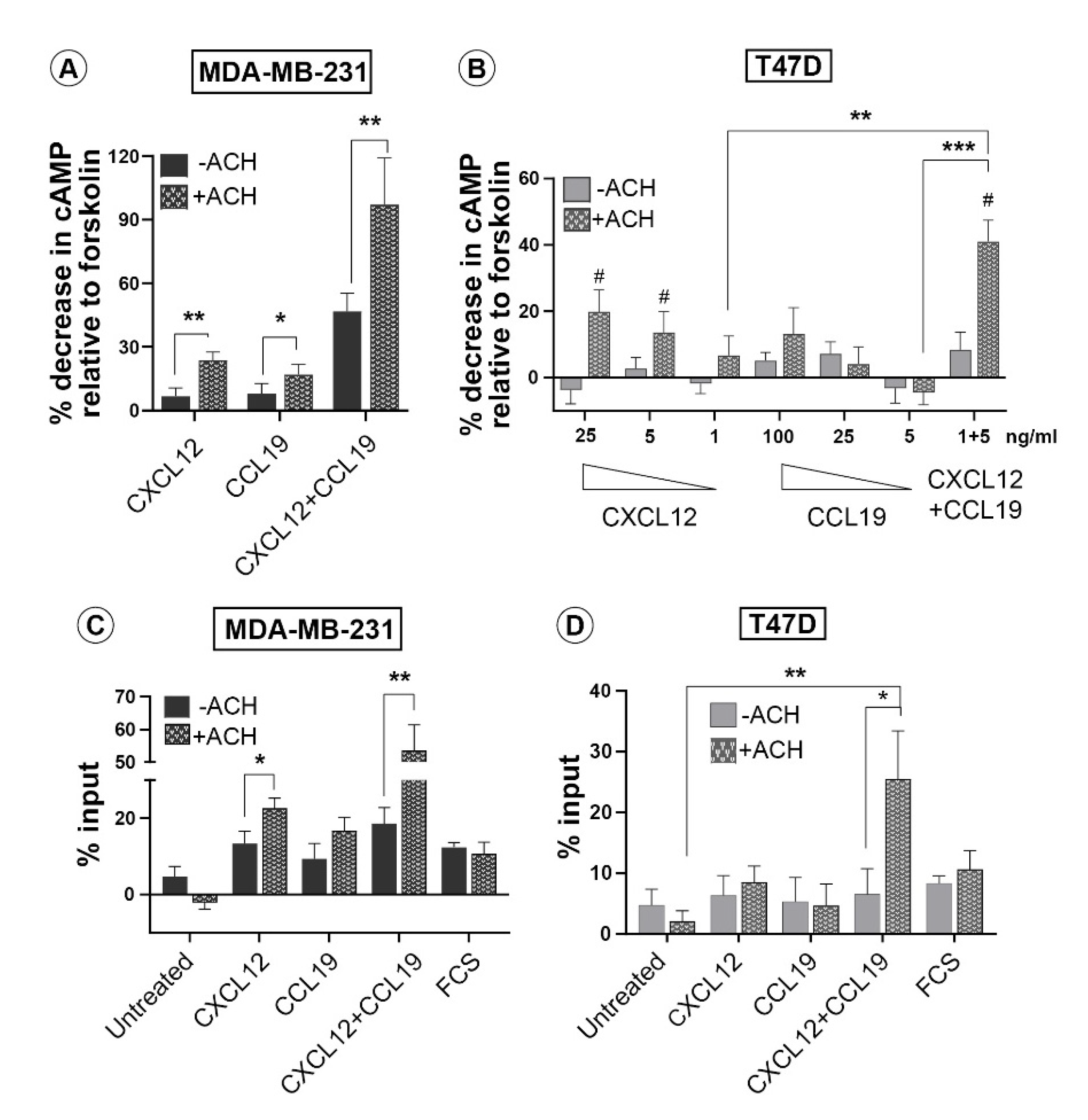
3.1. CXCL12 and CCL19 Synergize in the Cell Surface Binding and Signaling Response Only in Invasive Breast Cancer Cells
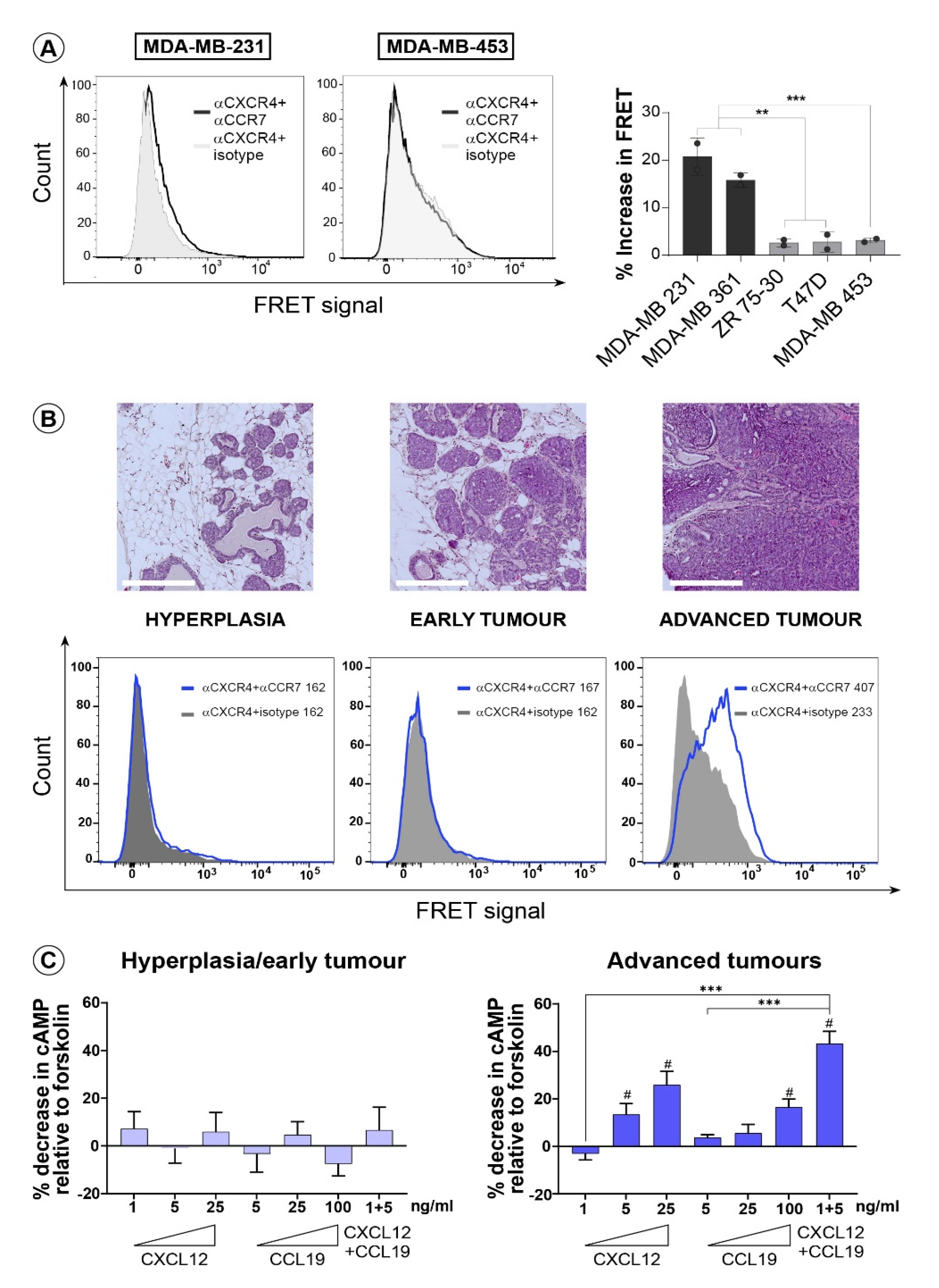
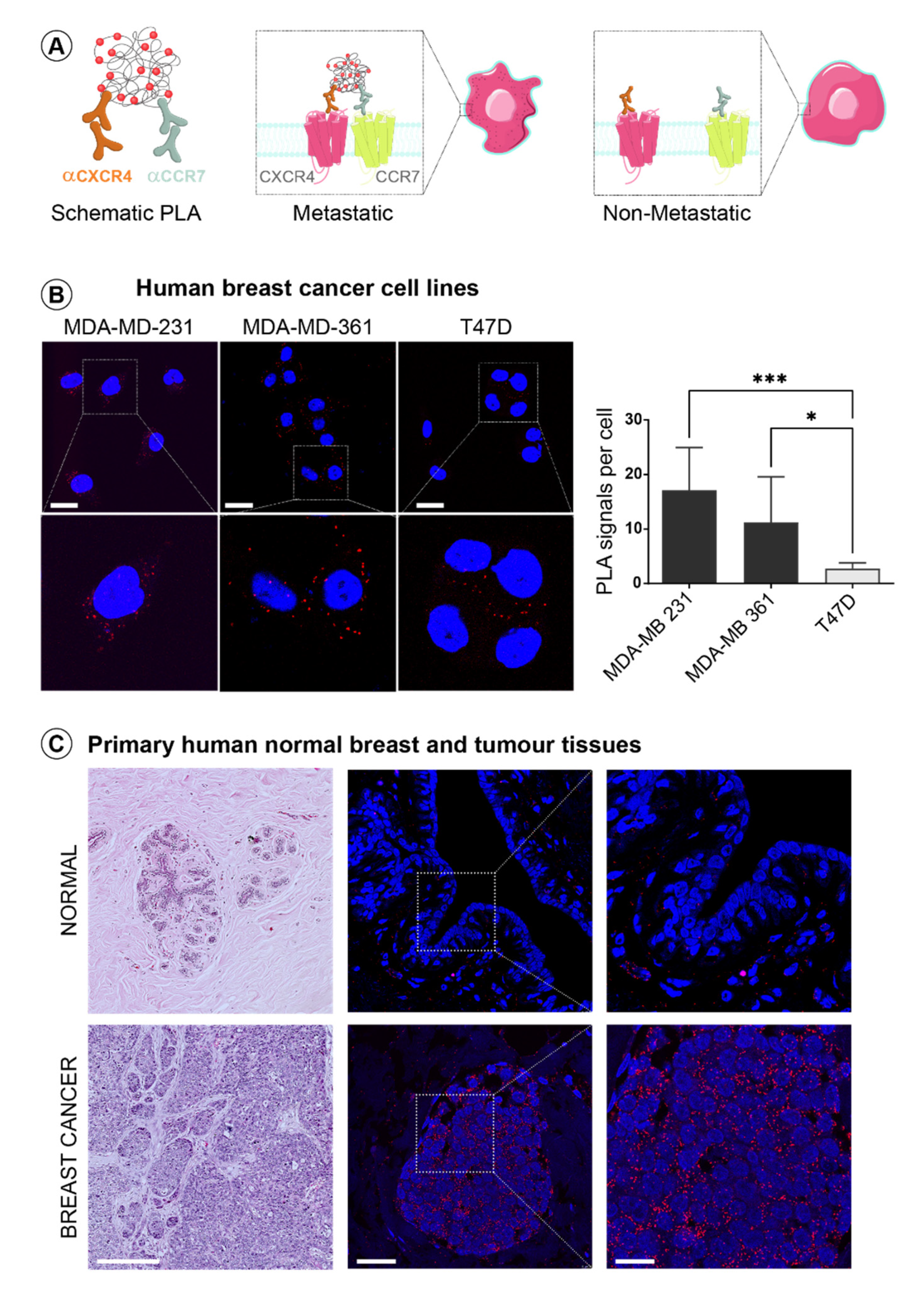
3.2. CXCR4 and CCR7 Interact on the Surface of Invasive Breast Cancer Cells to Form Functional Heterodimeric Receptors
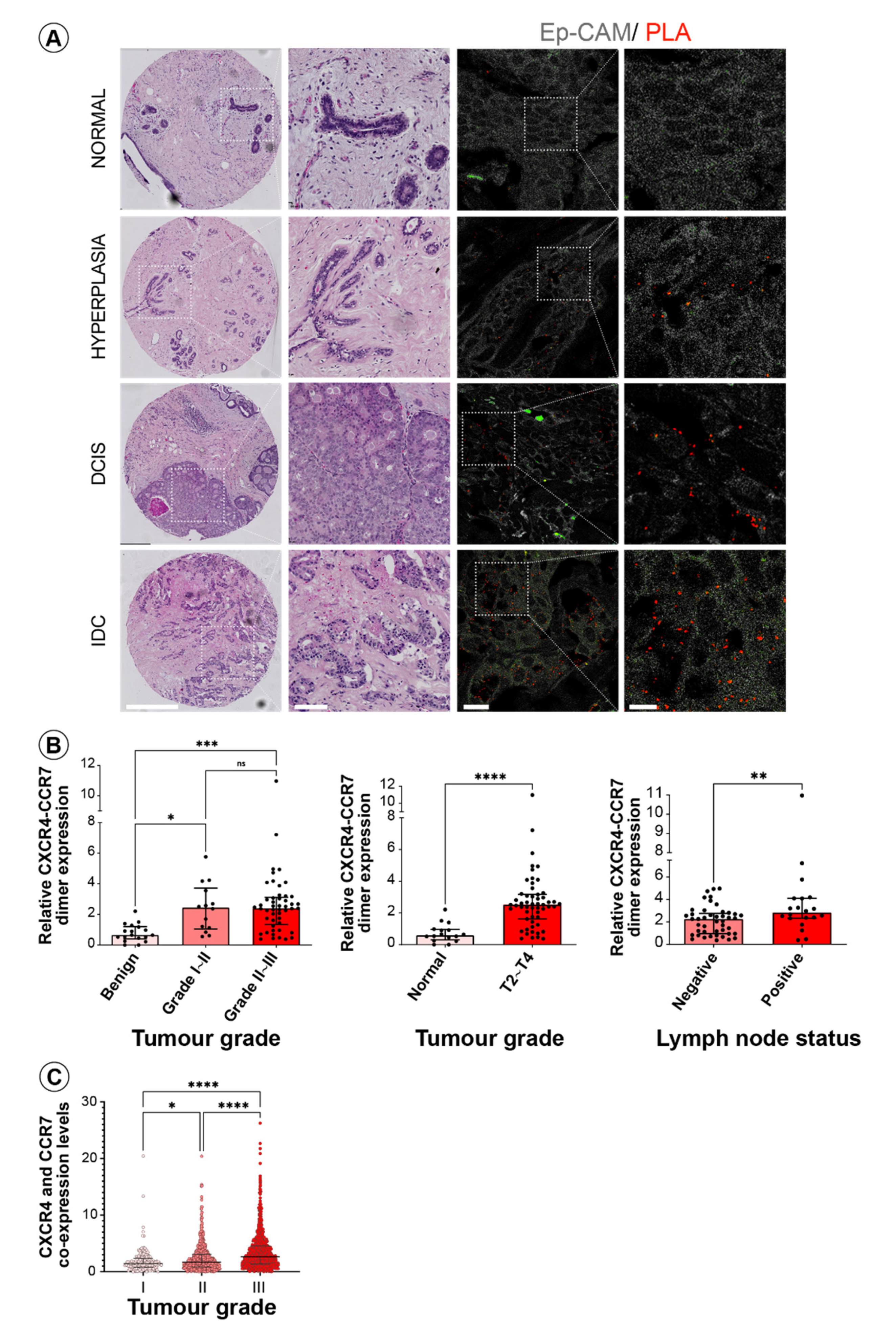
3.3. CXCR4 and CCR7 Association Marks Breast Cancer Progression to Invasive Disease
3.4. Forced Dimerization of CXCR4 and CCR7 Leads to Their Functional Activation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Hojo, S.; Akashi, T.; Yasumoto, K.; Saiki, I. Chemokine receptors in cancer metastasis and cancer cell-derived chemokines in host immune response. Cancer Sci. 2007, 98, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Zu, G.; Luo, B.; Yang, Y.; Tan, Y.; Tang, T.; Zhang, Y.; Chen, X.; Sun, D. Meta-analysis of the prognostic value of C-C chemokine receptor type 7 in patients with solid tumors. Cancer Manag. Res. 2019, 11, 1881–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, A.; Alotaibi, M.; Mroueh, R.; Basheer, H.A.; Afarinkia, K. CCR7 as a therapeutic target in Cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875. [Google Scholar] [CrossRef]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef] [Green Version]
- Luker, G.D.; Yang, J.; Richmond, A.; Scala, S.; Festuccia, C.; Schottelius, M.; Wester, H.J.; Zimmermann, J. At the Bench: Pre-clinical evidence for multiple functions of CXCR4 in cancer. J. Leukoc. Biol. 2021, 109, 969–989. [Google Scholar] [CrossRef]
- Rizeq, B.; Malki, M.I. The Role of CCL21/CCR7 Chemokine Axis in Breast Cancer Progression. Cancers 2020, 12, 1036. [Google Scholar] [CrossRef]
- Mishan, M.A.; Ahmadiankia, N.; Bahrami, A.R. CXCR4 and CCR7: Two eligible targets in targeted cancer therapy. Cell Biol. Int. 2016, 40, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. GPCRs and Signal Transducers: Interaction Stoichiometry. Trends Pharmacol. Sci. 2018, 39, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Abadir, P.M.; Periasamy, A.; Carey, R.M.; Siragy, H.M. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension 2006, 48, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quitterer, U.; AbdAlla, S. Discovery of Pathologic GPCR Aggregation. Front. Med. 2019, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Agostino, G.; Garcia-Cuesta, E.M.; Gomariz, R.P.; Rodriguez-Frade, J.M.; Mellado, M. The multilayered complexity of the chemokine receptor system. Biochem. Biophys. Res. Commun. 2020, 528, 347–358. [Google Scholar] [CrossRef]
- Martinez-Munoz, L.; Villares, R.; Rodriguez-Fernandez, J.L.; Rodriguez-Frade, J.M.; Mellado, M. Remodeling our concept of chemokine receptor function: From monomers to oligomers. J. Leukoc. Biol. 2018, 104, 323–331. [Google Scholar] [CrossRef]
- Mellado, M.; Rodriguez-Frade, J.M.; Vila-Coro, A.J.; Fernandez, S.; de Ana, A.M.; Jones, D.R.; Toran, J.L.; Martinez-A, C. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001, 20, 2497–2507. [Google Scholar] [CrossRef] [Green Version]
- Contento, R.L.; Molon, B.; Boularan, C.; Pozzan, T.; Manes, S.; Marullo, S.; Viola, A. CXCR4-CCR5: A couple modulating T cell functions. Proc. Natl. Acad. Sci. USA 2008, 105, 10101–10106. [Google Scholar] [CrossRef] [Green Version]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef] [Green Version]
- Gahbauer, S.; Pluhackova, K.; Bockmann, R.A. Homo- and heterodimerization of G protein coupled chemokine receptors. Eur. Biophys. J. Biophys. Lett. 2017, 46, S375. [Google Scholar]
- Gahbauer, S.; Pluhackova, K.; Bockmann, R.A. Closely related, yet unique: Distinct homo- and heterodimerization patterns of G protein coupled chemokine receptors and their fine-tuning by cholesterol. PLoS Comput. Biol. 2018, 14, e1006062. [Google Scholar] [CrossRef]
- Hayasaka, H.; Kobayashi, D.; Yoshimura, H.; Nakayama, E.E.; Shioda, T.; Miyasaka, M. The HIV-1 Gp120/CXCR4 axis promotes CCR7 ligand-dependent CD4 T cell migration: CCR7 homo- and CCR7/CXCR4 hetero-oligomer formation as a possible mechanism for up-regulation of functional CCR7. PLoS ONE 2015, 10, e0117454. [Google Scholar] [CrossRef]
- McHeik, S.; Van Eeckhout, N.; De Poorter, C.; Gales, C.; Parmentier, M.; Springael, J.Y. Coexpression of CCR7 and CXCR4 During B Cell Development Controls CXCR4 Responsiveness and Bone Marrow Homing. Front. Immunol. 2019, 10, 2970. [Google Scholar] [CrossRef]
- Luker, K.E.; Lewin, S.A.; Mihalko, L.A.; Schmidt, B.T.; Winkler, J.S.; Coggins, N.L.; Thomas, D.G.; Luker, G.D. Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene 2012, 31, 4750–4758. [Google Scholar] [CrossRef] [Green Version]
- Decaillot, F.M.; Kazmi, M.A.; Lin, Y.; Ray-Saha, S.; Sakmar, T.P.; Sachdev, P. CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. J. Biol. Chem. 2011, 286, 32188–32197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; An, S.; Ward, R.; Yang, Y.; Liu, Y.; Guo, X.X.; Hao, Q.; Xu, T.R. Methods used to study the oligomeric structure of G-protein-coupled receptors. Biosci. Rep. 2017, 37, BSR20160547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, J.D.; Kochetkova, M.; Akekawatchai, C.; Dottore, M.; Lopez, A.; McColl, S.R. Differential functional activation of chemokine receptor CXCR4 is mediated by G proteins in breast cancer cells. Cancer Res. 2006, 66, 4117–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochetkova, M.; Kumar, S.; McColl, S.R. Chemokine receptors CXCR4 and CCR7 promote metastasis by preventing anoikis in cancer cells. Cell Death Differ. 2009, 16, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Shee, K.; Jiang, A.; Varn, F.S.; Liu, S.; Traphagen, N.A.; Owens, P.; Ma, C.X.; Hoog, J.; Cheng, C.; Golub, T.R.; et al. Cytokine sensitivity screening highlights BMP4 pathway signaling as a therapeutic opportunity in ER+ breast cancer. FASEB J. 2019, 33, 1644–1657. [Google Scholar] [CrossRef]
- Boyle, S.T.; Poltavets, V.; Kular, J.; Pyne, N.T.; Sandow, J.J.; Lewis, A.C.; Murphy, K.J.; Kolesnikoff, N.; Moretti, P.A.B.; Tea, M.N.; et al. ROCK-mediated selective activation of PERK signalling causes fibroblast reprogramming and tumour progression through a CRELD2-dependent mechanism. Nat. Cell Biol. 2020, 22, 882–895. [Google Scholar] [CrossRef]
- Boyle, S.T.; Ingman, W.V.; Poltavets, V.; Faulkner, J.W.; Whitfield, R.J.; McColl, S.R.; Kochetkova, M. The chemokine receptor CCR7 promotes mammary tumorigenesis through amplification of stem-like cells. Oncogene 2016, 35, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Koos, B.; Andersson, L.; Clausson, C.M.; Grannas, K.; Klaesson, A.; Cane, G.; Soderberg, O. Analysis of Protein Interactions in situ by Proximity Ligation Assays. Curr. Top. Microbiol. 2014, 377, 111–126. [Google Scholar] [CrossRef]
- Klaesson, A.; Grannas, K.; Ebai, T.; Heldin, J.; Koos, B.; Leino, M.; Raykova, D.; Oelrich, J.; Arngarden, L.; Soderberg, O.; et al. Improved efficiency of in situ protein analysis by proximity ligation using UnFold probes. Sci. Rep.-UK 2018, 8, 5400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, D.; Endo, M.; Ochi, H.; Hojo, H.; Miyasaka, M.; Hayasaka, H. Regulation of CCR7-dependent cell migration through CCR7 homodimer formation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ejendal, K.; Przybyla, J.; Watts, V. Adenylyl cyclase isoform-specific signaling of GPCRs. In G Protein-Coupled Receptors: Structure, Signaling, and Physiology; Siehler, G.M.S., Ed.; Cambridge University Press: Cambridge, UK, 2010; pp. 189–216. [Google Scholar] [CrossRef]
- Milligan, G.; Smith, N.J. Allosteric modulation of heterodimeric G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 615–620. [Google Scholar] [CrossRef]
- Chan, F.K.; Holmes, K.L. Flow cytometric analysis of fluorescence resonance energy transfer: A tool for high-throughput screening of molecular interactions in living cells. Methods Mol. Biol. 2004, 263, 281–292. [Google Scholar] [CrossRef]
- Bagchi, S.; Fredriksson, R.; Wallen-Mackenzie, A. In Situ Proximity Ligation Assay (PLA). Methods Mol. Biol. 2015, 1318, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Xiang, B.; Muthuswamy, S.K. Controlled activation of ErbB1/ErbB2 heterodimers promote invasion of three-dimensional organized epithelia in an ErbB1-dependent manner: Implications for progression of ErbB2-overexpressing tumors. Cancer Res. 2006, 66, 5201–5208. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Guo, L.; Zhao, H.; Zhao, J.; Weng, H.; Zhao, B. CXCR4 over-expression and survival in cancer: A system review and meta-analysis. Oncotarget 2015, 6, 5022–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gahbauer, S.; Bockmann, R.A. Membrane-Mediated Oligomerization of G Protein Coupled Receptors and Its Implications for GPCR Function. Front. Physiol. 2016, 7, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poltavets, V.; Faulkner, J.W.; Dhatrak, D.; Whitfield, R.J.; McColl, S.R.; Kochetkova, M. CXCR4-CCR7 Heterodimerization Is a Driver of Breast Cancer Progression. Life 2021, 11, 1049. https://doi.org/10.3390/life11101049
Poltavets V, Faulkner JW, Dhatrak D, Whitfield RJ, McColl SR, Kochetkova M. CXCR4-CCR7 Heterodimerization Is a Driver of Breast Cancer Progression. Life. 2021; 11(10):1049. https://doi.org/10.3390/life11101049
Chicago/Turabian StylePoltavets, Valentina, Jessica W. Faulkner, Deepak Dhatrak, Robert J. Whitfield, Shaun R. McColl, and Marina Kochetkova. 2021. "CXCR4-CCR7 Heterodimerization Is a Driver of Breast Cancer Progression" Life 11, no. 10: 1049. https://doi.org/10.3390/life11101049
APA StylePoltavets, V., Faulkner, J. W., Dhatrak, D., Whitfield, R. J., McColl, S. R., & Kochetkova, M. (2021). CXCR4-CCR7 Heterodimerization Is a Driver of Breast Cancer Progression. Life, 11(10), 1049. https://doi.org/10.3390/life11101049