
Microfluidic Platforms to Unravel Mysteries of Alzheimer’s Disease: How Far Have We Come?

 ,
,  ,
, 
 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
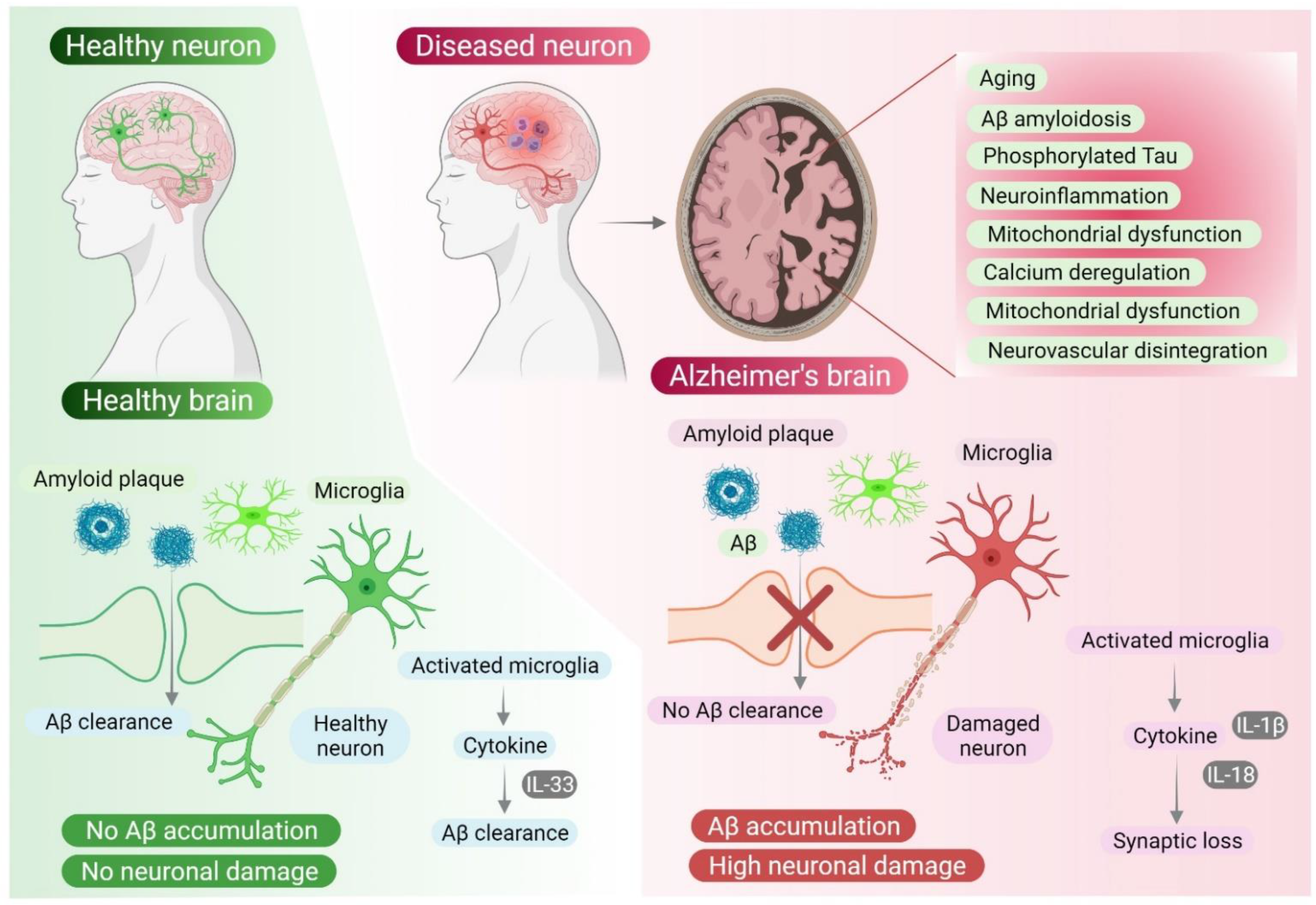
2. Revisiting Alzheimer’s Disease: What Is Known?
2.1. History
2.2. Causes
2.3. Diagnostic Biomarkers and Therapeutics
3. Unsolved Mysteries in Alzheimer’s Disease Research
4. Cellular and Animal Models of AD
4.1. In Vitro Models
4.1.1. Primary Cell Lines
4.1.2. Human Neuroblastoma (SH-SY5Y) Cell Lines
4.1.3. iPSCs-Based Models of AD
4.2. In Vivo Models
4.2.1. Transgenic Animal Models of AD
4.2.2. Non-Transgenic Animal Models of AD
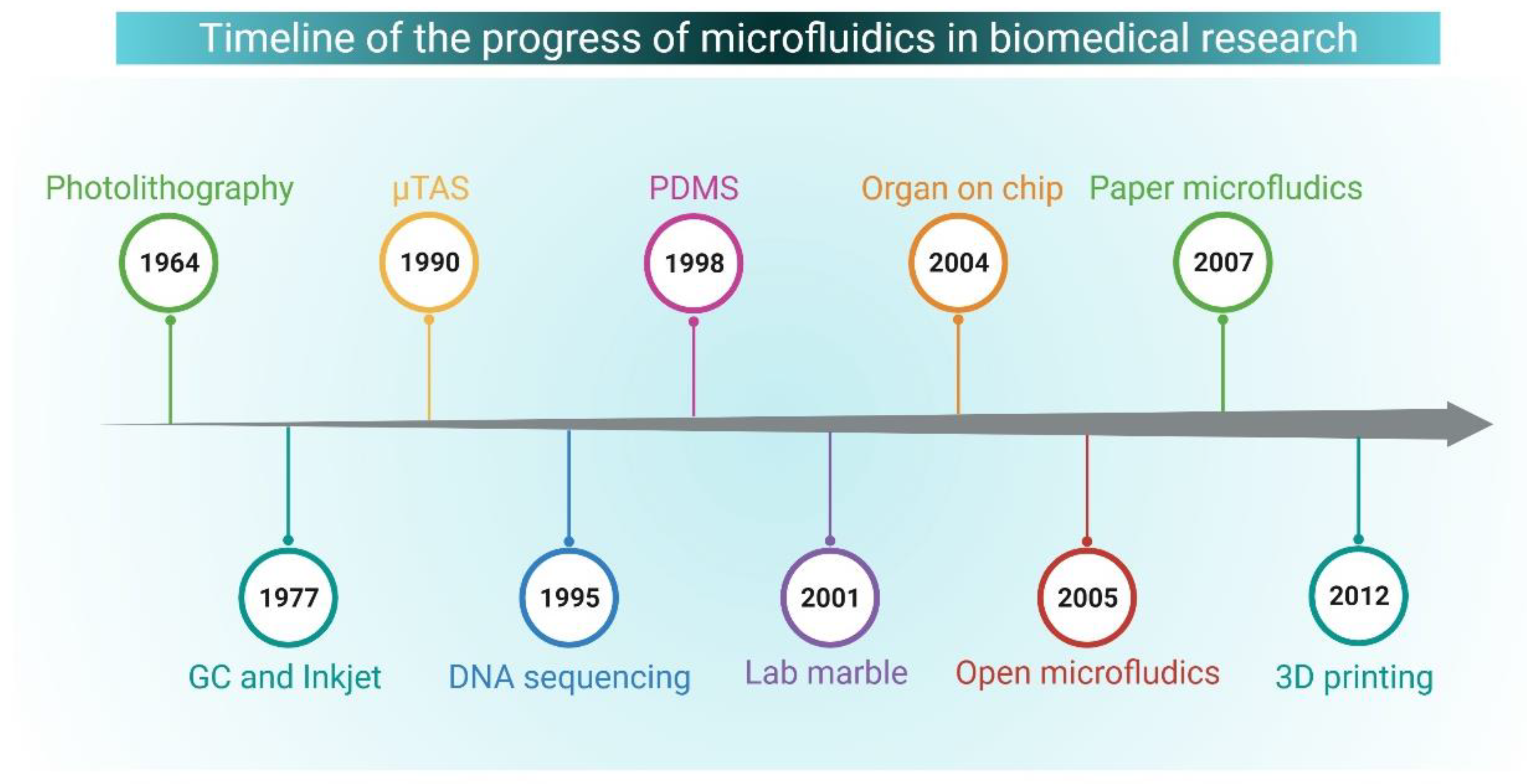
5. Microfluidics: An Overview and Biological Applications

{kind=link}
{kind=link}
{kind=link}
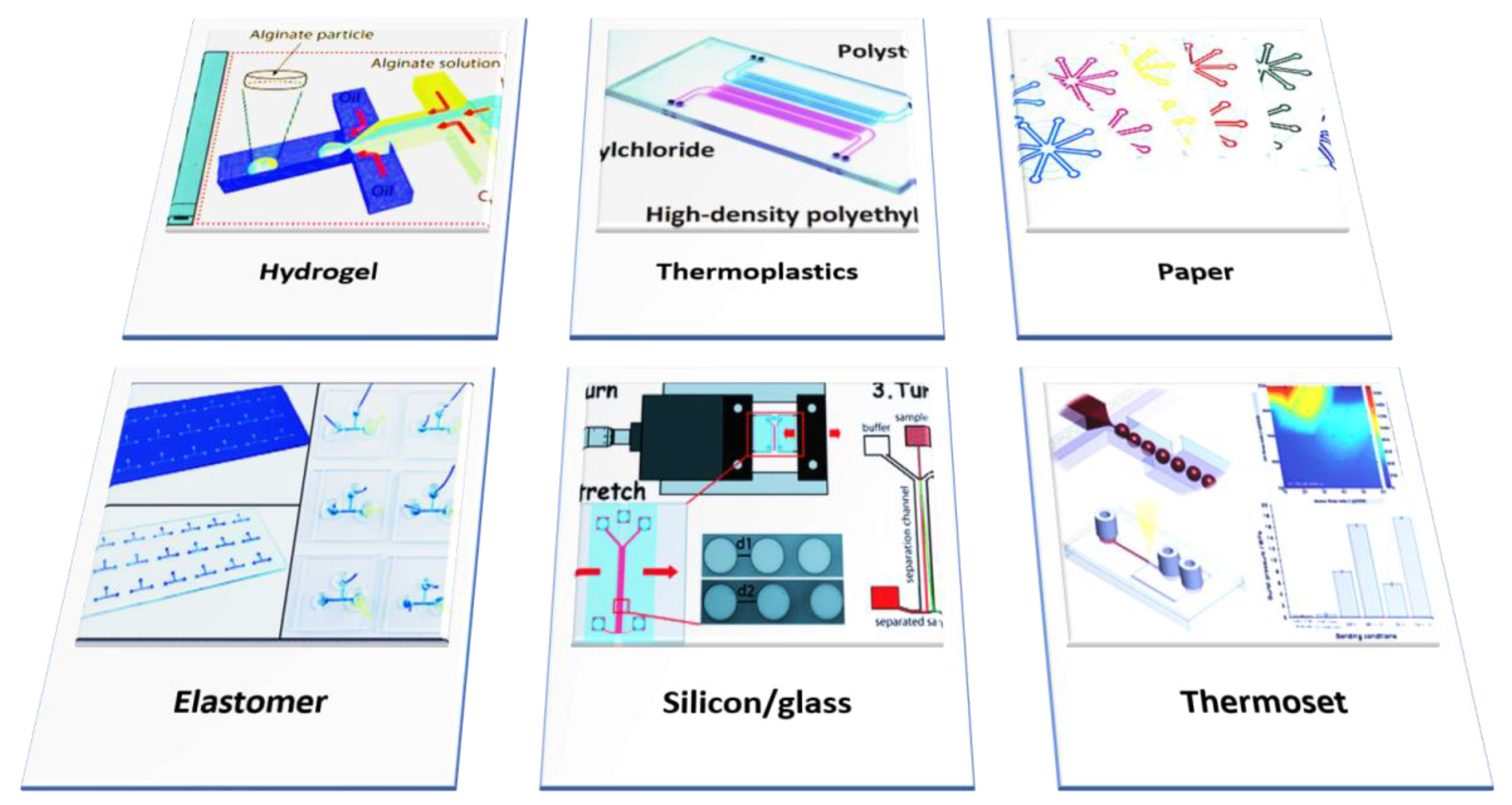
| Properties | Inorganic Materials | Elastomers | Thermoset | Thermoplastics | Hydrogel | Paper |
|---|---|---|---|---|---|---|
| Examples | Silicon/Glass | PDMS | Polyester | Polyethylene, Polystyrene Polycarbonate Polyurethane, Teflon, Cyclic Olefin Co-polymer (COC/COP) | Hyaluronic Acid, Agarose, PEG-DA, Alginate, PMMA, And Chitosan | - |
| Biological Use | Drug Screening, Assays | Assays, Cell Culture | Capillary | Electrophoresis, DNA Sequencing, PCR | Study Cell-Cell and Cell-Matrix Interaction | Diagnostics |
| Young’s Modulus | 130–180/50–90 | ~0.0005 | 2.0–2.7 | 1.4–4.1 | Low | 0.0003–0.0025 |
| Fabrication Technique | Photolithography | Casting, 3D Printing | Casting/ Photopolymerization | Thermomoulding | Casting/Photopolymerization | Photolithography, Printing |
| Valving | Yes | Yes | No | No | Yes | Yes |
| Channel Dimension/Profile | <100 nm/3D | <1 µm/3D | <100 nm/Arbitrary 3D | ~100 nm/3D | ~10 µm/3D | ~200 µm/2D |
| Thermostability | Very High | Medium | High | Medium-High | Low | Medium |
| Oxygen Permeability | <0.01 | ~500 | 0.03-1 | 0.05–5 | >1 | >1 |
| Solvent Compatibility | Very High | Low | High | Medium-High | Low | Medium |
| Hydrophobicity | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophilic | Amphiphilic |
| Surface Charge | Very Stable | Stable | Stable | Stable | - | - |
| Transparency | No/High | High | High | Medium-High | Low-Medium | Low |
| Cost | High | Low | High | Low | Medium | Low |
| Disadvantage | High Cost, Brittle | Protein Adsorption, Permeability, Autofluorescence | Rigid, Poor Conductivity, Non-Recyclable | Low Melting Point, Brittle | Non-Adherent, Low Mechanical Strength | Porous, Sample Consumption |
| Reference(s) | [121,122] | [123,124] | [125] | [126,127,128] | [129,130] | [131] |
6. Application of Microfluidics in Neurodegenerative Studies
7. Impact of Microfluidic Tools in Alzheimer’s Disease Research: Recent Developments
| Cells/Peptide | Flow Control Device | Flow Surface | Active/Passive | Application | Reference(s) |
|---|---|---|---|---|---|
| Axon | NA | Glass | P | Study axonal function | [154] |
| Neural Progenitor Cell | Osmotic micropump | - | A | Study the neurotoxicity of amyloid beta | [36] |
| Neuron | Osmotic micropump | Glass | A & P | in vitro brain model, high-throughput drug screening | [143] |
| Brain Cells | Pneumatically-driven pumps | Polysulfone | P | To provide MPSs for in vitro drug discovery | [175] |
| Aβ42 Peptide | Precision pump | Glass | A | Aβ (1–42) detection | [168] |
| Aβ Peptide | Syringe | - | A | - | [176] |
| Axons | N/A | Glass | P | Study impaired axonal deficit | [156] |
| Axons | N/A | MEA | P | Investigate axonal signals in developmental stage | [177] |
| Neurites | Syringe | Glass | A | Study durotactic behavior of cells and neurite growth | [161] |
| Axons | Gravity/Hydrostatic pressure | PCB/Glass | P | Study axonal physiology and modeling CNS injury | [178] |
| Soma and Axon | N/A | Glass | P | Compartmentalizing the network structure into interconnected sub-populations | [179] |
| Hippocampal Neuronal/Glia Cells | Pressure gradient | Glass | P | Probing the functional synaptic connectivity between mixed primary hippocampal co-cultures | [163] |
| Dendrite | N/A | PDMS | NM | Investigate dendrite-to-nucleus signaling | [170] |
| Oligodendrocyte | N/A | Glass | P | - | [172] |
| Drg/Mc3t3-E1 | N/A | Glass | NM | Mimicking the in vivo scenario to study the interaction between the peripheral nervous system and bone cells | [160] |
| Nmj | Pipette | Glass | N/A | Study subcellular microenvironments, NMJ formation, maintenance, and disruption | [162] |
| Axons | Pipette | Glass | P | Perform drug screening assays | [180] |
| Dendrites and Somata | Syringe | Glass | A | Manipulate synaptic regions and presynaptic and postsynaptic compartments in vitro | [101] |
| Glial Cells/Motor Neurons | N/A | Glass | P | Study interactions with glial cells and other skeletal cells in the chamber | [159] |
| Astrocyte | N/A | acrylic plate | P | AD triculture model showing beta-amyloid aggregation, phosphorylated tau accumulation, and neuroinflammatory activity | [144] |
| Tau | N/A | Glass | P | Study effects of tau on mitochondrial transport | [181] |
| (Aβ) Peptides | N/A | Glass | P | Study effects of local Aβ stress on neuronal sub-compartments and networks | [182] |
| ADAM10 | Syringe | N/A | A | ADAM10 biomarker detection in plasma and cerebrospinal fluid | [167] |
| Tau | N/A | Glass | P | Quantify AD-derived Tau propagation | [147] |
| Aβ | N/A | Glass | P | Study roles of Aβ on microglial accumulation | [183] |
| Aβ | Syringe | Overflow microfluidic networks | A | Study cell-to-cell communication, role of astrocytes derived from cortex and hippocampus on neuronal viability | [146] |
| Axons | - | Glass | - | Study mechanisms of indirect axonal excitotoxicity | [174] |
| Neurites | Hydrostatic pressure | Glass and Polystyrene | P | Grow neuronal culture | [142] |
| Cortical Neurons | Pressure difference | Glass | P | Synthesize experimental models emulating pathological states | [173] |
| Ren-WT/Ren-AD Cells | N/A | Glass | P | Grow 3D human neural cell culture, screen novel drugs capable of passing through the BBB to reach deeper neural tissues | [148] |
| Protein | N/A | Glass | P | Detect protein aggregation | [184] |
| Axons | Hydrostatic pressure | Glass or Polystyrene | P | Study localized axon-glia interaction and signaling | [185] |
| Axons | N/A | Glass | P | Examine axonal trauma in neuronal networks | [166] |
| Axons-glia | Hydrostatic pressure | Glass | P | Study axon-glia interactions | [186] |
| Neurites | Syringe | Glass | A | Investigating chemotaxis of neutrophils | [187] |
8. Challenges in the Application of Microfluidics in the Alzheimer’s Disease Research
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADAM10 | A Disintegrin and Metalloprotease 10 |
| Apo E | Apolipoprotein E |
| Aβ | Amyloid-beta |
| CD33 | Complementary determinant 33 |
| IL | Interleukin |
| MMP | Matrix metallopeptidase 9 |
| NMJ | Neuromuscular junction |
| PCR | Polymerase chain reaction |
| PDMS | Polydimethylsiloxane |
| PK/PD | Pharmacokinetic/Pharmacodynamic |
| TNF-α | Tumor necrosis factor-alpha |
| TREM2 | Triggering receptor expressed on myeloid cells2 |
References
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor-kappa β as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef] [Green Version]
- Crimins, J.L.; Pooler, A.; Polydoro, M.; Luebke, J.I.; Spires-Jones, T.L. The Intersection of Amyloid Beta and Tau in Glutamatergic Synaptic Dysfunction and Collapse in Alzheimer’s Disease. Ageing Res. Rev. 2013, 12, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Jha, N.K.; Jha, S.K.; Ramani, K.; Ambasta, R.K. Tau phosphorylation, molecular chaperones, and ubiquitin E3 ligase: Clinical relevance in Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 341–461. [Google Scholar] [CrossRef]
- Lee, J.C.; Kim, S.J.; Hong, S.; Kim, Y.S. Diagnosis of Alzheimer’s Disease Utilizing Amyloid and Tau as Fluid Biomarkers. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, S.K.; Jha, N.K.; Kumar, D.; Sharma, R.; Shrivastava, A.; Ambasta, R.K.; Kumar, P. Stress-Induced Synaptic Dysfunction and Neurotransmitter Release in Alzheimer’s Disease: Can Neurotransmitters and Neuromodulators be Potential Therapeutic Targets? J. Alzheimers Dis. 2017, 57, 1017–1039. [Google Scholar] [CrossRef] [PubMed]
- Jha, N.K.; Jha, S.K.; Kumar, D.; Kejriwal, N.; Sharma, R.; Ambasta, R.K.; Kumar, P. Impact of Insulin Degrading Enzyme and Neprilysin in Alzheimer’s Disease Biology: Characterization of Putative Cognates for Therapeutic Applications. J. Alzheimers Dis. 2015, 48, 891–917. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiuo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- D’Argenio, V.; Sarnataro, D. New Insights into the Molecular Bases of Familial Alzheimer’s Disease. J. Pers. Med. 2020, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Lynch, C. World Alzheimer Report 2019: Attitudes to Dementia, a Global Survey. Alzheimers Dement. 2020, 16, e038255. [Google Scholar] [CrossRef]
- 2021 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2021, 17, 327–406. [CrossRef]
- International, D. World Alzheimer Report 2020–Design Dignity Dementia: Dementia-Related Design and the Built Environment. Alzheimers Dis. Int. 2020, 1, 106–113. [Google Scholar]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s Disease: The Challenge of the Second Century. Sci. Transl. Med. 2011, 3, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocahan, S.; Doğan, Z. Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-Methyl-D-Aspartate Receptors, Tau Protein and Other Risk Factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments for Alzheimer’s Disease. Ther. Adv. Neurol. Disord. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and Cellular Mechanisms Underlying the Pathogenesis of Alzheimer’s Disease. Mol. Neurodegener. 2020, 15, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Strassnig, M.; Ganguli, M. About a Peculiar Disease of the Cerebral Cortex: Alzheimer’s Original Case Revisited. Psychiatry 2005, 2, 30–33. [Google Scholar] [PubMed]
- You, Y.; Perkins, A.; Cisternas, P.; Muñoz, B.; Taylor, X.; You, Y.; Garringer, H.J.; Oblak, A.L.; Atwood, B.K.; Vidal, R.; et al. Tau as a Mediator of Neurotoxicity Associated to Cerebral Amyloid Angiopathy. Acta Neuropathol. Commun. 2019, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.C.; Jiang, Z.F. Accumulated Amyloid-β Peptide and Hyperphosphorylated Tau Protein: Relationship and Links in Alzheimer’s Disease. J. Alzheimers Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Zhao, P.; Nandakumar, K.; Wang, L.; Song, Y. Microfluidics-Based Systems in Diagnosis of Alzheimer’s Disease and Biomimetic Modeling. Micromachines 2020, 11, 787. [Google Scholar] [CrossRef]
- Velve-Casquillas, G.; Le Berre, M.; Piel, M.; Tran, P.T. Microfluidic Tools for Cell Biological Research. Nano Today 2010, 5, 28–47. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. ASSAY Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, D.; Enright, H.A.; Cadena, J.; Peters, S.K.; Sales, A.P.; Osburn, J.J.; Soscia, D.A.; Kulp, K.S.; Wheeler, E.K.; Fischer, N.O. Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Sci. Rep. 2019, 9, 1–5. [Google Scholar]
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deture, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, C.C.; Zheng, H.; Huang, T.Y. Amyloid, Tau, Pathogen Infection and Antimicrobial Protection in Alzheimer’s Disease -Conformist, Nonconformist, and Realistic Prospects for AD Pathogenesis. Transl. Neurodegener. 2018, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s Disease. Subcell. Biochem. 2012, 65, 329–352. [Google Scholar]
- Jack, C.R., Jr.; Holtzman, D.M. Biomarker modeling of Alzheimer’s disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s Disease Pathology Propagation by Exosomes Containing Toxic Amyloid-Beta Oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; LeVine, H., III. Alzheimer’s Disease and the $β$-Amyloid Peptide. J. Alzheimers Dis. 2010, 19, 311. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Tanzi, R.E.; Bertram, L. Twenty Years of the Alzheimer’s Disease Amyloid Hypothesis: A Genetic Perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Mobley, W.C. Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons:Converging Insights from Alternative Hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenowitz, W.D.; Nelson, P.T.; Besser, L.M.; Heller, K.B.; Kukull, W.A. Cerebral Amyloid Angiopathy and Its Co-Occurrence with Alzheimer’s Disease and Other Cerebrovascular Neuropathologic Changes. Neurobiol. Aging 2015, 36, 2702–2708. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and Inflammation in Alzheimers Disease. CNS Neurol Disord. Drug Targets 2012, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Chae, S.; Kim, J.H.; Barald, K.F.; Park, J.Y.; Lee, S.H. Neurotoxic Amyloid Beta Oligomeric Assemblies Recreated in Microfluidic Platform with Interstitial Level of Slow Flow. Sci. Rep. 2013, 3, 1921. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.T.; Tan, L. Role of Pro-Inflammatory Cytokines Released from Microglia in Alzheimer’s Disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Ismail, R.; Parbo, P.; Madsen, L.S.; Hansen, A.K.; Hansen, K.V.; Schaldemose, J.L.; Kjeldsen, P.L.; Stokholm, M.G.; Gottrup, M.G.; Eskildsen, S.F.; et al. The Relationships between Neuroinflammation, Beta-Amyloid and Tau Deposition in Alzheimer’s Disease: A Longitudinal PET Study. J. Neuroinflammation 2020, 17, 151. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzeriat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1956. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s Disease: Current Evidence and Future Directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix Metalloproteinases in the Brain and Blood–Brain Barrier: Versatile Breakers and Makers. Br. J. Pharmacol. 2016, 36, 1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Bird, T.D. Genetic Aspects of Alzheimer Disease. Genet. Med. 2009, 10, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Birks, J.S.; Chong, L.Y.; Evans, J.G. Rivastigmine for Alzheimer’s Disease. Cochrane Database Syst. Rev. 2015, 9, CD001191. [Google Scholar]
- Woodruff-Pak, D.S.; Vogel, R.W.; Wenk, G.L. Galantamine: Effect on Nicotinic Receptor Binding, Acetylcholinesterase Inhibition, and Learning. Proc. Natl. Acad. Sci. USA 2001, 98, 2089–2094. [Google Scholar] [CrossRef]
- Lilienfeld, S. Cholinesterase Inhibitors for Alzheimer Disease. JAMA 2003, 289, 2359. [Google Scholar] [CrossRef]
- Govind, N. Donepezil for Dementia Due to Alzheimer’s Disease. Br. J. Community Nurs. 2020, 25, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.A.; Antimisiaris, D.; O’Brien, J. Drugs for Alzheimer’s Disease: Are They Effective? Pharm. Ther. 2010, 35, 208–211. [Google Scholar]
- Villa, C. Biomarkers for Alzheimer’s Disease: Where Do We Stand and Where Are We Going? J. Pers. Med. 2020, 10, 238. [Google Scholar] [CrossRef]
- Ray, S.; Britschgi, M.; Herbert, C.; Takeda-Uchimura, Y.; Boxer, A.; Blennow, K.; Friedman, L.F.; Galasko, D.R.; Jutel, M.; Karydas, A.; et al. Classification and Prediction of Clinical Alzheimer’s Diagnosis Based on Plasma Signaling Proteins. Nat. Med. 2007, 13, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- O’Bryant, S.E.; Xiao, G.; Barber, R.; Reisch, J.; Hall, J.; Cullum, C.M.; Doody, R.; Fairchild, T.; Adams, P.; Wilhelmsen, K.; et al. A Blood-Based Algorithm for the Detection of Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2011, 32, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galimberti, D.; Villa, C.; Fengolio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.G.; Arighi, A.; Fumagalli, G.; Scrapini, E. Circulating MiRNAs as Potential Biomarkers in Alzheimer’s Disease. J. Alzheimers Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Konietzko, U. Amyloid-β Immunisation for Alzheimer’s Disease. Lancet Neurol. 2008, 7, 805. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.Y.; Cheng, I.H.; Lee, C.C.; Chiu, M.J.; Lee, M.J.; Chen, T.F.; Hsu, J.L. Clinical phenotype of G206D mutation in the presenilin 1 gene in pathologically confirmed familial Alzheimer’s disease. J. Alzheimers Dis. 2011, 25, 145–150. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and Progress of Hypotheses and Clinical Trials for Alzheimer’s Disease. Signal Transduct. Target. Ther. 2019, 4, 1–22. [Google Scholar]
- Takeda, S. Tau Propagation as a Diagnostic and Therapeutic Target for Dementia: Potentials and Unanswered Questions. Front. Neurosci. 2019, 13, 1274. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-Mediated Neurodegeneration in Alzheimer’s Disease and Related Disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Marshall, K.E.; Vadukul, D.M.; Dahal, L.; Theisen, A.; Fowler, M.W.; Al-Hilaly, Y.; Ford, L.; Kemenes, G.; Day, I.J.; Staras, K.; et al. A Critical Role for the Self-Assembly of Amyloid-Β1-42 in Neurodegeneration. Sci. Rep. 2016, 6, 30182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broersen, K.; Rousseau, F.; Schymkowitz, J. The Culprit behind Amyloid Beta Peptide Related Neurotoxicity in Alzheimer’s Disease: Oligomer Size or Conformation? Alzheimers Res. Ther. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbito, A.; Dulewicz, M.; Kulczyńska-Przybik, A.; Mroczko, B. Biochemical Markers in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1989. [Google Scholar] [CrossRef] [Green Version]
- O’Bryant, S.E.; Mielke, M.M.; Rissman, R.A.; Lista, S.; Vanderstichele, H.; Zetterberg, H.; Lewczuk, P.; Posner, H.; Hall, J.; Johnson, L.; et al. Blood-Based Biomarkers in Alzheimer Disease: Current State of the Science and a Novel Collaborative Paradigm for Advancing from Discovery to Clinic. Alzheimers Dement. 2017, 13, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal Models of Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, Biologic Function and Clinical Potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Yin, Q.; Ji, X.; Lv, R.; Pei, J.J.; Du, Y.; Shen, C.; Hou, X. Targetting Exosomes as a New Biomarker and Therapeutic Approach for Alzheimer’s Disease. Clin. Interv. Aging 2020, 15, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Howitt, J.; Hill, A.F. Exosomes in the Pathology of Neurodegenerative Diseases. J. Biol. Chem. 2016, 291, 26589–26597. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.G.; Gray, E.; Heman-Ackah, S.M.; Mäger, I.; Talbot, K.; El Andaloussi, S.; Wood, M.J.; Turner, M.R. Extracellular Vesicles in Neurodegenerative Disease-Pathogenesis to Biomarkers. Nat. Rev. Neurol. 2016, 12, 346–357. [Google Scholar] [CrossRef]
- Saraceno, C.; Musardo, S.; Marcello, E.; Pelucchi, S.; Luca, M.D. Modeling Alzheimer’s Disease: From Past to Future. Front. Pharmacol. 2013, 4, 77. [Google Scholar] [CrossRef] [Green Version]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-β and Alzheimer’s Disease Type Pathology Differentially Affects the Calcium Signalling Toolkit in Astrocytes from Different Brain Regions. Cell Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional Impairment of Microglia Coincides with Beta-Amyloid Deposition in Mice with Alzheimer-like Pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Nguyen, T.L.X.; Kim, C.K.; Cho, J.-H.; Lee, K.-H.; Ahn, J.-Y. Neuroprotection Signaling Pathway of Nerve Growth Factor and Brain-Derived Neurotrophic Factor against Staurosporine Induced Apoptosis in Hippocampal H19-7/IGF-IR [Corrected]. Exp. Mol. Med. 2010, 42, 583–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, L.M.D.; Bastiani, M.A.D.; Rico, E.P.; Schonhofen, P.; Pfaffenseller, B.; Wollenhaupt-Aguiar, B.; Grun, L.; Barbe-Tuana, F.; Zimmer, E.R.; Castro, M.A.A.; et al. Cholinergic Differentiation of Human Neuroblastoma SH-SY5Y Cell Line and Its Potential Use as an In vitro Model for Alzheimer’s Disease Studies. Mol. Neurobiol. 2019, 56, 7355–7367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, H.; Zhao, X.; Lin, X.; Tan, C.; Cao, G.; Wang, Z. Neuroprotective Effects of Salidroside against Beta-Amyloid-Induced Oxidative Stress in SH-SY5Y Human Neuroblastoma Cells. Neurochem. Int. 2010, 57, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Forster, J.I.; Koglsberger, S.; Trefois, C.; Boyd, O.; Baumuratov, A.S.; Buck, L.; Balling, R.; Antony, P.M.A. Characterization of Differentiated SH-SY5Y as Neuronal Screening Model Reveals Increased Oxidative Vulnerability. J. Biomol. Screen. 2016, 21, 496–509. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Li, Y.; Lin, L.; Cao, Y. Anti-Autophagic and Anti-Apoptotic Effects of Memantine in a SH-SY5Y Cell Model of Alzheimer’s Disease via Mammalian Target of Rapamycin-Dependent and -Independent Pathways. Mol. Med. Rep. 2015, 12, 7615–7622. [Google Scholar] [CrossRef] [Green Version]
- Dafnis, I.; Argyri, L.; Sagnou, M.; Tzinia, A.; Tsilibary, E.C.; Stratikos, E.; Chroni, A. The Ability of Apolipoprotein E Fragments to Promote Intraneuronal Accumulation of Amyloid Beta Peptide 42 Is Both Isoform and Size-Specific. Sci. Rep. 2016, 6, 30654. [Google Scholar] [CrossRef] [Green Version]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling Familial Alzheimer’s Disease with Induced Pluripotent Stem Cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.P.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T. The Familial Alzheimer’s Disease APPV717I Mutation Alters APP Processing and Tau Expression in IPSC-Derived Neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunda, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s Disease with IPSCs Reveals Stress Phenotypes Associated with Intracellular Aβ and Differential Drug Responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Gorp, S.V.; Nazor, K.L.; Boscolo, F.S.; et al. Probing Sporadic and Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Jones, V.C.; Atkinson, R.-D.; Verkharatsky, A.; Mohamet, L. Aberrant IPSC-Derived Human Astrocytes in Alzheimer’s Disease. Cell Death Dis. 2017, 8, e2696. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerakis, Y.; Hetz, C. Brain Organoids: A next Step for Humanized Alzheimer’s Disease Models? Mol. Psychiatry 2019, 24, 474–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Hunsberger, J.G.; Simeonov, A.; Malik, N.; Pei, Y.; Rao, M. Concise review: Modeling central nervous system diseases using induced pluripotent stem cells. Stem Cells Transl. Med. 2014, 3, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bao, X.; Wang, R. Experimental Models of Alzheimer’s Disease for Deciphering the Pathogenesis and Therapeutic Screening (Review). Int. J. Mol. Med. 2016, 37, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet, A.-B.; Ohno, M.; Disterhoft, J.; Eldik, L.V.; et al. Intraneuronal Beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Grootendorst, J.; Bour, A.; Vogel, E.; Kelche, C.; Sullivan, P.M.; Dodart, J.-C.; Bales, K.; Mathis, C. Human ApoE Targeted Replacement Mouse Lines: H-ApoE4 and h-ApoE3 Mice Differ on Spatial Memory Performance and Avoidance Behavior. Behav. Brain Res. 2005, 159, 1–14. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE Isoform-Dependent Effects on Brain β-Amyloid Levels in PDAPP Transgenic Mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef]
- Flores, J.A.-C.; Madrid, A.; Fernandez, P.L.; Perez, A.R.-L.; Oviedo, D.C.; Britton, G.B.; Carreira, M. Critical Review of the Alzheimer’s Disease Non-Transgenic Models: Can They Contribute to Disease Treatment? J. Alzheimers Dis. 2021, 82, S227–S250. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, J.; Shi, J.-S. SAMP8 Mice as a Model of Age-Related Cognition Decline with Underlying Mechanisms in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 75, 385–395. [Google Scholar] [CrossRef]
- Whitesides, G.M. The Origins and the Future of Microfluidics. Nat. Cell Biol. 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Xia, Y.; Whitesides, G.M. Soft Lithography. Angew. Chem. Int. Ed. 1998, 37, 550–575. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Ostuni, E.; Takayama, S.; Jiang, X.; Ingber, D.E. Soft Lithography in Biology and Biochemistry. Annu. Rev. Biomed. Eng. 2001, 3, 335–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, A.; Toner, M. Cellular Micropatterns on Biocompatible Materials. Biotechnol. Prog. 1998, 14, 388–392. [Google Scholar] [CrossRef]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.A.; Whitesides, G.M. Rapid Prototyping of Microfluidic Systems in Poly(Dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef]
- Taylor, A.M.; Dieterich, D.C.; Ito, H.T.; Kim, S.A.; Schuman, E.M. Microfluidic Local Perfusion Chambers for the Visualization and Manipulation of Synapses. Neuron 2010, 66, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Midwoud, P.M.; Janse, A.; Merema, M.T.; Groothuis, G.M.M.; Verpoorte, E. Comparison of Biocompatibility and Adsorption Properties of Different Plastics for Advanced Microfluidic Cell and Tissue Culture Models. Anal. Chem. 2012, 84, 3938–3944. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Lee, S.H.; Suh, K.Y. Cell Research with Physically Modified Microfluidic Channels: A Review. Lab Chip 2008, 8, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnen, K.F.; Merten, C.A. Microfluidics as an Emerging Precision Tool in Developmental Biology. Dev. Cell 2019, 48, 293–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncombe, T.A.; Tentori, A.M.; Herr, A.E. Microfluidics: Reframing Biological Enquiry. Nat. Rev. Mol. Cell Biol. 2015, 16, 554–567. [Google Scholar]
- Dittrich, P.S.; Manz, A. Lab-on-a-Chip: Microfluidics in Drug Discovery. Nat. Rev. Drug Discov. 2006, 5, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Wang, Q. Microfluidic Chip-Based Technologies: Emerging Platforms for Cancer Diagnosis. BMC Biotechnol. 2013, 13, 76. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.D.; Laksanasopin, T.; Cheung, Y.K.; Steinmiller, D.; Linder, V.; Parsa, H.; Wang, J.; Moore, H.; Rouse, R.; Umviligihozo, G.; et al. Microfluidics-Based Diagnostics of Infectious Diseases in the Developing World. Nat. Med. 2011, 17, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Tay, A.; Pavesi, A.; Yazdi, S.R.; Lim, C.T.; Warkiani, M.E. Advances in Microfluidics in Combating Infectious Diseases. Biotechnol. Adv. 2016, 34, 404–421. [Google Scholar] [CrossRef]
- Reboud, J.; Xu, G.; Garrett, A.; Adriko, M.; Yang, Z.; Tukahebwa, E.M.; Rowell, C.; Cooper, J.M. Paper-Based Microfluidics for DNA Diagnostics of Malaria in Low Resource Underserved Rural Communities. Proc. Natl. Acad. Sci. USA 2019, 116, 4834–4842. [Google Scholar] [CrossRef] [Green Version]
- Vadivelu, R.; Kashaninejad, N.; Sreejith, K.R.; Bhattacharjee, R.; Cock, I.; Nguyen, N.T. Cryoprotectant-Free Freezing of Cells Using Liquid Marbles Filled with Hydrogel. ACS Appl. Mater. Interfaces 2018, 10, 43439–43449. [Google Scholar] [CrossRef]
- Avrămescu, R.E.; Ghica, M.V.; Dinu-Pîrvu, C.; Udeanu, D.I.; Popa, L. Liquid Marbles: FromIndustrial to Medical Applications. Molecules 2018, 23, 1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Chen, L.; Sun, L. Digital microfluidics: A promising technique for biochemical applications. Front. Mech. Eng. 2017, 12, 510–525. [Google Scholar] [CrossRef]
- Shembekar, N.; Chaipan, C.; Utharala, R.; Merten, C.A. Droplet-based microfluidics indrug discovery, transcriptomics and high-throughput molecular genetics. Lab Chip 2016, 16, 1314–1331. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Williams, J.C.; Johnson, S.M. Brain slice on a chip: Opportunities and challenges of applying microfluidic technology to intact tissues. Lab Chip 2012, 12, 2103–2117. [Google Scholar] [CrossRef]
- Vedarethinam, I.; Avaliani, N.; Tonnesen, J.; Hansen, L.; Sabourin, D.; Dimaki, M.; Kokaia, M.; Dufva, M.; Svendsen, W.E.; Emneus, J.; et al. Long-term brain slice culturing in a microfluidic platform. In Proceedings of the 15th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Seattle, WA, USA, 2–6 October 2011. [Google Scholar]
- Astolfi, M.; Péant, B.; Lateef, M.A.; Rousset, N.; Kendall-Dupont, J.; Carmona, E.; Monet, F.; Saad, F.; Provencher, D.; Mes-Masson, A.M.; et al. Micro-dissected tumor tissues on chip: An ex vivo method for drug testing and personalized therapy. Lab Chip 2016, 16, 312–325. [Google Scholar] [CrossRef]
- Yamada, A.; Vignes, M.; Bureau, C.; Mamane, A.; Venzac, B.; Descroix, S.; Viovy, J.L.; Villard, C.; Peyrin, J.M.; Malaquin, L. In-mold patterning and actionable axo-somatic compartmentalization for on-chip neuron culture. Lab Chip 2016, 16, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Moriguchi, H.; Sato, A.; Kawai, T.; Shimba, K.; Jimbo, Y.; Tanaka, Y. Microcasting with agarose gel via degassed polydimethylsiloxane molds for repellency-guided cell patterning. RSC Adv. 2016, 6, 54754–54762. [Google Scholar] [CrossRef] [Green Version]
- Knöll, B.; Weinl, C.; Nordheim, A.; Bonhoeffer, F. Stripe assay to examine axonal guidance and cell migration. Nat. Protoc. 2007, 2, 1216–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.B.; Xu, L.; Xu, Y.; Zhong, J.; Abedini, A.; Cheng, X.; Sinton, D. Disposable Silicon-Glass Microfluidic Devices: Precise, Robust and Cheap. Lab Chip 2018, 18, 3872–3880. [Google Scholar] [CrossRef]
- Qian, J.Y.; Hou, C.W.; Li, X.J.; Jin, Z.J. Actuation Mechanism of Microvalves: A Review. Micromachines 2020, 11, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friend, J.; Yeo, L. Fabrication of Microfluidic Devices Using Polydimethylsiloxane. Biomicrofluidics 2010, 4, 026502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T. PDMS-Based Microfluidic Devices for Biomedical Applications. Microelectron. Eng. 2002, 61–62, 907–914. [Google Scholar] [CrossRef]
- Fiorini, G.S.; Jeffries, G.D.M.; Lim, D.S.W.; Kuyper, C.L.; Chiu, D.T. Fabrication of Thermoset Polyester Microfluidic Devices and Embossing Masters Using Rapid Prototyped Polydimethylsiloxane Molds. Lab Chip 2003, 3, 158–163. [Google Scholar] [CrossRef]
- Wu, W.I.; Sask, K.N.; Brash, J.L.; Selvaganapathy, P.R. Polyurethane-Based Microfluidic Devices for Blood Contacting Applications. Lab Chip 2012, 12, 960–970. [Google Scholar] [CrossRef]
- Liu, K.; Fan, Z.H. Thermoplastic Microfluidic Devices and Their Applications in Protein and DNA Analysis. Analyst 2011, 136, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Gencturk, E.; Mutlu, S.; Ulgen, K.O. Advances in microfluidic devices made fromthermoplastics used in cell biology and analyses. Biomicrofluidics 2017, 11, 051502. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P. Recent Advances of Biologically Inspired 3D Microfluidic Hydrogel Cell Culture Systems. Cell Biol. Cell Metab. 2015, 2, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Morteza, B.; Mohseni, N.; Moghtader, M. An Introduction to Hydrogels and Some Recent Applications; IntechOpen, 2016. [Google Scholar]
- Li, X.; Ballerini, D.R.; Shen, W. A Perspective on Paper-Based Microfluidics: Current Status and Future Trends. Biomicrofluidics 2012, 6, 011301. [Google Scholar] [CrossRef] [Green Version]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-Chips at the Frontiers of Drug Discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, N.; Urrios, A.; Kang, S.; Folch, A. The Upcoming 3D-Printing Revolution in Microfluidics. Lab Chip 2016, 16, 1720–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luni, C.; Serena, E.; Elvassore, N. Human-on-Chip for Therapy Development and Fundamental Science. Curr. Opin. Biotechnol. 2014, 25, 45–50. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Z.; Abdul Rahim, N.A.; Van Noort, D.; Yu, H. Towards a Human-on-Chip: Culturing Multiple Cell Types on a Chip with Compartmentalized Microenvironments. Lab Chip 2009, 9, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, R.; Corlu, A.; Griscom, L.; Legallais, C.; Leclerc, E. Trends in the Development of Microfluidic Cell Biochips for in vitro Hepatotoxicity. Toxicol. Vitr. 2007, 21, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Osaki, T.; Shin, Y.; Sivathanu, V.; Campisi, M.; Kamm, R.D. In vitro Microfluidic Models for Neurodegenerative Disorders. Adv. Healthc. Mater. 2018, 7, 1700489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, K.I.W.; Jarazo, J.; Moreno, E.L.; Fleming, R.M.T.; Schwamborn, J.C. Passive Controlled Flow for Parkinson’s Disease Neuronal Cell Culture in 3D Microfluidic Devices. Org. Chip 2020, 2, 100005. [Google Scholar] [CrossRef]
- Perestrelo, A.R.; Águas, A.C.P.; Rainer, A.; Forte, G. Microfluidic Organ/Body-on-a-Chip Devices at the Convergence of Biology and Microengineering. Sensors 2015, 15, 31142–31170. [Google Scholar] [CrossRef] [Green Version]
- Haring, A.P.; Sontheimer, H.; Johnson, B.N. Microphysiological Human Brain and Neural Systems-on-a-Chip: Potential Alternatives to Small Animal Models and Emerging Platforms for Drug Discovery and Personalized Medicine. Stem Cell Rev. Rep. 2017, 13, 381–406. [Google Scholar] [CrossRef]
- Zheng, F.; Fu, F.; Cheng, Y.; Wang, C.; Zhao, Y.; Gu, Z. Organ-on-a-Chip Systems: Microengineering to Biomimic Living Systems. Small 2016, 12, 2253–2282. [Google Scholar] [CrossRef]
- Taylor, A.M.; Rhee, S.W.; Tu, C.H.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. Microfluidic Multicompartment Device for Neuroscience Research. Langmuir 2003, 19, 1551–1556. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, B.K.; Jeong, G.S.; Hyun, J.K.; Lee, C.J.; Lee, S.H. Three-Dimensional Brain-on-a-Chip with an Interstitial Level of Flow and Its Application as an in vitro Model of Alzheimer’s Disease. Lab Chip 2015, 15, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D Human Triculture System Modeling Neurodegeneration and Neuroinflammation in Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.I.; Amaral, M.H.; Costa, P.C.; Lopes, C.M.; Lamprou, D.A. Recent Developments in Microfluidic Technologies for Central Nervous System Targeted Studies. Pharmaceutics 2020, 12, 1–37. [Google Scholar] [CrossRef]
- McDonald, J.C.; Whitesides, G.M. Poly(dimethylsiloxane) as a material for fabricating microfluidic devices. Acc. Chem. Res. 2002, 35, 491–499. [Google Scholar] [CrossRef]
- Katsikoudi, A.; Ficulle, E.; Cavallini, A.; Sharman, G.; Guyot, A.; Zagnoni, M.; Eastwood, B.J.; Hutton, M.; Bose, S. Quantitative Propagation of Assembled Human Tau from Alzheimer’s Disease Brain in Microfluidic Neuronal Cultures. J. Biol. Chem. 2020, 295, 13079–13093. [Google Scholar] [CrossRef]
- Shin, Y.; Choi, S.H.; Kim, E.; Bylykbashi, E.; Kim, J.A.; Chung, S.; Kim, D.Y.; Kamm, R.D.; Tanzi, R.E. Blood–Brain Barrier Dysfunction in a 3D In vitro Model of Alzheimer’s Disease. Adv. Sci. 2019, 6, 1900962. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Liu, J.R.; Patel, B.; Solomon, D.E.; Vaidya, B.; Gupta, V. Microfluidics-based 3D Cell Culture Models: Utility in Novel Drug Discovery and Delivery Research. Bioeng. Transl. Med. 2016, 1, 63–81. [Google Scholar] [CrossRef]
- Natarajan, A.; Sethumadhavan, A.; Krishnan, U.M. Toward Building the Neuromuscular Junction: In vitro Models to Study Synaptogenesis and Neurodegeneration. ACS Omega 2019, 4, 12969–12977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Peyrin, J.M.; Deleglise, B.; Saias, L.; Vignes, M.; Gougis, P.; Magnifico, S.; Betuing, S.; Pietri, M.; Caboche, J.; Vanhoutte, P.; et al. Axon Diodes for the Reconstruction of Oriented Neuronal Networks in Microfluidic Chambers. Lab Chip 2011, 11, 3663–3673. [Google Scholar] [CrossRef]
- Gutruf, P.; Rogers, J.A. Implantable, Wireless Device Platforms for Neuroscience Research. Curr. Opin. Neurobiol. 2018, 50, 42–49. [Google Scholar] [CrossRef]
- Tsantoulas, C.; Farmer, C.; Machado, P.; Baba, K.; McMahon, S.B.; Raouf, R. Probing Functional Properties of Nociceptive Axons Using a Microfluidic Culture System. PLoS ONE 2013, 8, 80722. [Google Scholar] [CrossRef] [PubMed]
- Douville, N.J.; Tung, Y.C.; Li, R.; Wang, J.D.; El-Sayed, M.E.H.; Takayama, S. Fabrication of Two-Layered Channel System with Embedded Electrodes to Measure Resistance across Epithelial and Endothelial Barriers. Anal. Chem. 2010, 82, 2505–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, W.W.; Blurton-Jones, M.; Tu, C.H.; Feinberg, L.M.; Chabrier, M.A.; Harris, J.W.; Jeon, N.L.; Cotman, C.W. β-Amyloid Impairs Axonal BDNF Retrograde Trafficking. Neurobiol. Aging 2011, 32, 821–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, E.L.; Lu, H. Three-Dimensional Models for Studying Development and Disease: Moving on from Organisms to Organs-on-a-Chip and Organoids. Integr. Biol. 2016, 8, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Kunze, A.; Renaud, P. Compartmentalized microfluidics for in vitro Alzheimer’s disease studies. Microfluidic and Compartmentalized Platforms for Neurobiological Research. Neuromethods 2015, 103, 197–215. [Google Scholar]
- Southam, K.A.; King, A.E.; Blizzard, C.A.; McCormack, G.H.; Dickson, T.C. Microfluidic Primary Culture Model of the Lower Motor Neuron-Neuromuscular Junction Circuit. J. Neurosci. Methods 2013, 218, 164–169. [Google Scholar] [CrossRef]
- Neto, E.; Alves, C.J.; Sousa, D.M.; Alencastre, I.S.; Lourenço, A.H.; Leitão, L.; Ryu, H.R.; Jeon, N.L.; Fernandes, R.; Aguiar, P.; et al. Sensory Neurons and Osteoblasts: Close Partners in a Microfluidic Platform. Integr. Biol. 2014, 6, 586–595. [Google Scholar] [CrossRef]
- Sundararaghavan, H.G.; Monteiro, G.A.; Firestein, B.L.; Shreiber, D.I. Neurite Growth in 3D Collagen Gels with Gradients of Mechanical Properties. Biotechnol. Bioeng. 2009, 102, 632–643. [Google Scholar] [CrossRef]
- Ionescu, A.; Zahavi, E.E.; Gradus, T.; Ben-Yaakov, K.; Perlson, E. Compartmental Microfluidic System for Studying Muscle-Neuron Communication and Neuromuscular Junction Maintenance. Eur. J. Cell Biol. 2016, 95, 69–88. [Google Scholar] [CrossRef]
- Robertson, G.; Bushell, T.J.; Zagnoni, M. Chemically Induced Synaptic Activity between Mixed Primary Hippocampal Co-Cultures in a Microfluidic System. Integr. Biol. 2014, 6, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, F.; Tonna, N.; Lovchik, R.D.; Mastrangelo, R.; Morini, R.; Ruiz, A.; Delamarche, E.; Matteoli, M. Overflow Microfluidic Networks: Application to the Biochemical Analysis of Brain Cell Interactions in Complex Neuroinflammatory Scenarios. Anal. Chem. 2012, 84, 9833–9840. [Google Scholar] [CrossRef] [PubMed]
- Deleglise, B.; Lassus, B.; Soubeyre, V.; Alleaume-Butaux, A.; Hjorth, J.J.; Vignes, M.; Schneider, B.; Brugg, B.; Viovy, J.L.; Peyrin, J.M. Synapto-Protective Drugs Evaluation in Reconstructed Neuronal Network. PLoS ONE 2013, 8, e71103. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, T.R.; Erbereli, C.R.; Manzine, P.R.; Magalhães, T.N.C.; Balthazar, M.L.F.; Cominetti, M.R.; Faria, R.C. Early Diagnosis of Alzheimer’s Disease in Blood Using a Disposable Electrochemical Microfluidic Platform. ACS Sens. 2020, 5, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.; Jang, Y.C.; Chand, R.; Jha, S.K.; Lee, H.H.; Kim, Y.S. Microfluidic Biosensor for β-Amyloid(1-42) Detection Using Cyclic Voltammetry. J. Nanosci. Nanotechnol. 2011, 11, 5657–5662. [Google Scholar] [CrossRef]
- Yang, F.; Liao, X.; Tian, Y.; Li, G. Exosome Separation Using Microfluidic Systems: Size-Based, Immunoaffinity-Based and Dynamic Methodologies. Biotechnol. J. 2017, 12, 12. [Google Scholar] [CrossRef]
- Cohen, M.S.; Orth, C.B.; Kim, H.J.; Jeon, N.L.; Jaffrey, S.R. Neurotrophin-Mediated Dendrite-to-Nucleus Signaling Revealed by Microfluidic Compartmentalization of Dendrites. Proc. Natl. Acad. Sci. USA 2011, 108, 11246–11251. [Google Scholar] [CrossRef] [Green Version]
- Jorfi, M.; D’Avanzo, C.; Kim, D.Y.; Irimia, D. Three-Dimensional Models of the Human Brain Development and Diseases. Adv. Healthc. Mater. 2018, 7, 1002. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.H.; Gary, D.; Malone, M.; Dria, S.; Houdayer, T.; Belegu, V.; McDonald, J.W.; Thakor, N. Axon Myelination and Electrical Stimulation in a Microfluidic, Compartmentalized Cell Culture Platform. NeuroMolecular Med. 2012, 14, 112–118. [Google Scholar] [CrossRef]
- Kunze, A.; Meissner, R.; Brando, S.; Renaud, P. Co-Pathological Connected Primary Neurons in a Microfluidic Device for Alzheimer Studies. Biotechnol. Bioeng. 2011, 108, 2241–2245. [Google Scholar] [CrossRef]
- Hosie, K.A.; King, A.E.; Blizzard, C.A.; Vickers, J.C.; Dickson, T.C. Chronic Excitotoxin-Induced Axon Degeneration in a Compartmented Neuronal Culture Model. ASN Neuro 2012, 4, 47–57. [Google Scholar] [CrossRef]
- Edington, C.D.; Chen, W.L.K.; Geishecker, E.; Kassis, T.; Soenksen, L.R.; Bhushan, B.M.; Freake, D.; Kirschner, J.; Maass, C.; Tsamandouras, N.; et al. Interconnected Microphysiological Systems for Quantitative Biology and Pharmacology Studies. Sci. Rep. 2018, 8, 4530. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Park, C.B. Microfluidic Dissociation and Clearance of Alzheimer’s β-Amyloid Aggregates. Biomaterials 2010, 31, 6789–6795. [Google Scholar] [CrossRef] [PubMed]
- Hong, N.; Joo, S.; Nam, Y. Characterization of Axonal Spikes in Cultured Neuronal Networks Using Microelectrode Arrays and Microchannel Devices. IEEE Trans. Biomed. Eng. 2017, 64, 492–498. [Google Scholar]
- Lewandowska, M.K.; Bakkum, D.J.; Rompani, S.B.; Hierlemann, A. Recording Large Extracellular Spikes in Microchannels along Many Axonal Sites from Individual Neurons. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Habibey, R.; Latifi, S.; Mousavi, H.; Pesce, M.; Arab-Tehrany, E.; Blau, A. A Multielectrode Array Microchannel Platform Reveals Both Transient and Slow Changes in Axonal Conduction Velocity. Sci. Rep. 2017, 7, 8558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworak, B.J.; Wheeler, B.C. Novel MEA Platform with PDMS Microtunnels Enables the Detection of Action Potential Propagation from Isolated Axons in Culture. Lab Chip 2009, 9, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Stoothoff, W.; Jones, P.B.; Spires-Jones, T.L.; Joyner, D.; Chhabra, E.; Bercury, K.; Fan, Z.; Xie, H.; Bacskai, B.; Edd, J.; et al. Differential Effect of Three-Repeat and Four-Repeat Tau on Mitochondrial Axonal Transport. J. Neurochem. 2009, 111, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Deleglise, B.; Magnifico, S.; Duplus, E.; Vaur, P.; Soubeyre, V.; Belle, M.; Vignes, M.; Viovy, J.L.; Jacotot, E.; Peyrin, J.M.; et al. β-Amyloid Induces a Dying-Back Process and Remote Trans-Synaptic Alterations in a Microfluidic-Based Reconstructed Neuronal Network. Acta Neuropathol. Commun. 2014, 2, 145. [Google Scholar]
- Cho, H.; Hashimoto, T.; Wong, E.; Hori, Y.; Wood, L.B.; Zhao, L.; Haigis, K.M.; Hyman, B.T.; Irimia, D. Microfluidic Chemotaxis Platform for Differentiating the Roles of Soluble and Bound Amyloid-β on Microglial Accumulation. Sci. Rep. 2013, 3, 1823. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Huh, Y.S.; Erickson, D. Size-Selective Concentration and Label-Free Characterization of Protein Aggregates Using a Raman Active Nanofluidic Device. Lab Chip 2011, 11, 632–638. [Google Scholar] [CrossRef]
- Park, J.; Koito, H.; Li, J.; Han, A. Microfluidic Compartmentalized Co-Culture Platform for CNS Axon Myelination Research. Biomed. Microdevices 2009, 11, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Hosmane, S.; Yang, I.H.; Ruffin, A.; Thakor, N.; Venkatesan, A. Circular Compartmentalized Microfluidic Platform: Study of Axon-Glia Interactions. Lab Chip 2010, 10, 741–747. [Google Scholar] [CrossRef]
- Taylor, A.M.; Rhee, S.W.; Jeon, N.L. Microfluidic Chambers for Cell Migration and Neuroscience Research. Methods Mol. Biol. 2006, 321, 167–177. [Google Scholar] [PubMed]
- Goral, V.; Hsieh, Y.-C.; Petzold, O.; Faris, R.; Yuen, P.K. Hot Embossing of Plastic Microfluidic Devices Using Poly(Dimethylsiloxane) Molds. J. Micromech. Microeng. 2011, 21, 017002. [Google Scholar] [CrossRef]
- Chumbimuni-Torres, K.Y.; Coronado, R.E.; Mfuh, A.M.; Castro-Guerrero, C.; Silva, M.F.; Negrete, G.R.; Bizios, R.; Garcia, C.D. Adsorption of Proteins to Thin-Films of PDMS and Its Effect on the Adhesion of Human Endothelial Cells. RSC Adv. 2011, 1, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sjoberg, R.; Leyrat, A.; Houseman, B.; Shokat, K.; Quake, S. Biocompatibility and Reduced Drug Absorption of Sol-Gel-Treated Poly(Dimethyl Siloxane) for Microfluidic Cell Culture Applications. Anal. Chem. 2010, 82, 8954–8960. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yi, L.; Mukhitov, N.; Schrell, A.M.; Dhumpa, R.; Roper, M.G. Microfluidics-to-Mass Spectrometry: A Review of Coupling Methods and Applications. J. Chromatogr. A 2015, 1382, 98–116. [Google Scholar] [CrossRef] [Green Version]
- Ward, K.; Fan, Z.H. Mixing in Microfluidic Devices and Enhancement Methods. J. Micromech. Microeng. 2015, 25, 094001. [Google Scholar] [CrossRef] [PubMed]
- Oxford, A.E.; Stewart, E.S.; Rohn, T.T. Clinical Trials in Alzheimer’s Disease: A Hurdle in the Path of Remedy. Int. J. Alzheimers Dis. 2020, 2020, 5380346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasanna, P.; Rathee, S.; Rahul, V.; Mandal, D.; Chandra Goud, M.S.; Yadav, P.; Hawthorne, S.; Sharma, A.; Gupta, P.K.; Ojha, S.; et al. Microfluidic Platforms to Unravel Mysteries of Alzheimer’s Disease: How Far Have We Come? Life 2021, 11, 1022. https://doi.org/10.3390/life11101022
Prasanna P, Rathee S, Rahul V, Mandal D, Chandra Goud MS, Yadav P, Hawthorne S, Sharma A, Gupta PK, Ojha S, et al. Microfluidic Platforms to Unravel Mysteries of Alzheimer’s Disease: How Far Have We Come? Life. 2021; 11(10):1022. https://doi.org/10.3390/life11101022
Chicago/Turabian StylePrasanna, Pragya, Shweta Rathee, Vedanabhatla Rahul, Debabrata Mandal, Macherla Sharath Chandra Goud, Pardeep Yadav, Susan Hawthorne, Ankur Sharma, Piyush Kumar Gupta, Shreesh Ojha, and et al. 2021. "Microfluidic Platforms to Unravel Mysteries of Alzheimer’s Disease: How Far Have We Come?" Life 11, no. 10: 1022. https://doi.org/10.3390/life11101022
APA StylePrasanna, P., Rathee, S., Rahul, V., Mandal, D., Chandra Goud, M. S., Yadav, P., Hawthorne, S., Sharma, A., Gupta, P. K., Ojha, S., Jha, N. K., Villa, C., & Jha, S. K. (2021). Microfluidic Platforms to Unravel Mysteries of Alzheimer’s Disease: How Far Have We Come? Life, 11(10), 1022. https://doi.org/10.3390/life11101022