Tau Oligomers Neurotoxicity



{kind=link}
{kind=link}
Abstract
1. Introduction
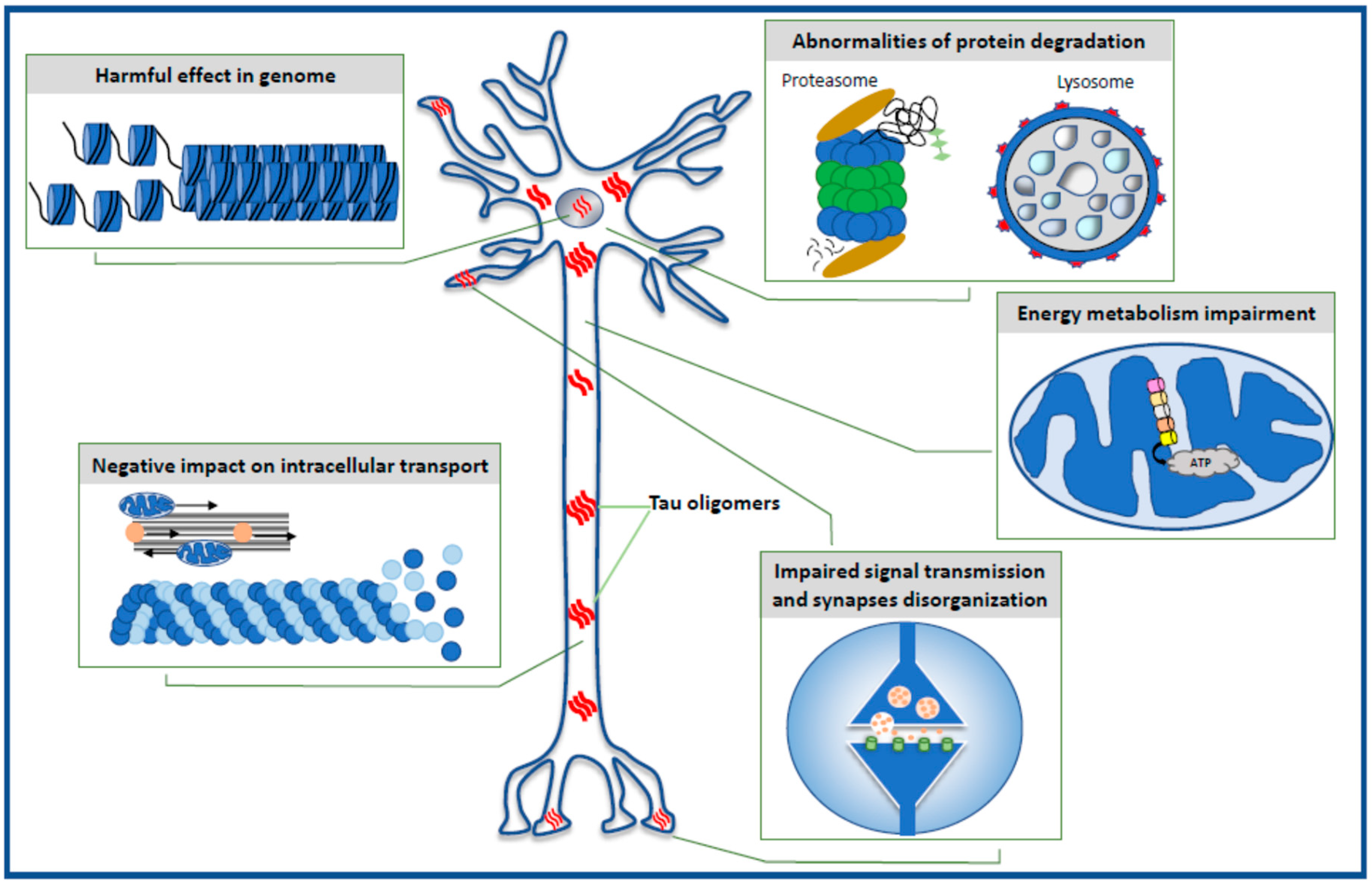
2. Harmful Effects in Genome
3. Impairment of Energy Production in the Neuron
4. Selective Effect of Tau Oligomers Treatment on Synaptic Integrity and Function
5. Negative Impact of Tau Oligomers on Microtubule Assembly, Neuronal Cytoskeleton, and Axonal Transport
6. Defective Protein Degradation
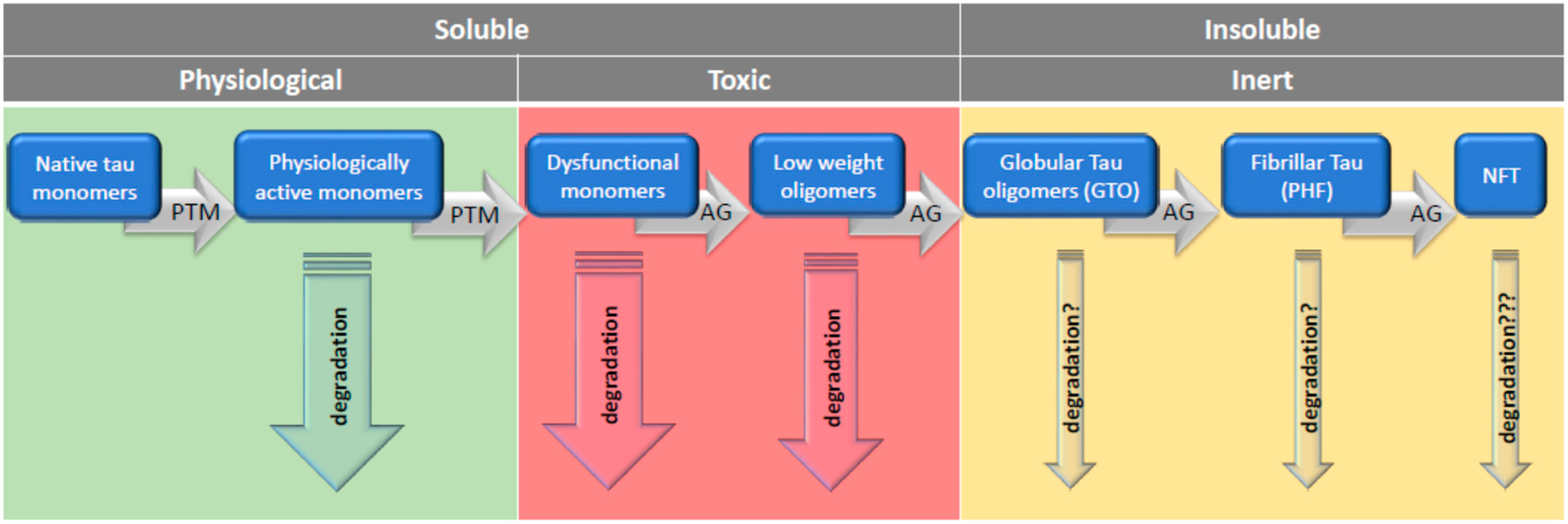
7. Rescue from Tau Oligomers: Degradation or Polymerization?
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buée, L.; Bussičre, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Stoothoff, W.H.; De Calingnon, A.; Jones, P.B.; Hyman, B.T. Tau pathophysiology in neurodegeneration: A tangled issue. Trends Neurosci. 2009, 32, 150–159. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Schmitt, F.A.; Kryscio, R.J.; Davis, D.G.; Smith, C.D.; Wekstein, D.R. Neuropathologic substrate of mild cognitive impairment. Arch. Neurol. 2006, 63, 38–46. [Google Scholar] [CrossRef]
- Feuillette, S.; Miguel, L.; Frébourg, T.; Campion, D.; Lecourtois, M. Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J. Neurochem. 2010, 113, 895–903. [Google Scholar] [CrossRef]
- Bonda, D.J.; Castellani, R.J.; Zhu, X.; Nunomura, A.; Lee, H.G.; Perry, G.; Smith, M.A. A Novel Perspective on Tau in Alzheimer Disease. Curr. Alzheimer Res. 2011, 8, 639–642. [Google Scholar] [CrossRef]
- Li, H.L.; Wang, H.H.; Liu, S.J.; Deng, Y.Q.; Zhang, Y.J.; Tian, Q.; Wang, X.C.; Chen, X.Q.; Yang, Y.; Zhang, J.Y.; et al. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 3591–3596. [Google Scholar] [CrossRef]
- Castellani, R.J.; Nunomura, A.; Rolston, R.K.; Moreira, P.I.; Takeda, A.; Perry, G.; Smith, M.A. Sublethal RNA oxidation as a mechanism for neurodegenerative disease. Int. J. Mol. Sci. 2008, 9, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A. The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer’s Disease (AD). Int. J. Mol. Sci. 2019, 20, 4661. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 2010, 49, 10039–10041. [Google Scholar] [CrossRef]
- Fox, L.M.; William, C.M.; Adamowicz, D.H.; Pitstick, R.; Carlson, G.A.; Spires-Jones, T.L.; Hyman, B.T. Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. J. Neuropathol. Exp. Neurol. 2011, 70, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Davidowitz, E.; Lopez, P.; Emadi, S.; Moe, J.; Sierks, M. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell Biol. 2013. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res. 2006, 54, 197–201. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular Tau oligomers as intermediates of Tau filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef]
- Sugino, E.; Nishiura, C.; Minoura, K.; In, Y.; Sumida, M.; Taniguchi, T.; Tomoo, K.; Ishida, T. Three-/four-repeat-dependent aggregation profile of tau microtubule-binding domain clarified by dynamic light scattering analysis. Biochem. Biophys. Res. Commun. 2009, 385, 236–240. [Google Scholar] [CrossRef]
- Zilka, N.; Filipcik, P.; Koson, P.; Fialova, L.; Skrabana, R.; Zilkova, L.; Rolkowa, G.; Kontsekova, E.; Nova, M. Truncated tau from sporadic Alzheimer’s diseases suffices to drive neurofibryllary degeneration in vivo. FEBS Lett. 2006, 580, 3582–3588. [Google Scholar] [CrossRef]
- Sahara, N.; Maeda, S.; Murayama, M.; Suzuki, T.; Dohmae, N.; Yen, S.H.; Takashima, A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 2007, 25, 3020–3029. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Takashima, A. Tau Oligomers. Adv. Exp. Med. Biol. 2019, 1184, 373–380. [Google Scholar] [PubMed]
- Manassero, G.; Guglielmotto, M.; Monteleone, D.; Vasciaveo, V.; Butenko, O.; Tamagno, E.; Arancio, O.; Tabaton, M. Dual Mechanism of Toxicity for Extracellular Injection of Tau Oligomers versus Monomers in Human Tau Mice. J. Alzheimers Dis. 2017, 59, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Pallas-Bazarra, N.; Terreros-Roncal, J.; Perea, J.R.; Jurado-Arjona, J.; Avila, J.; Llorens-Martin, M. Soluble tau has devastating effects on the structural plasticity of hippocampal granule neurons. Transl. Psychiatry 2017, 7, 1267. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Fischer, S.; Johnson, G.V.W.; Nehrke, K. Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 2020, 15, 65. [Google Scholar] [CrossRef]
- Florenzano, F.; Veronica, C.; Ciasca, G.; Ciotti, M.T.; Pittaluga, A.; Olivero, G.; Feligioni, M.; Iannuzzi, F.; Latina, V.; Maria Sciacca, M.F.; et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 2017, 8, 64745–64778. [Google Scholar] [CrossRef]
- Gómez-Ramos, A.; Díaz-Hernández, M.; Rubio, A.; Díaz-Hernández, J.I.; Miras-Portugal, M.T.; Avila, J. Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 2009, 19, 708–717. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Dujardin, S.; Bégard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.C.; Bousset, L.; Melki, R.; et al. Ectosomes: A New Mechanism for Non-Exosomal Secretion of Tau Protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef]
- Chai, X.; Dage, J.L.; Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 2012, 48, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016, 10, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Pampuscenko, K.; Morkuniene, R.; Krasauskas, L.; Smirnovas, V.; Tomita, T.; Borutaite, V. Distinct Neurotoxic Effects of Extracellular Tau Species in Primary Neuronal-Glial Cultures. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Gómez-Ramos, A.; Díaz-Hernández, M.; Rubio, A.; Miras-Portugal, M.T.; Avila, J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell Neurosci. 2008, 37, 673–681. [Google Scholar] [CrossRef]
- Morozova, V.; Cohen, L.S.; Makki, A.E.; Shur, A.; Pilar, G.; El Idrissi, A.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef]
- Wysocka, A.; Palasz, E.; Steczkowska, M.; Niewiadomska, G. Dangerous Liaisons: Tau Interaction with Muscarinic Receptors. Curr. Alzheimer Res. 2020, 17, 224–237. [Google Scholar] [CrossRef]
- Díaz-Hernández, M.; Gómez-Ramos, A.; Rubio, A.; Gómez-Villafuertes, R.; Naranjo, J.R.; Miras-Portugal, M.T.; Avila, J. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 2010, 285, 32539–32548. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, M.K.; Shestakova, E.; Mansuroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci. 2006, 119, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; He, R.Q.; Haque, N.; Qu, M.H.; Del Carmen Alonso, A.; Grundke-Iqbal, I.; Iqbal, K. Microtubule associated protein tau binds to double-stranded but not single-stranded DNA. Cell. Mol. Life Sci. 2003, 60, 413–421. [Google Scholar] [CrossRef]
- Wei, Y.; Qu, M.H.; Wang, X.S.; Chen, L.; Wang, D.L.; Liu, Y.; Hua, Q.; He, R.Q. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS ONE 2008, 3, e2600. [Google Scholar] [CrossRef]
- Qi, H.; Cantrelle, F.X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.C.; Landrieu, I. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef]
- Malmanche, N.; Dourlen, P.; Gistelinck, M.; Demiautte, F.; Link, N.; Dupont, C.; Vanden Broeck, L.; Werkmeister, E.; Amouyel, P.; Bongiovanni, A.; et al. Developmental expression of 4-Repeat-Tau induces neuronal aneuploidy in drosophila tauopathy models. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Violet, M.; Chauderlier, A.; Delattre, L.; Tardivel, M.; Sendid Chouala, M.; Sultan, A.; Marciniak, E.; Humez, S.; Binder, L.; Kayed, R.; et al. Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiol. Dis. 2015, 82, 540–551. [Google Scholar] [CrossRef]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef]
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Bégard, S.; et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 8. [Google Scholar] [CrossRef]
- Bukar Maina, M.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.E.; Amoils, S.; Horn, J.M.; Buckle, V.J.; Higgs, D.R.; Merkenschlager, M.; Fisher, A.G. Expression of alpha- and beta-globin genes occurs within different nuclear domains in haemopoietic cells. Nat. Cell Biol. 2001, 3, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Guenatri, M.; Bailly, D.; Maison, C.; Almouzni, G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 2004, 166, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Josse, T.; Mokrani-Benhelli, H.; Benferhat, R.; Shestakova, E.; Mansuroglu, Z.; Kakanakou, H.; Billecocq, A.; Bouloy, M.; Bonnefoy, E. Association of the interferon-β gene with pericentromeric heterochromatin is dynamically regulated during virus infection through a YY1-dependent mechanism. Nucleic Acids Res. 2012, 40, 4396–4411. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.J.; Elgin, S.C. Epigenetic codes for heterochromatin formation and silencing: Rounding up the usual suspects. Cell 2002, 108, 489–500. [Google Scholar] [CrossRef]
- Sasaki, T.; Lynch, K.L.; Mueller, C.V.; Friedman, S.; Freitag, M.; Lewis, Z.A. Heterochromatin controls γH2A localization in Neurospora crassa. Eukaryot. Cell 2014, 13, 990–1000. [Google Scholar] [CrossRef]
- Mastroeni, D.; Delvaux, E.; Nolz, J.; Tan, Y.; Grover, A.; Oddo, S.; Coleman, P.D. Aberrant intracellular localization of H3k4me3 demonstrates an early epigenetic phenomenon in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3121–3129. [Google Scholar] [CrossRef]
- Benhelli-Mokrani, H.; Mansuroglu, Z.; Chauderlier, A.; Albaud, B.; Gentien, D.; Sommer, S.; Schirmer, C.; Laqueuvre, L.; Josse, T.; Buée, L.; et al. Genome-wide identification of genic and intergenic neuronal DNA regions bound by Tau protein under physiological and stress conditions. Nucleic Acids Res. 2018, 46, 11405–11422. [Google Scholar] [CrossRef]
- Schindowski, K.; Bretteville, A.; Leroy, K.; Begard, S.; Brion, J.-P.; Hamdane, M.; Buee, L. Alzheimer’s disease-like tau ’neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 2016, 169, 599–616. [Google Scholar] [CrossRef]
- Hondius, D.C.; van Nierop, P.; Li, K.W.; Hoozemans, J.J.; van der Schors, R.C.; van Haastert, E.S.; van der Vies, S.M.; Rozemuller, A.J.; Smit, A.B. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimers Dement. 2016, 12, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, Guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [PubMed]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 132. [Google Scholar] [CrossRef]
- Lee, H.; Mok, K.H.; Muhandiram, R.; Park, K.H.; Suk, J.E.; Kim, D.H.; Chang, J.; Sung, Y.C.; Choi, K.Y.; Han, K.H. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem. 2000, 275, 29426–29432. [Google Scholar] [CrossRef]
- Dawson, R.; Muller, L.; Dehner, A.; Klein, C.; Kessler, H.; Buchner, J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003, 332, 1131–1141. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Giannakakou, P.; Sackett, D.L.; Ward, Y.; Webster, K.; Blagosklonny, M.; Fojo, T. P53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat. Cell Biol. 2000, 2, 709–717. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Clos, A.L.; Castillo-Carranza, D.; Sengupta, U.; Guerrero-Muñoz, M.; Kelly, B.; Wagner, R.; Kayed, R. Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem. Biophys. Res. Commun. 2013, 430, 963–968. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau protein disrupts Nucleocytoplasmic transport in Alzheimer’s disease. Neuron 2018, 99, 925–940. [Google Scholar] [CrossRef]
- Diez, L.; Wegmann, S. Nuclear Transport Deficits in Tau-Related Neurodegenerative Diseases. Front. Neurol. 2020, 11, 1056. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Tan, Y.; Wang, Z.; Wang, K.S.; Zhang, X.; Chen, X.; Li, C.S.; Wang, T.; Luo, X. Long noncoding RNAs in psychiatric disorders. Psychiatr. Genet. 2016, 26, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.; Dubreucq, B. Evolution goes GAGA: GAGA binding protein across kingdoms. Biochim. Biophys. Acta 2012, 1819, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Gohl, D.; Aoki, T.; Blanton, J.; Shanower, G.; Kappes, G.; Schedl, P. Mechanism of chromosomal boundary action: Roadblock, sink, or loop? Genetics 2011, 187, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Zullo, J.M.; Demarco, I.A.; Pique-Regi, R.; Gaffney, D.J.; Epstein, C.B.; Spooner, C.J.; Luperchio, T.R.; Bernstein, B.E.; Pritchard, J.K.; Reddy, K.L.; et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 2012, 149, 1474–1487. [Google Scholar] [CrossRef]
- Frost, B.; Bardai, F.H.; Feany, M.B. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr. Biol. 2016, 26, 129–136. [Google Scholar] [CrossRef]
- Winick-Ng, W.; Rylett, R.J. Into the fourth dimension: Dysregulation of genome architecture in aging and Alzheimer’s disease. Front. Mol. Neurosci. 2018, 11, 60. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Puangmalai, N.; Sengupta, U.; Bhatt, N.; Johnson, O.D.; Kharas, M.G.; Kayed, R. RNA-binding proteins Musashi and tau soluble aggregates initiate nuclear dysfunction. Nat. Commun. 2020, 11, 4305. [Google Scholar] [CrossRef]
- Benito, E.; Barco, A. CREB’s control of intrinsic and synaptic plasticity: Implications for CREB-dependent memory models. Trends Neurosci. 2010, 33, 230–240. [Google Scholar] [CrossRef]
- Bartolotti, N.; Bennett, D.A.; Lazarov, O. Reduced pCREB in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol. Psychiatry 2016, 21, 1158–1166. [Google Scholar] [CrossRef]
- Yin, Y.; Gao, D.; Wang, Y.; Wang, Z.-H.; Wang, X.; Ye, J.; Wu, D.; Fang, L.; Pi, G.; Yang, Y.; et al. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E3773–E3781. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, R.; Ochoa Thomas, E.; Ramirez, P.; Miller, H.E.; Beckmann, A.; Zuniga, G.; Dobrowolski, R.; Frost, B. Pathogenic Tau Causes a Toxic Depletion of Nuclear Calcium. Cell Rep. 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.L.; Kril, J.J.; Stevens, C.H.; Kwok, J.B.; Hallupp, M.; Kim, W.S.; Huang, Y.; McGinley, C.V.; Werka, H.; Kiernan, M.C.; et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 2018, 141, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer ’s disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef]
- Neumann, K.; Farías, G.; Slachevsky, A.; Perez, P.; Maccioni, R.B. Human platelets tau: A potential peripheral marker for Alzheimer’s disease. J. Alzheimers Dis. 2011, 25, 103–109. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Drose, S.; Brandt, U.; Muller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Sengupta, U.; Castillo-Carranza, D.; Gerson, J.E.; GuerreroMunoz, M.; Troncoso, J.C.; Jackson, G.R.; Kayed, R. The formation of tau pore-like structures is prevalent and cell specific: Possible implications for the disease phenotypes. Acta Neuropathol. Commun. 2014, 2, 56. [Google Scholar] [CrossRef]
- Amorim, J.A.; Canas, P.M.; Tome, A.R.; Rolo, A.P.; Agostinho, P.; Palmeira, C.M.; Cunha, R.A. Mitochondria in excitatory and inhibitory synapses have similar susceptibility to amyloid-beta peptides modeling Alzheimer’s disease. J. Alzheimers Dis. 2017, 60, 525–536. [Google Scholar] [CrossRef]
- Camilleri, A.; Ghio, S.; Caruana, M.; Weckbecker, D.; Schmidt, F.; Kamp, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Giese, A.; et al. Tau-induced mitochondrial membrane perturbation is dependent upon cardiolipin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183064. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta 2009, 1788, 2022–2031. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 16, 1434–1455. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Koppel, S.; Weidling, I.; Hayley, C.; Ji, Y.; Wilkins, H.M. Mitochondria, Cybrids, Aging, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 259–302. [Google Scholar]
- Weidling, I.W.; Wilkins, H.M.; Koppel, S.J.; Hutfles, L.; Wang, X.; Kalani, A.; Menta, B.W.; Ryan, B.; Perez-Ortiz, J.; Gamblin, T.C.; et al. Mitochondrial DNA Manipulations Affect Tau Oligomerization. J. Alzheimers Dis. 2020, 77, 149–163. [Google Scholar] [CrossRef]
- Zheng, J.; Akbari, M.; Schirmer, C.; Reynaert, M.L.; Loyens, A.; Lefebvre, B.; Croteau, D.L.; Galas, M.C.; Bohr, V.A. Hippocampal tau oligomerization early in tau pathology coincides with a transient alteration of mitochondrial homeostasis and DNA repair in a mouse model of tauopathy. Acta Neuropathol. Commun. 2020, 8, 25. [Google Scholar] [CrossRef]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef]
- Dai, S.H.; Chen, T.; Wang, Y.H.; Zhu, J.; Luo, P.; Rao, W.; Yang, Y.F.; Fei, Z.; Jiang, X.F. Sirt3 protects cortical neurons against oxidative stress via regulating mitochondrial Ca2+ and mitochondrial biogenesis. Int. J. Mol. Sci. 2014, 15, 14591–14609. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Wu, Y.X.; Zhang, T.; Qiu, Y.; Ding, Z.J.; Zha, D.J. Sirt3 confers protection against acrolein-induced oxidative stress in cochlear nucleus neurons. Neurochem. Int. 2018, 114, 1–9. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Gan, L. Tau-mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 51, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Niroomand, S.; Drolet, R.E.; Yao, L.; Gaspar, R.C.; Hatcher, N.G.; Schachter, J.; Renger, J.J.; Parmentier-Batteur, S. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J. Neurosci. 2015, 35, 14234–14250. [Google Scholar] [CrossRef] [PubMed]
- Fá, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce an Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Ondrejcak, T.; Klyubin, I.; Corbett, G.T.; Fraser, G.; Hong, W.; Mably, A.J.; Gardener, M.; Hammersley, J.; Perkinton, M.S.; Billinton, A.; et al. Cellular Prion Protein Mediates the Disruption of Hippocampal Synaptic Plasticity by Soluble Tau In Vivo. J. Neurosci. 2018, 38, 10595–10606. [Google Scholar] [CrossRef] [PubMed]
- Hill, E.; Karikari, T.K.; Moffat, K.G.; Richardson, M.J.E.; Wall, M.J. Introduction of Tau Oligomers into Cortical Neurons Alters Action Potential Dynamics and Disrupts Synaptic Transmission and Plasticity. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Decker, J.M.; Krüger, L.; Sydow, A.; Zhao, S.; Frotscher, M.; Mandelkow, E.; Mandelkow, E.M. Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca++ dysregulation. Acta Neuropathol. Commun. 2015, 3, 23. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Redeker, V.; Pieri, L.; Bousset, L.; Renner, M.; Madiona, K.; Mailhes-Hamon, C.; Coens, A.; Buée, L.; Hantraye, P.; et al. Clustering of tau fibrils impairs the synaptic composition of alpha3-Na+/K+-ATPase and AMPA receptors. EMBO J. 2019, 38, e99871. [Google Scholar] [CrossRef]
- Regan, P.; Cho, K. The Role of Tau in the Post-synapse. Adv. Exp. Med. Biol. 2019, 1184, 113–121. [Google Scholar]
- Afreen, S.; Riherd Methner, D.N.; Ferreira, A. Tau45–230 association with the cytoskeleton and membrane-bound organelles: Functional implications in neurodegeneration. Neuroscience 2017, 362, 104–117. [Google Scholar] [CrossRef]
- Kounakis, K.; Tavernarakis, N. The Cytoskeleton as a Modulator of Aging and Neurodegeneration. Adv. Exp. Med. Biol. 2019, 1178, 227–245. [Google Scholar]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef]
- Swanson, E.; Breckenridge, L.; McMahon, L.; Som, S.; McConnell, I.; Bloom, G.S. Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction. J. Alzheimers Dis. 2017, 58, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Yvon, A.M.; Wadsworth, P.; Jordan, M.A. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol. Biol. Cell 1999, 10, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Makrides, V.; Shen, T.E.; Bhatia, R.; Smith, B.L.; Thimm, J.; Lal, R.; Feinstein, S.C. Microtubule-dependent oligomerization of tau: Implications for physiological tau function and tauopathies. J. Biol. Chem. 2003, 278, 33298–33304. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.F.; Leboeuf, A.C.; Massie, M.R.; Jordan, M.A.; Wilson, L.; Feinstein, S.C. Three- and four-repeat tau regulate the dynamic instability of two distinct microtubule subpopulations in qualitatively different manners. Implications for neurodegeneration. J. Biol. Chem. 2005, 280, 13520–13528. [Google Scholar] [CrossRef] [PubMed]
- Gyparaki, M.T.; Arab, A.; Sorokina, E.M.; Santiago-Ruiz, A.N.; Bohrer, C.H.; Xiao, J.; Lakadamyali, M. Tau forms oligomeric complexes on microtubules that are distinct from pathological oligomers in disease. BioRxiv 2020. [Google Scholar] [CrossRef]
- Ward, S.M.; Himmelstein, D.S.; Lancia, J.K.; Binder, L.I. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 2012, 40, 667–671. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Morfini, G.A.; Lapointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef]
- Combs, B.; Mueller, R.L.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Tau and Axonal Transport Misregulation in Tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95. [Google Scholar]
- Cox, K.; Combs, B.; Abdelmesih, B.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Analysis of isoform-specific tau aggregates suggests a common toxic mechanism involving similar pathological conformations and axonal transport inhibition. Neurobiol. Aging 2016, 47, 113–126. [Google Scholar] [CrossRef]
- LaPointe, N.E.; Morfini, G.; Pigino, G.; Gaisina, I.N.; Kozikowski, A.P.; Binder, L.I.; Brady, S.T. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J. Neurosci. Res. 2009, 87, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Moreno, H.; Morfini, G.; Buitrago, L.; Ujlaki, G.; Choi, S.; Yu, E.; Moreira, J.E.; Avila, J.; Brady, S.T.; Pant, H.; et al. Tau pathology-mediated presynaptic dysfunction. Neuroscience 2016, 325, 30–38. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tiernan, C.T.; Combs, B.; Cox, K.; Morfini, G.; Brady, S.T.; Counts, S.E.; Kanaan, N.M. Pseudophosphorylation of tau at S422 enhances SDS-stable dimer formation and impairs both anterograde and retrograde fast axonal transport. Exp. Neurol. 2016, 283, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Dubey, M.; Chaudhury, P.; Kabiru, H.; Shea, T.B. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instability. Cell Motil. Cytoskelet. 2008, 65, 89–99. [Google Scholar] [CrossRef]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Götz, J. Phosphorylated Tau Interacts with c-Jun N-terminal Kinase-interacting Protein 1 (JIP1) in Alzheimer Disease. J. Biol. Chem. 2009, 284, 20909–20916. [Google Scholar] [CrossRef]
- Ittner, L.M.; Fath, T.; Ke, Y.D.; Bi, M.; van Eersel, J.; Li, K.M.; Gunning, P.; Gotz, J. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2008, 105, 15997–16002. [Google Scholar] [CrossRef]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau Accumulation Causes Mitochondrial Distribution Deficits in Neurons in a Mouse Model of Tauopathy and in Human Alzheimer’s Disease Brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef]
- Rodríguez-Martín, T.; Pooler, A.M.; Lau, D.H.W.; Mórotz, G.M.; De Vos, K.J.; Gilley, J.; Coleman, M.P.; Hanger, D.P. Reduced number of axonal mitochondria and tau hypophosphorylation in mouse P301L tau knockin neurons. Neurobiol. Dis. 2016, 85, 1–10. [Google Scholar] [CrossRef]
- Iovino, M.; Agathou, S.; González-Rueda, A.; Del Castillo Velasco-Herrera, M.; Borroni, B.; Alberici, A.; Lynch, T.; O’Dowd, S.; Geti, I.; Gaffney, D.; et al. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 2015, 138, 3345–3359. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Shiozawa, S.; Tsuboi, D.; Amano, M.; Watanabe, H.; Maeda, S.; Kimura, T.; Yoshimatsu, S.; Kisa, F.; Karch, C.M.; et al. Pathological Progression Induced by the Frontotemporal Dementia-Associated R406W Tau Mutation in Patient-Derived iPSCs. Stem Cell Rep. 2019, 13, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Friedman, R.; Shaw, G.; Chau, V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc. Natl. Acad. Sci USA 1987, 84, 3033–3036. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 1993, 10, 1151–1160. [Google Scholar] [CrossRef]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- López Salon, M.; Morelli, L.; Castaño, E.M.; Soto, E.F.; Pasquini, J.M. Defective ubiquitination of cerebral proteins in Alzheimer’s disease. J. Neurosci. Res. 2000, 62, 302–310. [Google Scholar] [CrossRef]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Liu, S.J.; Li, H.L.; Wang, J.Z. Microtubule-associated protein tau is a substrate of ATP/Mg2+-dependent proteasome protease system. J. Neural. Transm. 2005, 112, 547–555. [Google Scholar] [CrossRef]
- David, D.C.; Layfield, R.; Serpell, L.; Narain, Y.; Goedert, M.; Spillantini, M.G. Proteasomal degradation of tau protein. J. Neurochem. 2002, 83, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Schwartz, D.; Gygi, S.P.; Kosik, K.S. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004, 279, 4869–4876. [Google Scholar] [CrossRef] [PubMed]
- Sahara, N.; Murayama, M.; Mizoroki, T.; Urushitani, M.; Imai, Y.; Takahashi, R.; Murata, S.; Tanaka, K.; Takashima, A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005, 94, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 2007, 117, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Dou, F.; Rodina, A.; Chip, S.; Kim, J.; Zhao, Q.; Moulick, K.; Aguirre, J.; Wu, N.; Greengard, P.; et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.J.; Johnson, G.V. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J. Biol. Chem. 2010, 285, 21978–21987. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, B.R.; Kim, J.; LaFerla, F.M.; Park, J.H.Y.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane Upregulates the Heat Shock Protein Co-Chaperone CHIP and Clears Amyloid-β and Tau in a Mouse Model of Alzheimer’s Disease. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef]
- Webster, J.M.; Darling, A.L.; Uversky, V.N.; Blair, L.J. Small Heat Shock Proteins, Big Impact on Protein Aggregation in Neurodegenerative Disease. Front. Pharmacol. 2019, 10, 1047. [Google Scholar] [CrossRef]
- Björkdahl, C.; Sjögren, M.J.; Zhou, X.; Concha, H.; Avila, J.; Winblad, B.; Pei, J.J. Small heat shock proteins Hsp27 or alphaB-crystallin and the protein components of neurofibrillary tangles: Tau and neurofilaments. J. Neurosci. Res. 2008, 86, 1343–1352. [Google Scholar] [CrossRef]
- Abisambra, J.F.; Blair, L.J.; Hill, S.E.; Jones, J.R.; Kraft, C.; Rogers, J.; Koren, J., III; Jinwal, U.K.; Lawson, L.; Johnson, A.G.; et al. Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J. Neurosci. 2010, 30, 15374–15382. [Google Scholar] [CrossRef]
- Baughman, H.E.R.; Clouser, A.F.; Klevit, R.E.; Nath, A. HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J. Biol. Chem. 2018, 293, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Bhaskar, K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Krüger, U.; Wang, Y.; Kumar, S.; Mandelkow, E.M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17, e12692. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Peterhoff, C.M.; Schmidt, S.D.; Terio, N.B.; Duff, K.; Beard, M.; Mathews, P.M.; Nixon, R.A. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277. [Google Scholar] [CrossRef]
- Lim, F.; Hernández, F.; Lucas, J.J.; Gómez-Ramos, P.; Morán, M.A.; Avila, J. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol. Cell Neurosci. 2001, 18, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z. Regulation of neuronal autophagy in axon: Implication of autophagy in axonal function and dysfunction/degeneration. Autophagy 2007, 3, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Roney, J.C.; Cheng, X.T.; Li, S.; Cuddy, S.R.; Sheng, Z.H. Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Rep. 2019, 28, 51–64. [Google Scholar] [CrossRef]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Hamano, T.; Gendron, T.F.; Causevic, E.; Yen, S.H.; Lin, W.L.; Isidoro, C.; Deture, M.; Ko, L.W. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur. J. Neurosci. 2008, 27, 1119–1130. [Google Scholar] [CrossRef]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Colacurcio, D.J.; Pensalfini, A.; Jiang, Y.; Nixon, R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic. Biol. Med. 2018, 114, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; La Spada, A.R. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: Molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol. Dis. 2019, 122, 83–93. [Google Scholar] [CrossRef]
- Song, J.X.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.B.; Tong, B.C.; Iyaswamy, A.; Shang, W.B.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 2020, 19, e13069. [Google Scholar] [CrossRef]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Quehenberger, O.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef]
- Maher, P. Preventing and Treating Neurological Disorders with the Flavonol Fisetin. Brain Plast. 2020. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Konstantinidou, M.; Li, J.; Zhang, B.; Wang, Z.; Shaabani, S.; Ter Brake, F.; Essa, K.; Dömling, A. PROTACs- a game-changing technology. Expert Opin. Drug Discov. 2019, 14, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.T.; Gao, N.; Li, Q.Q.; Chen, P.G.; Yang, X.F.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell. Chem. Biol. 2016, 23, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Ferguson, F.M.; Cai, Q.; Donovan, K.A.; Nandi, G.; Patnaik, D.; Zhang, T.; Huang, H.T.; Lucente, D.E.; Dickerson, B.C.; et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8, e45457. [Google Scholar] [CrossRef]
- Kargbo, R.B. Treatment of Alzheimer’s by PROTAC-Tau Protein Degradation. Acs Med. Chem. Lett. 2019, 10, 699–700. [Google Scholar] [CrossRef]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell. 2019, 76, 797–810.e10. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Wegmann, S.; Kopeikina, K.J.; Hawkes, J.; Rudinskiy, N.; Andermann, M.L.; Spires-Jones, T.L.; Bacskai, B.J.; Hyman, B.T. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 510–514. [Google Scholar] [CrossRef]
- Morsch, R.; Simon, W.; Coleman, P.D. Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1999, 58, 188–197. [Google Scholar] [CrossRef]
- Gómez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997, 41, 17–24. [Google Scholar] [CrossRef]
- Park, S.; Lee, J.H.; Jeon, J.H.; Lee, M.J. Degradation or aggregation: The ramifications of post-translational modifications on tau. BMB Rep. 2018, 51, 265–273. [Google Scholar] [CrossRef]
- Zhao, K.; Ippolito, G.; Wang, L.; Price, V.; Kim, M.H.; Cornwell, G.; Fulenchek, S.; Breen, G.A.; Goux, W.J.; D’Mello, S.R. Neuron-selective toxicity of tau peptide in a cell culture model of neurodegenerative tauopathy: Essential role for aggregation in neurotoxicity. J. Neurosci. Res. 2010, 88, 3399–3413. [Google Scholar] [CrossRef]
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hübschmann, S.; Redecke, L.; Mandelkow, E.M.; Müller, D.J.; Mandelkow, E. Oligomer formation of tau protein hyperphosphorylated in cells. J. Biol. Chem. 2014, 289, 34389–343407. [Google Scholar] [CrossRef]
- Liu, M.; Sui, D.; Dexheimer, T.; Hovde, S.; Deng, X.; Wang, K.W.; Lin, H.L.; Chien, H.T.; Kweon, H.K.; Kuo, N.S.; et al. Hyperphosphorylation Renders Tau Prone to Aggregate and to Cause Cell Death. Mol. Neurobiol. 2020, 57, 4704–4719. [Google Scholar] [CrossRef]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Gerson, J.E.; Singh, G.; Estes, D.M.; Barrett, A.D.; Dineley, K.T.; Jackson, G.R.; et al. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J. Neurosci. 2014, 34, 4260–4272. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Gerson, J.E.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Kayed, R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J. Alzheimers Dis. 2014, 40 (Suppl. 1), S97–S111. [Google Scholar] [CrossRef]
- Cowan, C.M.; Quraishe, S.; Hands, S.; Sealey, M.; Mahajan, S.; Allan, D.W.; Mudher, A. Rescue from tau-induced neuronal dysfunction produces insoluble tau oligomers. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- De Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef]
- Guzmán-Martinez, L.; Farías, G.A.; Maccioni, R.B. Tau oligomers as potential targets for Alzheimer’s diagnosis and novel drugs. Front. Neurol. 2013, 4, 167. [Google Scholar] [CrossRef]
- Martinez, A.; Gil, C.; Perez, D.I. Glycogen synthase kinase 3 inhibitors in the next horizon for Alzheimer’s disease treatment. Int. J. Alzheimers Dis. 2011. [Google Scholar] [CrossRef]
- Llorach-Pares, L.; Rodriguez-Urgelles, E.; Nonell-Canals, A.; Alberch, J.; Avila, C.; Sanchez-Martinez, M.; Giralt, A. Meridianins and Lignarenone B as Potential GSK3β Inhibitors and Inductors of Structural Neuronal Plasticity. Biomolecules 2020, 10, 639. [Google Scholar] [CrossRef]
- Soeda, Y.; Saito, M.; Maeda, S.; Ishida, K.; Nakamura, A.; Kojima, S.; Takashima, A. Methylene Blue Inhibits Formation of Tau Fibrils but not of Granular Tau Oligomers: A Plausible Key to Understanding Failure of a Clinical Trial for Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1677–1686. [Google Scholar] [CrossRef]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial. J. Alzheimers Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef]
- Schelter, B.O.; Shiells, H.; Baddeley, T.C.; Rubino, C.M.; Ganesan, H.; Hammel, J.; Vuksanovic, V.; Staff, R.T.; Murray, A.D.; Bracoud, L.; et al. Concentration-Dependent Activity of Hydromethylthionine on Cognitive Decline and Brain Atrophy in Mild to Moderate Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 931–946. [Google Scholar] [CrossRef]
- Shiells, H.; Schelter, B.O.; Bentham, P.; Baddeley, T.C.; Rubino, C.M.; Ganesan, H.; Hammel, J.; Vuksanovic, V.; Staff, R.T.; Murray, A.D.; et al. Concentration-Dependent Activity of Hydromethylthionine on Clinical Decline and Brain Atrophy in a Randomized Controlled Trial in Behavioral Variant Frontotemporal Dementia. J. Alzheimers Dis. 2020, 75, 501–519. [Google Scholar] [CrossRef]
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jové, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 2014, 23, 3716–3732. [Google Scholar] [CrossRef]
- Riedel, G.; Klein, J.; Niewiadomska, G.; Kondak, C.; Schwab, K.; Lauer, D.; Magbagbeolu, M.; Steczkowska, M.; Zadrozny, M.; Wydrych, M.; et al. Mechanisms of Anticholinesterase Interference with Tau Aggregation Inhibitor Activity in a Tau-Transgenic Mouse Model. Curr. Alzheimer Res. 2020, 17, 285–296. [Google Scholar] [CrossRef]
- Dominguez-Meijide, A.; Vasili, E.; Outeiro, T.F. Pharmacological Modulators of Tau Aggregation and Spreading. Brain Sci. 2020, 10, 858. [Google Scholar] [CrossRef]
- Zhang, F.; Zhong, R.J.; Cheng, C.; Li, S.; Le, W.D. New therapeutics beyond amyloid-β and tau for the treatment of Alzheimer’s disease. Acta Pharm. Sin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. Biomed. Res. Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G.; Kayed, R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006, 66 (Suppl. 1), S74–S748. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niewiadomska, G.; Niewiadomski, W.; Steczkowska, M.; Gasiorowska, A. Tau Oligomers Neurotoxicity. Life 2021, 11, 28. https://doi.org/10.3390/life11010028
Niewiadomska G, Niewiadomski W, Steczkowska M, Gasiorowska A. Tau Oligomers Neurotoxicity. Life. 2021; 11(1):28. https://doi.org/10.3390/life11010028
Chicago/Turabian StyleNiewiadomska, Grazyna, Wiktor Niewiadomski, Marta Steczkowska, and Anna Gasiorowska. 2021. "Tau Oligomers Neurotoxicity" Life 11, no. 1: 28. https://doi.org/10.3390/life11010028
APA StyleNiewiadomska, G., Niewiadomski, W., Steczkowska, M., & Gasiorowska, A. (2021). Tau Oligomers Neurotoxicity. Life, 11(1), 28. https://doi.org/10.3390/life11010028