Abstract
Amyloids are highly ordered fibrous cross-β protein aggregates that are notorious primarily because of association with a variety of incurable human and animal diseases (termed amyloidoses), including Alzheimer’s disease (AD), Parkinson’s disease (PD), type 2 diabetes (T2D), and prion diseases. Some amyloid-associated diseases, in particular T2D and AD, are widespread and affect hundreds of millions of people all over the world. However, recently it has become evident that many amyloids, termed “functional amyloids,” are involved in various activities that are beneficial to organisms. Functional amyloids were discovered in diverse taxa, ranging from bacteria to mammals. These amyloids are involved in vital biological functions such as long-term memory, storage of peptide hormones and scaffolding melanin polymerization in animals, substrate attachment, and biofilm formation in bacteria and fungi, etc. Thus, amyloids undoubtedly are playing important roles in biological and pathological processes. This review is focused on functional amyloids in mammals and summarizes approaches used for identifying new potentially amyloidogenic proteins and domains.
1. Introduction
For a long time, the term “amyloid” was used to describe extracellular tissue deposits of protein fibrils with a characteristic appearance in electron microscope (EM), typical X-ray diffraction pattern, and an affinity to Congo red dye (CR) resulting in green-yellow birefringence [1]. It was thought that amyloids are primarily associated with human and animal diseases (termed “amyloidoses”). Later studies broadened the term “amyloid” and emphasized its structural characteristics so that now it is generally used for the cross β-sheet non-covalent unbranched fibrous protein polymers formed both in vivo and in vitro. In an amyloid, β strands of repeated units are placed perpendicular to the fiber axis, forming an intermolecular cross-β sheet [2]. An amyloid polymer can immobilize non-amyloid monomeric protein molecules of the same sequence, and thus grow via a process of nucleated polymerization. Due to its highly ordered structure, amyloid fibrils as well as oligomers are characterized by resistance to ionic detergents (such as sodium dodecyl sulfate (SDS) or sarcosyl) and some proteases. In addition, they demonstrate an affinity to dyes such as CR, thioflavin T (ThT), and thioflavin S (ThS). Amyloids can also be detected with some amyloid-specific antibodies [3] or aptamers [4]. To date, 37 amyloidogenic proteins that are associated with about 70 different human diseases are known [5]. The most important amyloid-associated diseases include Alzheimer’s and Parkinson’s diseases, type 2 diabetes, and transmissible spongiform encephalopathies (TSEs), or prion diseases, such as Creutzfeldt-Jakob disease [6]. Recent data indicate that amyloids are also associated with preeclampsia and some forms of cancer, although this is unclear whether amyloids cause these diseases or arise as a consequence of the disease and serve as biomarkers [7,8,9,10,11].
In addition to pathogenic amyloids, amyloids participating in a wide range of physiological functions have been identified in various organisms, from bacteria to higher eukaryotes such as vertebrates, plants, and humans. These amyloids are termed as functional amyloids [12,13,14]. For example, the CPEB proteins of Aplysia californica, as well as its orthologs in Drosophila melanogaster and Mus musculus (see below) form amyloid-like SDS-resistant oligomers that are involved in the maintenance of long-term memory [15,16,17]. Spider spidroins form amyloid-based insoluble silk fibrils that are stronger than steel [18]. The protein Luminidependens of the plant Arabidopsis thaliana, that is capable of forming amyloid-like oligomers, is involved in the regulation of flowering by temperature [19]. Notably, this protein is a chromatin remodeler [20], albeit a connection of this function to amyloidogenecity is still hypothetical [21]. Curli proteins in Escherichia coli are assembled on a bacterial cell surface as a part of the extracellular matrix during biofilm formation and control resistance to a variety of environmental stresses [22,23].
In yeast and filamentous fungi, amyloid- based protein polymers can be transmitted from cell to cell during a cell division or by a cytoplasm exchange and control phenotypically detectable traits [24,25,26]. These self-perpetuating amyloids provide a basis for protein-based inheritance and are termed yeast (or fungal) prions. While some of them are clearly pathogenic, others are hypothesized or (in a very few cases) shown to be associated with adaptive roles [27]. Specifically, prion [Het-s] of the fungus Podospora anserina is involved in the control of vegetative incompatibility through the destruction of a mycelium not containing the prion at the position of contact in a manner similar to programmed cell death [28,29].
Constructs based on the translation termination factor Sup35, that can convert into the prion form termed [PSI+], are frequently applied to studying amyloid properties of mammalian and human proteins (see [30] and below).
A significant fraction of proteins involved in transcriptional regulation both in yeast and mammalian cells contain sequences, enriched by Q and/or N residues that are similar to prion domains (PrDs) of yeast prions [31,32,33] and therefore termed PrD-like domains (PrDL). However, the abilities of the majority of these proteins to form an amyloid and their potential role as amyloids are not studied.
To conclude, literature data indicate that the ability to form amyloids under physiological conditions is a characteristic feature of many proteins (including a variety of human proteins) [34], that could be linked to both pathological processes and normal biological functions. This review summarizes data on functional amyloids in mammals and describes approaches for identifying new potentially amyloidogenic proteins and domains.
2. Functional Amyloids in Mammals
2.1. Peptide Hormones in Secretory Granules
Storage of hormones inside the cell can be challenging due to their chemical activity. Some protein and peptide hormones of secretory cells such as neuroendocrine cells and exocrine cells can be stored for a long time in high concentration in membrane-enclosed secretory storage granules (StGs) until an outside signal for their excretion to the extracellular space is received. The first direct evidence for the storage of hormones and pro-hormones in granules has been reported for insulin by Steiner [35]. Further investigation indicated that growth hormone (GH) [36], prolactin [37], adrenocorticotropic hormone (ACTH) [38], and parathyroid hormone [39] form storage granules as well. Maji and colleagues later showed that over 30 of pro-hormones form amyloid structures during storage [40].
The formation of the StG starts from the self-association of hormone molecules in the Golgi complex [41]. As a rule, each StG contains insoluble aggregates of one secretory protein or peptide [42]. Amyloid properties of proteins forming these aggregates have been studied by Riek lab [14,40], by employing a set of approaches such as ThT and CR binding, luminescent conjugated polyelectrolyte probes (LCP), electron microscopy, circular dichroism (CD) spectroscopy, X-ray diffraction. 42 peptide hormones associated with StGs of multiple species and organs were analyzed. It has been shown that most of them can form amyloid aggregates [40]. Aggregation of some peptide hormones and/or prohormones can be initiated spontaneously after reaching a critical concentration threshold at pH 5.5, reflecting conditions in StGs. However, the presence of helper molecules such as glycosaminoglycans, GAGs (for example, heparin), was required for most peptide hormones, presumably for stabilizing the amyloid conformation [40]. Prolactin did not form aggregates in the presence of heparin but showed an aggregation in the presence of chondroitin sulfate A [40], which is a GAG compound found in prolactin-specific granules [43]. An aggregation of ACTH required the presence of an amyloid form of β-endorphin. ACTH and β-endorphin are processed from the same prohormone (proopiomelanocortin) and are stored in StGs together. Besides, aggregation of peptide hormones was also affected by bivalent metal cations [44]. In particular, Zn(II) was shown to initiate an early oligomerization of GH in the pituitary of rat, which may facilitate GH aggregation and amyloid formation [45].
Studies of StGs of the mouse pituitary tumor neuroendocrine cell line AtT20 confirmed their amyloid nature. Purified granules from AtT20 cells reacted to amyloid-specific OC antibodies, and were stained with ThT and CR dyes (also showing the birefringence in the latter case) [45]. In addition, X-ray diffraction of the membrane-less secretory StGs pattern was typical for the cross-β structure. Moreover, β-endorphin, GH, oxytocin, prolactin, vasopressin, and ACTH were stained by ThS or reacted to fibril-specific antibody OC in the mouse pituitary tissue [40]. It was also shown that amyloid fibrils formed by hormones can release monomers [46] if pH is increased to 7.4, the same pH to which hormones are exposed during secretion.
Some mammalian amyloids, including aggregates of the Aβ peptide (associated with Alzheimer’s disease), are toxic to neuronal cells [47]. Maji et al. demonstrated that typically, amyloid aggregates of peptide hormones formed in vitro were less toxic to culture neuronal cells than Aβ aggregates, although some aggregated hormones (for example, ovine CRH, mUcnIII, hUcnIII, human GRF, and glucagon-like peptide-2), demonstrated toxicity comparable to Aβ, whereas aggregated glucagon was much more toxic than Aβ in 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [40]. Apparently, for these hormones condensation in StGs serves as a tool counteracting the toxicity of aggregated hormones to the cell.
Taken together, in vitro and in vivo data confirm the amyloid nature of protein and peptide hormones stored in StGs. Due to the high stability of amyloids, StGs can exist for long periods until the cell receives a signal to excrete hormones, leading to the disaggregation of amyloid assemblies into a functional soluble hormone, that is promoted by an increase in pH from 5.5 (dormant StGs) to 7.4 (blood). In addition, extracellular chaperones may be involved in disaggregation [14,40].
2.2. PMEL Protein and Melanin Biosynthesis
Melanins are chemically heterogeneous pigment molecules found in most organisms. There are three major types of melanin in mammals: eumelanin, neuromelanin, and pheomelanin. Eumelanin and pheomelanin are the ubiquitous pigments present in skin, eye, and hairs that effectively absorb ultraviolet (UV) and visible light [48,49] and protect against sunlight, UV radiation, small toxic molecules as well as involved in thermoregulation and control of coloring. Neuromelanin is present in the human central nervous system [50,51,52], specifically, it is found in large quantities in neurons of the Substantia nigra, ventral tegmental area, and Locus coeruleus [53,54,55]. It has been linked to etiology and pathogenesis of Parkinson’s disease [56,57]. Neuromelanin is composed of lipid, melanine, and peptide components, with the melanic portion being a mixture of pheomelanin and eumelanin [58,59].
Melanin’s synthesis takes place in melanosomes, specialized acidic organelles of pigmented cells (melanocytes). Melanosomes are derived from lysosomes. Formation of luminal amyloid fibrils originated from the melanocyte-specific pre-melanosomal protein PMEL17 has been detected during melanosome biogenesis and maturation, confirmed by ThS and CR staining of melanosomes [60]. Amyloid matrix of PMEL fibrils apparently serves as a scaffold for melanin polymerization, thus promoting melanin synthesis; it is also implicated in sequestering the toxic intermediates of melanin biosynthetic pathway [60,61,62]. These toxic intermediates possess some structural similarity to the amyloid-binding dye ThT. Binding to PMEL17 accelerates the conversion of intermediate 5,6-dihydroxyindole into the covalent melanin polymers [60]. PMEL17-derived amyloid fibrils are mainly detected during the eumelanin production, while PMEL17 participation in the formation of neuromelanin and pheomelanin remains questionable, as PMEL17 fibrils were not detected in pheomelanosomes [62,63]. PMEL17 deficiency did not affect production of pheomelanin [64] and PMEL17 has not been found at significant levels in neuromelanin-producing organelles [65]. However isolated neuromelanin shows a typical cross-β sheet structure X-ray diffraction pattern of 4.7 Å, indicating the presence of amyloid fibrils [66]. The neuromelanin-producing organelles contain alpha-synuclein and a PMEL homolog, the glycoprotein NMB, that can produce amyloid fibrils, which probably starts neuromelanin synthesis [65].
Eumelanosome maturation includes four different stages, and it is accompanied by functional and morphological changes [67]. Stage I and stage II melanosomes are referred to as immature/premelanosomal compartments, and lack pigment. Melanosome maturation is accompanied by an increase in intraluminal pH, from pH 4 in stages I and II to near neutral pH upon full maturation [68]. PMEL17 fibrils start to form in stages I and II, and are organized into parallel sheets that elongate the compartment. An acidic environment of organelles at I and II stages is optimal for the assembly of PMEL17 amyloid fibrils. However, melanin biosynthesis is suppressed at low pH, therefore it begins at stage III melanosomes and is complete at stage IV [63,69].
PMEL is a melanocyte-specific type I transmembrane glycoprotein. PMEL contains a short cytoplasmic C-terminal domain, a transmembrane domain, a luminal N-terminal domain, and an N-terminal signal peptide. The long luminal domain consists of four sub-domains: (1) N-terminal region (NTR), that contains 3 highly conserved N-glycosylation sites and 3 cysteine residues that might form disulfide bonds; (2) polycystic kidney disease-like domain (PKD), that, as predicted, might adopt β-sheet conformation [70]; (3) repeat domain (RPT) containing one conserved cysteine residues and 10 imperfect direct repeats of 13 amino acids sequence rich in glutamic acid, proline, serine, and threonine; (4) Kringle-like domain (KLD), a cysteine-rich region that also has an N-linked glycosylation site essential for protein folding and secretion [71].
PMEL17 fibril formation is tightly regulated and restricted to melanosomes, that allows the protection of the cell from the potential toxic effect of an amyloid (Figure 1). During synthesis, PMEL17 protein is targeted to the endoplasmic reticulum (ER), and is modified via the removal of the signal peptide, the addition of N-linked core oligosaccharides, and formation of disulfide bonds [72,73,74]. Then PMEL17 is transferred from ER to the Golgi complex, where it is O-glycosylated [75,76,77,78] and transported to the premelanosomal vesicles [68,79]. Within the acidic premelanosomal compartment, PMEL17 is proteolytically cleaved by a proprotein convertase, resulting in the large luminal α fragment (Mα), encompassing residues 25‒467 that cover NTR, PKD, and RPT sub-domains, and smaller integral membrane β fragment (Mβ), encompassing residues 468‒668 that cover the KLD sub-domain as well as transmembrane and cytosolic domains. Mα and Mβ fragments remain linked by disulfide bonds [80,81]. The Mβ transmembrane fragment is then cleaved by protease β-secretase 2 (BACE2) that releases the Mα fragment, associated with a luminal part of Mβ (MβN), from the membrane [82,83,84]. Then a series of proteolytic cleavages of Mα produce smaller N-terminal MαN and C-terminal MαC fragments, forming amyloid fibrils in acidic pH [85]. MαN contains the NTR and the PKD sub-domains, and the subsequent truncation at NTR leads to the formation of the PKD-containing fragment [86]. MαC is also further processed, resulting in a ladder of fragments [85,87].
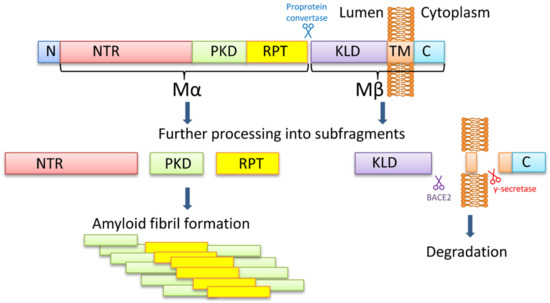
Figure 1.
The PMEL17 processing and formation of amyloid fibrils. The PMEL17 is cut in endoplasmic reticulum and then transported to Golgi apparatus for O-glycosylation. Later in the acidic environment of premelanosomes, the proprotein convertase cuts it to the Mα (an N-terminal ectodomain) and the Mβ (the C-terminal polypeptide containing the transmembrane domain) fragments. The fragments remain connected via the disulfide bond. The BACE2 cuts the Mβ fragment out of the membrane, and so the Mα with the attached KLD domain becomes luminal. Serial cleavages later process the Mα fragment into subfragments, which can create amyloid fibrils in premelanosome. NTR—N-terminal region; PKD—polycystic kidney disease domain; RPT—proline, serine, threonine-rich repeat domain; KLD—kringle-like domain; TM—transmembrane domain; C—cytoplasmic domain.
It is still being debated which PMEL17 region is a primary amyloid-forming region. The RPT sub-domain had initially been proposed to form the amyloid core in vivo [85,87], and this has indeed been shown that RPT fragment forms amyloid in vitro at acidic pH, resembling conditions at early stages of the melanosome biogenesis [87,88,89]. Moreover, preformed fibrils can be rapidly dissolved at neutral pH, which has been proposed as an evolutionary mechanism designed to prevent toxicity of PMEL17 fibrils occasionally released into cytosol [87]. However, other researchers showed that the RPT domain is dispensable for the amyloid formation in vivo, and point to the PKD domain (residues 201–314) [86] and/or core amyloid fragment (residues 148–223) [90] as potential amyloid-forming regions, considering the RPT subdomain as a regulator of the matrix morphology, that maximizes the surface area available for pigment [91]. Further work is needed to determine the which PMEL17 region plays a primary role in the amyloid formation in vivo, although it is possible that different regions of PMEL17 possess amyloid properties, that are involved in various aspects of melanosome biogenesis and melanin production.
2.3. CPEB3 Protein and Long-Term Memory
Cytoplasmic polyadenylation element-binding (CPEB) protein has been implicated as a regulator of local protein synthesis at active synapses in neurons of the mollusk Aplysia [92]. CPEB modulates translation of mRNAs by regulating cytosolic mRNA polyadenylation (see [93,94] for review). Aplysia CPEB [15] and the CPEB isoform Orb2 [16] in the fruit fly Drosophila are shown to be required for the maintenance, but not for the formation, long-term memory and synaptic plasticity [95,96]. Notably, Aplysia CPEB protein exhibits prion-like properties when expressed in yeast cells [97] and can form amyloid-like oligomers, assembled into punctate structures in active synapses [15].
Human and mice genomes contain four CPEB coding genes (CPEB1-4). All mammalian CPEB protein isoforms contain RNA-binding domains at the C-terminus, but only CPEB2 and CPEB3 contain a glutamine (Q)-rich domain at the N-terminus [98]. Aggregation properties and regulation of mouse CPEB3 have been studied in more detail. Similar to some other amyloidogenic proteins CPEB3 can exist either as soluble monomer form or as a self-sustained oligomer or aggregate. Recombinant CPEB3 isolated from bacteria forms fibrils that bind CR and exhibit a green/yellow birefringence in polarized light [99]. Expression of recombinant CPEB3 fused to yellow fluorescent protein or 3 x hemagglutinin tag in yeast cells results in the formation of detergent-resistant aggregates [99], controlled by the Q-rich domain of CPEB3 and depending on the chaperone Hsp104, required for the propagation of endogenous yeast prions (see [26] for review).
CPEB3 has been shown to bind specific neuronal mRNAs, such as mRNAs for GluR2 and beta-actin, and inhibit their translation [99,100]. Monoubiquitination of CPEB3 with Neuralized1 ubiquitin ligase (Neurl1) abolished CPEB3 dependent repression and activated translation [101]. The study of the CPEB3 conditional knockout (cKO) mice revealed that the CPEB3-modulated protein synthesis is necessary for the maintenance but not for the acquisition of long-term memory [17]. Likewise, CPEB3 cKO does not affect the early phase of long-term potentiation (E-LTP) but inhibits the formation of the late phase of long-term potentiation (L-LTP) that lasts more than 24 h and is dependent on protein synthesis. The most interesting property of CPEB3 is its ability to transition from the soluble to aggregated form in response to synaptic stimulation, which leads to the cessation of target RNAs repression, and local activation of translation in active synapses [102]. Removal of the amyloidogenic Q-rich domain of CPEB3 has been shown to repress the stimulation-induced changes in the CPEB3 activity; this repression has led to profound deficits in LTP and memory [17]. Taken together, these data indicate that CPEB3 dependent upregulation of translation (supposedly due to CPEB3 conformational changes in response to neuronal activity) is crucial for the retention of memories [102].
In its soluble form, CPEB3 is SUMOylated and acts as a translation inhibitor in cytoplasmic P-bodies, non-membrane cell compartments accumulating translationally repressed mRNAs and promoting their degradation [103,104,105]. SUMOylation of CPEB3 in hippocampal neurons is decreased in response to the stimulation of learning and to the dendritic activity, accompanied by an increase in the levels of Neurl1, which leads to ubiquitination of CPEB3 and its translocation to polysomes. Translational upregulation coincides with the appearance of detergent-resistant CPEB3 aggregates [101,103,104]. As a result, protein synthesis is induced and LTP is maintained for several days [17,99]. Interestingly, SUMO-2 mRNA is also found among mRNAs upregulated in these conditions, which suggests the existence of a negative feedback loop including CPEB3 and SUMO proteins, a possible regulatory mechanism preventing excessive aggregation of CPEB3 [103].
According to the model proposed by Si and Kandel [102], the CPEB-dependent activation of translation in active synapses is mediated by an aggregated form of CPEB, as aggregates contain the exposed RNA binding domain [102]. Binding of multiple mRNAs on the surface of the fibrous CPEB3 scaffold could allow for the coordinated translation of a variety of coregulated mRNAs, products of which are required for the stabilization of synaptic growth. It is however unclear whether self-sustaining fibrillar amyloids or small oligomers are crucial for translational activation. Existing data are also compatible with an alternative hypothesis, suggesting that aggregation of CPEB induces translation via downregulating the repressive activity, exhibited by the soluble form of CPEB [106]. Besides, it has been reported that the CPEB-dependent activation of translation in Aplysia is required only during the first 72 h after training; after that period, protein synthesis inhibitors and CPEB knockdowns no longer affect synaptic growth [107]. Thus, further studies are needed to decipher the exact mechanism by which CBEP3 oligomerization and/or aggregation modulate long-term potentiation and memory.
A recent study of Hervas and colleagues confirmed an amyloid structure, revealed a formation of the amyloid core, and thoroughly described a mechanism of activity [108]. According to the group, the Orb2 filament was composed of a hydrophilic core with a stabilization via interdigitated glutamines [108].
2.4. RNA-Binding Protein FXR1
Fragile X-related Proteins (FXR1 and FXR2) are RNA-binding proteins that regulate transcription, translation, and RNA stability [109,110,111]. FXRs are associated with ribosomes, predominantly with the large (60S) ribosomal subunits [112], and with the RNA-induced silencing complex via Argonaute 2 (AGO2) [113,114]. Small FXR1/AGO2-containing ribonucleoprotein (RNP) granules associate with TNFα AU-rich elements (ARE) facilitating reporter translation under conditions of growth inhibition. However, in dividing cells, FXR1 forms insoluble complexes and the TNFα 3′-UTR-containing reporter RNA translation is blocked [115,116]. The repression of translation is accompanied by translocation of FXR1 from the AGO2-bound polysomal mRNP to the P-body positive foci [115]. In addition, FXR1 is possibly involved in the formation of stress granules known to accumulate stalled translation initiation complexes during unfavorable conditions [117,118]. It was shown that the highly conserved N-terminus of human FXR proteins forms aggregates with amyloid properties in vitro [119].
Using PSIA-LC-MALDI approach (proteomic screening and identification of amyloids/liquid chromatography coupled with mass-spectrometry) for searching amyloids in the rat brain, Sopova et al. observed that FXR1 forms SDS-resistant aggregates of amyloid type [120]. Authors demonstrated that FXR1 is binding CR, ThS, and ThT in rat brain cells, as well as in human neuroblastoma cell culture. Formation of the amyloid form of FXR1 in tissues possibly depends on the conserved N-terminal fragment of the protein. It was suggested that an aggregated state of FXR1 is important for the inhibition of translation and protection of mRNA from degradation [120], which could be crucial for long-lived cells such as neurons.
2.5. Zona Pellucida Proteins
Zona Pellucida (ZP) is an extracellular fibrillar coat of oocytes that plays a vital role during fertilization and preimplantation development. ZP consists of three (mouse) or four (human) glycoproteins (ZP1–4) that are produced by growing oocytes [121,122]. Each ZP protein contains a ZP polymerization domain that controls the formation of fibrils and their assembly into a porous three-dimensional ZP matrix [123]. Mouse ZP1 and ZP2 are shown to form heterodimers with ZP3, which are assembled into long filaments cross-linking through ZP1 homodimers [124,125,126]. Isolated ZP possess amyloid features, as shown by their reaction to amyloid-specific antibodies, CR binding and green/yellow birefringence, ThS binding, and detergent resistance [127]. So, all three mice ZP proteins are probably included in the ZP matrix in an amyloid shape.
Comparison of the ZP3 polymerization domains among various vertebrates from fish to humans demonstrated the lack of overall sequence conservation; however, conserved stretches were found in sites that are predicted to be amyloidogenic [127]. Some of these sites are localized in regions for which the formation of β-strands is expected according to the crystal structure of monomeric chicken ZP3, including regions important for the interaction between ZP subdomains [128]. Other amyloidogenic sites have been found in regions located outside of the ZP polymerization domain, including ZP-N repeats, which mediate interactions with spermatozoid cells [129]. None of ZP proteins contains QN-rich prion-like domains that would be similar to aggregation-prone sequences of yeast prion proteins.
Like some other functional amyloids, ZP can switch between distinct functional states, as a part of the cell physiological response to external factors. After the sperm and egg fusion, a cascade of biochemical reactions is launched, leading to the exocytosis of cortical vesicles that release proteases and other enzymes into the perivitelline space between ZP and the cell membrane and modify ZP by cleaving the polypeptide fragment from ZP2 [130]. As a result, additional spermatozoa are no longer able to penetrate ZP. Transgenic female mice with truncated ZP2 are sterile [131]. The presence of 6–8 amyloidogenic sites in each of the ZP proteins generates a potential for a variety of cross-β structures [132].
ZP amyloids may protect the oocyte after fertilization when it is surrounded by proteases and other hydrolytic enzymes released from the sperm. The protective function of ZP amyloids is probably required in subsequent processes of early embryogenesis since ZP holds together the blastomeres devoid of cell contacts [133,134]. In addition, based on the fact that the acrosomal matrix (insoluble fraction in the acrosome of spermatozoon) has an amyloid cortex, which includes ZP binding proteins, it is suggested that sperm-ZP binding can also occur through amyloid-amyloid interactions [132].
2.6. RIP1 and RIP3 Proteins
RIP1 and RIP3 are critical receptor-interacting serine/threonine kinases responsible for mediating necroptosis [135,136,137], a process that has been implicated as an important driver of inflammation and pathology in certain human diseases, such as ischemic brain injury, immune system disorders, some forms of neurodegeneration, and cancer [138,139,140]. Necroptosis is one of the regulated forms of necrosis. Conventionally, necrosis was considered as unprogrammed cell death in contrast to the standard programmed cell death via apoptosis, however recent data uncovered multiple caspase-independent pathways of regulated necrosis, including RIP1/RIP3-mediated necroptosis [136,141,142]. Both RIP1 and RIP3 contain the N-proximal Ser/Thr kinase domains (KDs), encompassing aa residues 17–289 (RIP1), or 21–287 (RIP3), and RIP homotypic interaction motifs, (RHIMs) encompassing aa residues 531–547 (RIP1) or 450–466 (RIP3) [143]. RIP1 also contains a C-terminal death domain” (DD) located at aa positions 583–669 and implicated in the recruitment of RIP1 into the TNFR1 (tumor necrosis factor receptor 1) signaling complex [144,145,146]. RIP1 and RIP3 can form mixed amyloid fibrils mediated by their RHIM domains, and this amyloid-based complex actually serves as an inducer of necroptosis [137]. The amyloid nature of the RIP1/RIP3 complex was confirmed in vitro by its ability to bind ThT and CR, circular dichroism, infrared spectroscopy, X-ray diffraction pattern, and solid-state NMR analysis, as well as in vivo by using ThT staining of RIP1/RIP3-containing puncta in necrotic HeLa cells [147].
It appears that various modifications of RIP1 determine various cell fates, such as survival, apoptosis, or necroptosis. For example, ubiquitination of RIP1 promotes cell survival due to activation of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases, while non-ubiquitinated RIP1 induces the caspase-8-mediated apoptosis pathway [148]. Amyloid RIP1/RIP3 complex, also known as necrosome or ripoptosome is formed in the conditions when caspase-8 is inhibited [136]. During necrosome formation, RIP1 and RIP3 autophosphorylate and transphosphorylate each other [136,149]. Then, RIP3 phosphorylates the mixed lineage kinase domain-like protein (MLKL) [143], resulting in its oligomerization and translocation in the membrane that causes pore formation and disruption of membrane integrity [139,150]. Interestingly, some viruses such as Herpes Virus 1 [151] or Epstein–Barr Virus [152] interfere with amyloid formation via RHIM-less proteins mimicking RIP1 and RIP3.
Some mutations in the RHIM domains of RIP1 or RIP3 impair amyloid formation and protect cells from necroptosis in vitro, thus signifying the importance of an amyloid formation for the process of necroptosis [147]. Presumably, RIP1 and RIP3 form hetero-amyloid structures. High-resolution solid-state NMR structural studies of amyloids formed by RIP1, RIP3, or RIP1 and RIP3 together show that the RIP1/RIP3 heteroamyloid is energetically favorable in comparison to either homoamyloid [153]. To date, the immunoprecipitation method revealed that RIP1/RIP3 can compose a heteroamyloid [149], although the structural and functional relationships require further clarification. It is also worth noting that the RHIM motifs of RIP proteins are homologous to the prion domain of Het-s, a prion protein of the fungus Podospora anserina, that is also involved in the programmed mycelium destruction process, controlling the cytoplasmic incompatibility, a biologically advantageous process in fungi [154,155]. Proteins with similar domains and roles are also found in other fungi [156]. These findings point to the evolutionary conservation of the amyloid-based triggering of programmed cell death.
2.7. MBP-1 Protein
Eosinophil major basic protein 1 (MBP-1) is a component of eosinophil membrane-enclosed granules that are released in the inflammation foci. These granules also include two ribonucleases, ECP and EDN/RNase2, and peroxidase EPO, which together participate in the destruction of bacteria, viruses, and helminths [157,158]. Inactive MBP-1 is accumulated in granules in the form of amyloid-type aggregates that could be stained with ThT and conformation-specific antibodies [159]. The MBP-1 monomers aggregate in vitro, as visible by transmission electron microscopy [159].
When eosinophils are activated at the site of inflammation, granules are acidified, and their contents are released into the extracellular space by piecemeal degranulation [157,160]. Acidification of the granule contents promotes disassembly of insoluble aggregates into monomers and/or oligomers, and therefore, conversion of MBP-1 into an active toxic form [159]. This secretion into the neutral pH of cellular milieu favors binding of soluble MBP-1 to the surface of bacterial cells, resulting in the membrane damage and bacterial death. It is believed that after binding to the bacterial membrane, activated MBP-1 aggregates into toxic β-sheet-rich clusters.
In silico analysis reveals a 5-aa stretch in MBP-1 that shows a tendency to form β-crosslinks [159]. It was shown that MBP-1-derived peptide encompassing aa residues 18–45 [161] and including this amyloidogenic stretch, causes toxicity via thinning the keratinocyte layer and promoting DNA fragmentation in keratinocytes when injected into the mouse dermis [159].
Several diseases associated with massive eosinophil infiltration and degranulation, such as eosinophilic asthma, atopic dermatitis, etc., are caused by MBP-1 toxicity to the host cells in inflammatory loci [162,163]. Anti-amyloid antibodies and other amyloid-binding substances such as heparin accelerate MBP-1 aggregation and significantly suppress the toxic effect of MBP-1 [164]. In addition, large extracellular amyloid deposits of MBP-1 are often found both in tissues infiltrated with eosinophils and in those with relatively little evidence for eosinophil-associated tissue damage [165,166]. Heparin is known to be released by mast cells, granulated white blood cells, which are also present in the sites of inflammation. This could serve as a mechanism accelerating aggregation and subsequent inactivation of MBP-1, and therefore protecting tissues from the toxic effects of MBP-1 [159,167].
2.8. CRES and PAP
Functional amyloids have also been found in the epididymis, a convoluted tubule that is located in the testis. During migration through the epididymis, spermatozoa undergo maturation and acquire motility and the potential for fertility. The maturation of spermatozoa requires interactions with proteins secreted by the epididymal epithelium. The epididymis is also involved in protection against pathogens [168]. Proteins secreted by the epididymal epithelium produce an amyloid-based matrix, consisting of multiple members of the family 2 cystatins, including the cysteine protease inhibitors (such as cystatin C) and four members of the cystatin-related epididymal spermatogenic subgroup, namely CRES, CRES2, CRES3 and cystatin E2 [169].
Several standard techniques such as binding to ThS, ThT, and anti-amyloid antibodies, X-ray diffraction, and negative-stain transmission electron microscopy confirmed the amyloid nature of the CRES-based epidydimal matrix [168]. However, the physiological function of these amyloids is not clear. In mouse models, it has been demonstrated that disruption of the amyloid matrix can result in epididymal pathology, including infertility [170,171], lysosomal storage disease, and decreased sperm survival [172].
Amyloid structures were also found in the acrosomal matrix (AM) and an equatorial segment of a spermatozoon, as confirmed in vivo by ThT staining and by resistance to SDS and formic acid treatments [132]. AM contains fifty-nine proteins, including known amyloidogenic proteins such as cystatin C, CRES (same as in epididymal matrix), lysozyme, transglutaminase 3, zonadhesin, zona pellucida 3 receptor and others [173]. To date, it is not clear which proteins form amyloid in AM. Moreover, the functional role of the amyloid structures in AM is not determined. It was suggested that the AM amyloid could be crucial for the acrosome exosomal reaction during fertilization and/or for proper orientation of signaling complexes during fertilization [129,172,174].
Prostate acidic phosphatase (PAP) is a semen-derived protein, which is not studied well yet, but there is some evidence of the formation of amyloid structure by this protein and in particular, by its region encompassing aa residues 248–286, based on X-ray scanning [175]. It tends to form structured fibrils in the alkaline or neutral environment but produces amorphous agglomerates in the acidic environment [176]. PAP fibrils apparently produce a fibrous network in the vagina. Additional evidence of the amyloid nature of PAP fibrils is that they are destroyed by anti-amyloid β agents (such as (−)-Epigallocatechin-3-gallate [177]) or by yeast chaperone Hsp104 [178] involved in the fragmentation of yeast amyloid-based prions [26,179].
It is possible that the PAP network is involved in antibacterial protection during sexual intercourse [180]. Unfortunately for humankind, some viruses such as HIV or herpes virus have adapted to this mechanism, so the naturally occurred protection mechanism now provides an increase of virus infiltration 4–5 fold [181]. Locking the virus with the mechanism described above is one of the promising ways to decrease virus spreading [182].
2.9. MAVS Proteins
The system composed of the MDA5 (melanoma differentiation-associated 5) protein and MAVS (mitochondrial antiviral signaling protein) is involved in defense against viral agents and targets genetic material or replication intermediates of many viruses [183]. MDA5-MAVS system is a primary aim of the viral immune system. Hepatitis C [184], enterovirus 71 [185], and coxsackievirus [186] target MDA5 and inhibit downstream signaling.
MDA5 includes the N-proximal caspase recruitment domains (CARDs), middle DExD/H-box helicase domain, and C-terminal domain (CTD) [187]. MDA5 binds non-specifically to long nucleic acid molecules that are frequently associated with viral invasion (such as dsDNA viral genomes and viral replication products e.g., folded RNA) via its helicase and CTD domains and forms a polar helical structure with a twist around dsDNA [188]. This conformation allows exposure of CARDs, enabling them to bind MAVS proteins, anchored into membranes of mitochondria and peroxisomes [189]. Thus, CARDs of MDA5 seed the MAVS filaments formation. MAVS protein consists of the transmembrane domain, long cytosolic domain containing the CARD-binding site, and targeting domain (TM). Amyloid nature of MAVS fibrils was confirmed by electron microscopy, resistance to detergents (using semidenaturating detergent-agarose gel electrophoresis), and resistance to high concentrations of proteases; MAVS depositions were also studied using fluorescence microscopy [190]. Polymeric (but not monomeric) MAVS activates downstream RLR signaling pathway by inducing IRF3 and HF-kB via its TM domains [191,192]. Hou and colleagues report that one mitochondria with polymerized MAVS can initiate the same process on another MAVS protein in another mitochondria and so drastically increase signal propagation [190]. On the other hand, a cell can regulate the RLR signaling activity with a truncated alternatively spliced form of MAVS (mini MAVS) antagonizing activation by polymeric MAVS [193].
2.10. Fibrin
Fibrin is a polymeric protein component of blood clots, formed from fibrinogen via its cleavage by thrombine protease, followed by polymerization. Fibrin mesh is necessary for the prevention of blood loss and healing the wounds [194].
Fibrinogen is a hexamer of three pairs of polypeptide chains, Aα, Bβ, and γ [195,196]. During fibrin formation, thrombine cleaves the small fibrinopeptides A and B (FpA and FpB) of fibrinogen to yield the α, β and γ chains, forming the fibrin monomer. Then monomeric fibrin self-assembles spontaneously to yield polymeric fibrin mesh [194].
The first evidence of amyloidogenic properties of fibrin was reported by Kranenburg et al. [197], who demonstrated that in the conditions, mimicking the physiological situation, a peptide corresponding to the aa positions 148–160 within the fibrin α chain can form amyloid fibrils in vitro, as confirmed by ThT binding and X-ray diffraction patterns. The fibrin peptide and fibrin in the polymerized cross-β form are binding tissue plasminogen activator (tPa), thus stimulating tPA-mediated plasmin formation, and proteolysis of fibrin itself (fibrinolysis) in vitro [197]. Moreover, Kranenburg et al. showed that some other proteins in cross-β form (Aβ, Human islet amyloid polypeptide (hIAPP) and endostatin) also support tPA-mediated plasminogen activation [197].
However, it is still not clear whether or not fibrin forms the amyloid-type cross-β structure in vivo. Some authors suggested that polymerized fibrin retains α-helical secondary structure [198,199,200,201], but can undergo structural transformation into cross-β amyloids under certain conditions, such as mechanical stretching [202,203], in individuals with certain amyloidosis such as Alzheimer’s or Parkinson’s diseases, type 2 diabetes and others [204,205,206,207,208], or in a result of a mutation [11,209,210,211]. Thus, the α-helix to β-sheet transition could be a sign of an anomalous blood clotting, leading to hypercoagulability and hypofibrinolysis [212]. For example, Aβ (a peptide related to Alzheimer’s disease, which is also present in blood) binds fibrinogen with high affinity, and such an interaction induces abnormal fibrin clotting that is resistant to degradation [213,214,215]. These abnormal clots are stained with ThT, but it is not clear whether fibrin in clots is in amyloid form or not, and whether fibrinogen provokes aggregation of Aβ or vice versa [212]. According to these findings, amyloid formation by fibrin is mostly related to pathological amyloidosis rather than to the fibrin function. However, this issue remains unclear, and further investigation on the role of fibrin-based amyloid fibrils in vivo is needed.
2.11. TIA-1 Protein and Stress Granules
During stress, cells have to suppress particular metabolic pathways and block some functions, simultaneous increasing production of components needed for the defense against stress, such as heat shock proteins. For the rapid reorganization of the translational machinery, eukaryotic cells transiently assemble the non-essential translational pre-initiation complexes into formations, termed stress granules (SGs). SGs contain mRNAs (not coding for the stress response proteins), translational factors, and small ribosomal subunits, as well as some additional components, such as helicases, ribonucleases, kinases, and signaling molecules [216]. After return to normal conditions, SGs disassemble; otherwise, they can turn into solid aggregates [217].
SGs are non-membranous organelles, which possess features of liquid-liquid phase separation assemblies (biocondensates); however, they are nucleated by a protein that has been historically named T-cell intracellular antigen-1 (TIA-1) and is suspected to possess amyloid properties. TIA-1 contains RNA-recognition motifs at the N-terminus and a Q/N-rich domain, similar to prion domains (PrDs) of yeast proteins, and therefore termed the PrD-like domain (PrDL), at the C-terminus [218]. TIA-1 is associated with mRNAs via the RNA-recognition motifs, while PrDL mediates interactions between TIA-1 molecules. TIA-1 formed fibrillar structures in vitro; when expressed in yeast, mammalian TIA-1 produces filamentous detergent-resistant polymers with prion-like features and interacts with PrD of the yeast translation termination factor Sup35 [218]. Notably, Sup35 PrD can substitute for the TIA-1 PrDL domain in the process of SG nucleation in mammalian cells [219]. Additionally, Sup35 PrD fused to hemagglutinin tag is recruited into SGs in non-stressed cells of the N2a cell line [220]. Rayman and Kandel reported a transition of TIA-1 from the monomeric state into an SDS-resistant (possibly amyloid-like) structure in a mouse brain [221]. Notably, TIA-1 regulates the synthesis of stress-related chaperones Hsp40 and Hsp70 [219,222]. Still, it remains unclear whether TIA-1 polymers formed in the process of SG formation represent amyloids.
The TIA-1-deficient mice are viable, but exhibit high mortality at early stages of development and elevated susceptibility to lipopolysaccharide-associated toxicity [223], as well as alterations in lipid dynamics [224]. The depletion of the TIA-1 also increases susceptibility to a viral infection, in agreement with increased accumulation of SGs are the sites of viral assembly in wild-type cells [225]. Some viruses such as rhabdovirus modulate or inhibit SG formation [226]. There is also evidence of the association of the AD-related amyloidogenic proteins, Aβ, and tau with SGs [227]. Tau, in particular, plays an important role in the formation of solid aggregates via regulating TIA-1 distribution and promoting SG formation and transition to the solid aggregated state [228].
Information about all the proteins described in this section is summarized in Table 1.

Table 1.
Functional mammalian amyloids considered in this review.
3. Approaches for Identification of New Amyloids and Potentially Amyloidogenic Proteins
Most amyloids known to date were identified either due to their accumulation in specific diseases or through detailed studying of the mechanisms of the impact of specific proteins on particular biological or pathological processes. Unfortunately, the initial identification of many amyloids and potentially amyloidogenic proteins in biological samples cannot be carried out based on Congo red staining, EM or X-ray diffraction data, because these can be used for characterization of purified proteins or large deposits, but are not directly applicable to the detection of small qualities of fibrils or oligomers proteins in the cells, organisms or body fluids. Until recently, no unbiased approaches for the identification of amyloids and potentially amyloidogenic proteins existed. While the formation of insoluble aggregates is a characteristic feature of many amyloids, not all insoluble aggregates are amyloids. Various computational tools for amyloid prediction were developed, however most of them work well for short peptides and/or in vitro conditions, but not for full-length proteins in vivo (for review, see [229,230]). The matter is further complicated by the fact that many (if not most) proteins can form amyloid in vitro depending on concentrations and conditions (such as pH, etc.) [231]; however, amyloidogenic potential of the majority of protein sequences is suppressed in cells and organisms, as it interferes with normal protein functions. Therefore, there is a significant demand for developing unbiased approaches to identification of in vivo, amyloid potential, capable of composing actual and potential “amyloidomes” (i.e., complete cells of amyloid or potentially amyloidogenic proteins in proteomes) of living cells and organisms.
3.1. Identification of Amyloid Proteins Based on Their Biochemical Properties
One of the tools for universal identification of amyloids is based on a common biochemical property of most amyloid fibrils, that is, their high resistance to ionic detergents such as SDS or sarcosyl, which solubilize almost all non-amyloid complexes and disrupt lateral interactions between amyloid fibrils, but don’t monomerize fibrils per se [232]. Amyloids are sedimented from detergent containing solutions and further analyzed by either 2D gel electrophoresis, or liquid chromatography followed by mass-spectrometry (Figure 2) [117,233,234,235]. By using the latter combination, the proteome-wide approach termed PSIA (Proteomic Screening and Identification of Amyloids) has been developed [117,234,235]. PSIA method was applied to identification of new amyloid proteins (potential functional amyloids) in bacteria [236,237] and yeast [238], as well as in the rat hippocampus [120]. Amyloidogenic properties of some of these proteins, including bacterial proteins YghJ, RopA and RopB, yeast proteins Gas1, Toh1 and Ygp1, and the abovementioned rat Fxr1 have been then confirmed by using a variety of standard amyloid characterization techniques [117,237,238,239,240]. An approach similar to PSIA has been independently developed by F. Shewmaker lab and applied to identification of yeast prions [241].
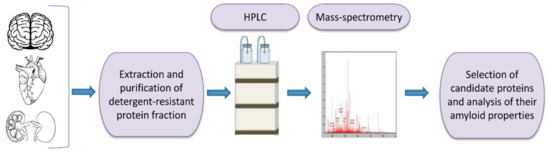
Figure 2.
A scheme of proteomic screening and identification of amyloids (PSIA) method for amyloid screening. The pellet fraction of a homogenized sample, that is resistant to sodium dodecyl sulfate or sarcosyl is analyzed by high performance liquid chromatography followed by mass-spectrometry. Individual candidate proteins, identified by PSIA, can be further investigated in vivo and in vitro using conventional approaches such as circular dichroism spectrum, X-ray fiber diffraction, ability to bind Congo red dye with green/yellow birefringence in polarized light, etc.
A disadvantage of this approach is that PSIA is not completely selective and can also detect some non-amyloid formations that are stable in detergent [120]. In addition, proteins forming amyloids only in certain physiological conditions would not be detected if proper conditions are not used.
3.2. C-DAG
The ability of E. coli cells to produce extracellular fibers known as “curli”, inspired the creation of a new system for amyloid detection called curli-dependent amyloid generator (C-DAG) [242]. When the export signal sequence of CsgA protein is fused to an amyloidogenic protein (or its amyloidogenic domain), the resulting chimeric protein is secreted and can form amyloid fibrils, anchored into the outer membrane and exposed to the outside environment (Figure 3). When plated on Congo red containing media, these amyloid-producing colonies become reddish due to the binding of CR to amyloids. Amyloid nature of CR-bound fibrils is further confirmed by apple-green birefringence in the polarized light, and fibrils can also be visualized by electron microscopy [243]. This approach was successfully tested using several known yeast amyloidogenic proteins (Sup35NM, Rnq1, Cyc8, and New1) and polyQ (aggregating) region of human huntingtin (Htt72Q), while the non-aggregating linker domain of yeast Sup35 protein (Sup35M) and non-aggregating derivative of human huntingtin (Htt25Q) were used as negative controls [242]. At subsequent stages, the C-DAG system was used to confirm the amyloidogenic properties of several yeast proteins (Mss11 and Pub1 [242], Gas1 and Ygp1 [238], and Toh1 [240]), some bacterial proteins (biofilm-associated proteins especially from Enterococcus faecalis and Bap from S. aureus [244], RopA and RopB from Rhizobium leguminosarum [237], and YghJ from E.coli [239]), and Fxr1 protein from rat Rattus norvegicus [120]. C-DAG approach was suggested as a convenient method for screening of potentially amyloidogenic proteins from DNA libraries [242]. This approach can also be used to identify mutations that influence the process of amyloidization, as shown for the Sup35NM variant lacking four oligopeptide repeat sequences [242]. However, the C-DAG approach has several limitations. First, colony coloration may vary depending on the particular protein [242]. Second, not all mammalian proteins are efficiently produced and exported in E. coli [245]. Third, an extracellular environment might potentially antagonize amyloid formation.
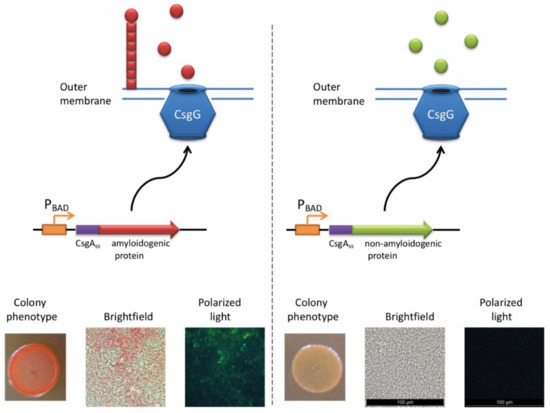
Figure 3.
The principle of C-DAG approach (for detail, see [244]). A chimeric protein, containing the export signal sequence of CsgA protein fused to the protein of interest is exposed to the extracellular space. If the protein of interest is amyloidogenic, it forms a “curli”-like fibril. Amyloid-producing colonies become reddish on the medium containing Congo red (CR). Moreover, binding of CR to amyloid fibrils results in apple-green birefringence in polarized light. Images are obtained by J.V. Sopova (unpublished data).
3.3. Approaches Based on Phenotypic Detection of Prion Formation in Yeast
The yeast Saccharomyces cerevisiae contains endogenous self-perpetuating amyloids (yeast prions) that serve as a convenient and reliable model for studying amyloid aggregation (for review, see [25,26]). Aggregation of a variety of mammalian human amyloidogenic proteins has also been reproduced and studied in yeast. These topics, especially in regard to studies of pathogenic mammalian amyloids in yeast, have been comprehensively covered in recent reviews (see [26,30]). Here, we only mention yeast-based approaches that can be applied to the identification of new amyloidogenic proteins (among them, potential functional amyloids).
The major advantage of the yeast model is that amyloid formation by yeast proteins can be detected phenotypically, via growth and/or color of specially engineered yeast strains on specific media. These phenotypes are usually resulting from the partial inactivation of a yeast protein in a prion form, thus chimeric constructs based on the fusion of a portion of the yeast protein to the potentially amyloidogenic region of a mammalian protein can utilize yeast protein as a reporter for the amyloid formation by a mammalian protein, allowing for easy phenotypic readout.
Prion-forming protein Sup35 that is most frequently employed in such studies is a yeast counterpart of the translation termination (release) factor, eRF3 [246,247]. Sup35 protein is composed of three major regions, namely: (a) N-terminal PrD, or Sup35N that is responsible for amyloid formation, (b) middle region or Sup35M that promotes Sup35 solubility in a pH-dependent manner, and (c) the C-proximal functional region, that is essential and sufficient for the role of Sup35 in termination of translation [25,248]. While Sup35N and Sup35M are dispensable for translation termination and cell viability, Sup35 can antagonize this function of Sup35C by incorporating the Sup35 protein into [PSI+] prion aggregates. Prion aggregation of Sup35 results in a decrease of its ability to access terminating ribosomes, thus causing readthrough of nonsense codons [25]. Therefore, formation of the Sup35 prion state can be phenotypically detected, for example in yeast strains with a premature stop codon in the ADE1 gene (the UGA mutation ade1-14). In normal conditions, such a strain is typically incapable of growing on the medium lacking adenine (-Ade) and accumulates red pigment (a polymerized intermediate of the adenine biosynthetic pathway) on complete organic (Yeast extract with Peptone and Dextrose, or YPD) medium. However, UGA readthrough due to partial inactivation of Sup35 in the prion ([PSI+]) form leads to growth on -Ade and more whitish color on YPD (Figure 4).
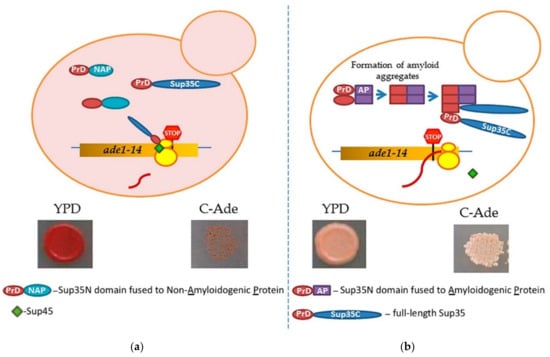
Figure 4.
A scheme of testing heterologous proteins for amyloidogenicity in yeast. Yeast strain contains an ade1-14 (UGA) reporter, allowing the detection of the Sup35 prion ([PSI+]) due to translational readthrough. (a) Overexpression of Sup35 PrD alone or in fusion to non-amyloidogenic protein (PrD-NAP) does not lead to efficient nucleation of the [PSI+] prion in the [pin−] cells yeast lacking any known pre-existing prions. Respectively, there is no growth on the medium lacking adenine, and red color is detected on complete (YPD) medium due to the accumulation and polymerization of an intermediate in the adenine biosynthetic pathway. (b) An amyloidogenic protein (AP) attached to Sup35 PrD forms an amyloid aggregate in yeast, thus bringing together the Sup35 PrD regions and facilitating the conversion of these regions into a cross-β nucleus. Complete Sup35 protein, present in the same cell, is then immobilized into such a nucleus via the PrD-PrD interaction, and converted into a prion form, thus producing the [PSI+] prion and allowing for a phenotypic detection. Respectively, there is growth on the medium lacking adenine, and a whitish color is detected on YPD medium.
One approach for uncovering and/or studying amyloidogenic potential of proteins of various origins (for example, see [249]), is based on the substitution of the whole Sup35N domain (or in some cases, its N-terminal aggregation-prone QN-rich stretch, e.g., see [250]) by the known or suspected amyloidogenic sequence. If this sequence can promote amyloid formation, Sup35 protein can be turned into a phenotypically detectable prion. While this approach has been and is continued to be used successfully for some mammalian proteins associated with amyloid diseases (for review, see [30,251]), it also has certain limitations. First, some fusions antagonize Sup35 function by mechanisms not related to amyloid formation; second, in some cases when partial inactivation of Sup35 occurs due to instant amyloid formation by a chimeric protein in yeast, this system becomes difficult to apply when studying transitions between non-amyloid and amyloid states.
Another approach, specifically targeting de novo amyloid nucleation in yeast [252], has been developed on the basis of the ability of yeast prions to promote formation of other prions [253,254,255]. This approach employs the fusion of a candidate protein or protein domain to Sup35N or NM fragment that is expressed in a yeast cell separately from the full-length Sup35 protein. Transient overproduction of Sup35 or its PrD-containing fragments is known to nucleate formation of the [PSI+] prion in yeast cells [256,257]. However, this process is efficient only in the presence of another preexisting prion, such as [PIN+], a prion form of Rnq1 protein [253,254,255]. Possibly Rnq1 prion (or another prion, typically with a QN-rich domain) is needed to cross-seed the initial nucleation of the Sup35 prion in trans. However, when Sup35N or Sup35NM fragment is fused to another amyloidogenic protein (not necessarily QN-rich) in cis, such a construct can nucleate a prion on its own when overproduced (and for some highly amyloidogenic proteins, even at moderate levels of expression) [252]. The most likely scenario is that the amyloidogenic protein or domain attached to Sup35 PrD forms an amyloid aggregate in yeast, thus bringing together the Sup35 PrD regions and facilitating promoting conversion of these regions into a cross-β nucleus. Complete Sup35 protein, present in the same cell, is then immobilized into such a nucleus via the PrD-PrD interaction, and converted into a prion form, thus producing the [PSI+] prion and allowing for a phenotypic detection.
This approach has worked successfully for known mammalian amyloidogenic proteins including mouse PrP (an agent of transmissible spongiform encephalopathies, or prion diseases), Aβ (associated with Alzheimer’s disease), α-synuclein (associated with Parkinson’s disease), and amylin, or IAPP (associated with type II diabetes) [252]. Moreover, sequence alterations in PrP and Aβ that are known to antagonize prion propagation or amyloid formation also decreased the ability of respective constructs to nucleate the [PSI+] prion in yeast model system, while to the sequence alterations associated with a heritable form of the disease promoted [PSI+] nucleation. The formation of detergent-resistant aggregates by chimeric proteins and immobilization of full-length Sup35 into an aggregated state has also been confirmed by biochemical approaches [252]. In contrast, non-amyloid proteins, including those known to form globular multimeric assemblies, failed to nucleate the [PSI+] prion in the absence of pre-existing prions when fused to Sup35 PrD. Even though further studies are needed to determine if some proteins forming more complex non-globular non-amyloid assemblies, such as liquid droplets, hydrogels, or hydrophobic agglomerates, are capable of nucleating prion in yeast, existing data already establish a fusion of the protein or domain of interest to Sup35 PrD as an assay for the initial phenotypic detection of amyloidogenic properties of the proteins of various origins in yeast.
This assay is amenable to the large-scale screening and has been applied to studying human-derived library and several human proteins whose amyloidogenicity has been predicted by a computational algorithm (A. Zelinsky, N. Romanova, D. Kachkin, A. Aksenova, A. Rubel, and Y. Chernoff, unpublished data). Several new amyloidogenic human proteins have indeed been identified and are currently under detailed investigation by using standard amyloid detection techniques and studies of aggregation properties in the native environment (human cells). Some of these proteins may represent candidates for new functional amyloids in humans.
4. Conclusions
A number of examples of mammalian functional amyloids are described to date (see Table 1). Recently developed unbiased approaches for amyloid detection can expand our knowledge of mammalian amyloidomes very quickly. While functions of amyloids vary between different proteins and tissues, some common features could be recognized as well. Typically, in contrast to pathogenic or heritable amyloids, functional amyloids are dynamic formations that undergo structural changes in response to physiological or external signals. In response to such signals, amyloidogenic proteins can be assembled into oligomers or polymers (CPEB), solubilized from polymers into monomers (MBP-1, peptide hormones) or undergo changes in structural and functional properties (ZP proteins). For some mammalian functional amyloids, such dynamic transitions could be promoted by covalent posttranslational modifications, such as SUMOylation/ubiquitination or site-specific proteolysis. Such modifications of proteins regulating amyloid assembly or disassembly are also described for other taxa [258,259,260].
Notably, some pathogenic amyloids have been hypothesized to also play biologically positive roles. For example, Aβ (related to AD) and PrP (associated with transmissible spongiform encephalopathies) were suggested to participate in the antimicrobial defense in brains [261,262]. This indicates that some amyloid-related pathologies could represent a by-product of the functional manifestations of certain amyloids. Mechanisms of pathological amyloidoses are still poorly understood, therefore the study of functional amyloids could provide additional insights into the processes leading to amyloid pathogenicity. While functional amyloids are formed (or disassembled) in a controlled manner for performing certain biological functions, pathogenic amyloids escape the cellular or organismal control, essentially becoming the spreading disease agents. Nevertheless, molecular mechanisms responsible for the conversion of proteins to the amyloid form or for the disassembly/clearance of amyloids could be similar for functional and pathological entities [263]. Thus, further investigation of functional amyloids may help us to elaborate on new approaches to the treatment of amyloidosis.
Author Contributions
Conceptualization, M.S.R., Y.O.C. and A.A.R.; validation, M.S.R., S.A.F., A.V.G., Y.O.C. and A.A.R.; writing—original draft preparation, A.A.R.; writing—review and editing, M.S.R., S.A.F., A.V.G., J.V.S., O.A.M., Y.O.C., A.A.R.; Visualization A.V.G., Y.V.S.; supervision, Y.O.C. and A.A.R.; project administration., Y.O.C. and A.A.R.; funding acquisition, S.A.F., Y.O.C. and A.A.R. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by grant 20-14-00148 from Russian Science Foundation. S.A.F. was supported by Postdoctoral Fellowship from St. Petersburg State University.
Acknowledgments
The authors acknowledge Resource Centers “Molecular and Cell Technologies,” “Biobank,” and “Chromas” (St. Petersburg State University) for technical support.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| aa | Amino acid |
| ACTH | Adrenocorticotropic hormone |
| AD | Alzheimer’s disease |
| AGO2 | Argonaute 2 complex |
| AM | Acrosomal matrix |
| AMP | Adenine mononucleotidephosphate |
| AP | Amyloidogenic protein |
| ARE | AU-rich elements |
| Aβ | Amyloid beta |
| C-DAG | Curli-dependent amyloid generator |
| CARD | Caspase recruitment domains |
| cKO | Conditional knockout |
| CPEB | Cytoplasmic polyadenylation element binding protein |
| CD | Circular dichroism |
| CR | Congo red |
| CRH | Corticotropin releasing hormone |
| CTD | C-terminal domain |
| DD | Death domain |
| E-LTP | Early phase of long-term potentiation |
| EM | Electron microscopy |
| ER | Endoplasmic reticulum, |
| FXR | Fragile X-related protein |
| GAG | Glycosaminoglycans |
| GH | Growth hormone |
| GRF | Growth-hormone releasing factor |
| hIAPP | Human islet amyloid polypeptide |
| IgG | Immunoglobulin G |
| Igl | Immunoglobulin light chains |
| KD | Kinase domain |
| KLD | Kringle-like domain |
| LCP | Luminescent conjugated polyelectrolyte probes |
| L-LTP | Late phase of long-term potentiation |
| LPS | Lipopolysaccharide |
| MAVS | Mitochondrial antiviral signaling protein |
| MBP-1 | Eosinophil major basic protein 1 |
| MDA5 | Melanoma differentiation-associated 5 |
| MLKL | Mixed lineage kinase domain-like protein |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromideformazan |
| NAP | Non-amyloidogenic protein |
| NF-κB | Nuclear factor kappa B |
| NMR | Nuclear magnetic resonance |
| NTR | N-terminal repeats |
| PAD | Protein aggregation disease |
| PAP | Prostate acidic phosphatase |
| PD | Parkinson’s disease |
| PE | Preeclampsia |
| PKD | Polycystic kidney disease-like domain |
| PrD | Prionogenic domain |
| PrDL | Prion-like domain |
| PSIA | Proteomic Screening for Identification of Amyloid proteins |
| Q-rich | Glutamine-rich |
| QN-rich | Glutamine/Asparagine-rich |
| RHIM | RIP homotypic interaction motifs |
| RNP | Ribonucleoprotein |
| RPT | Repeat domain |
| SDS | Sodium dodecyl sulfate |
| SGs | Stress granules |
| StG | Storage granule |
| T2D | Type 2 diabetes |
| ThS | Thioflavin-S |
| ThT | Thioflavin-T |
| TIA-1 | T-cell intracellular antigen-1 |
| TM | Targeting domain |
| TNF | Tumor necrosis factor |
| tPA | Tissue plasminogen activator |
| TSE | Transmissible spongiform encephalopathy |
| UTR | Untranslated terminal repeats |
| UV | Ultraviolet |
| ZP | Zona Pellucida |
References
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2018, 25, 215–219. [Google Scholar] [CrossRef]
- Fändrich, M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell. Mol. Life Sci. 2007, 64, 2066–2078. [Google Scholar] [CrossRef]
- Lee, C.C.; Julian, M.C.; Tiller, K.E.; Meng, F.; DuConge, S.E.; Akter, R.; Raleigh, D.P.; Tessier, P.M. Design and Optimization of Anti-amyloid Domain Antibodies Specific for β-Amyloid and Islet Amyloid Polypeptide. J. Biol. Chem. 2016, 291, 2858–2873. [Google Scholar] [CrossRef]
- Evangelista, B.A.; Kim, Y.-S.; Kolpashchikov, D.M. FaptaSyme: A Strategy for Converting a Monomer/Oligomer-Nonselective Aptameric Sensor into an Oligomer-Selective One. ChemBioChem 2018, 19, 1123–1126. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Gerasimova, E.M.; Fedotov, S.A.; Kachkin, D.V.; Vashukova, E.S.; Glotov, A.S.; Chernoff, Y.O.; Rubel, A.A. Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. Int. J. Mol. Sci. 2019, 20, 6183. [Google Scholar] [CrossRef]
- Mezencev, R.; Chernoff, Y.O. Risk of Alzheimer’s Disease in Cancer Patients: Analysis of Mortality Data from the US SEER Population-Based Registries. Cancers (Basel) 2020, 12, 796. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M.; Glabe, C.G.; Buhimschi, C.S. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci. Transl. Med. 2014, 6, 245ra92. [Google Scholar] [CrossRef] [PubMed]
- Picken, M.M. Fibrinogen amyloidosis: the clot thickens! Blood 2010, 115, 2985–2986. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid--from bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Riek, R. Biology of amyloid: structure, function, and regulation. Structure 2010, 18, 1244–1260. [Google Scholar] [CrossRef]
- Otzen, D.; Riek, R. Functional Amyloids. Cold Spring Harb. Perspect. Biol. 2019, 11, a033860. [Google Scholar] [CrossRef] [PubMed]
- Si, K.; Choi, Y.-B.; White-Grindley, E.; Majumdar, A.; Kandel, E.R. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 2010, 140, 421–435. [Google Scholar] [CrossRef]
- Majumdar, A.; Cesario, W.C.; White-Grindley, E.; Jiang, H.; Ren, F.; Khan, M.R.; Li, L.; Choi, E.M.-L.; Kannan, K.; Guo, F.; et al. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell 2012, 148, 515–529. [Google Scholar] [CrossRef]
- Fioriti, L.; Myers, C.; Huang, Y.-Y.; Li, X.; Stephan, J.S.; Trifilieff, P.; Colnaghi, L.; Kosmidis, S.; Drisaldi, B.; Pavlopoulos, E.; et al. The Persistence of Hippocampal-Based Memory Requires Protein Synthesis Mediated by the Prion-like Protein CPEB3. Neuron 2015, 86, 1433–1448. [Google Scholar] [CrossRef]
- Slotta, U.; Hess, S.; Spiess, K.; Stromer, T.; Serpell, L.; Scheibel, T. Spider silk and amyloid fibrils: a structural comparison. Macromol. Biosci. 2007, 7, 183–188. [Google Scholar] [CrossRef]
- Chakrabortee, S.; Byers, J.S.; Jones, S.; Garcia, D.M.; Bhullar, B.; Chang, A.; She, R.; Lee, L.; Fremin, B.; Lindquist, S.; et al. Intrinsically Disordered Proteins Drive Emergence and Inheritance of Biological Traits. Cell 2016, 167, 369–381.e12. [Google Scholar] [CrossRef]
- Lee, I.; Aukerman, M.J.; Gore, S.L.; Lohman, K.N.; Michaels, S.D.; Weaver, L.M.; John, M.C.; Feldmann, K.A.; Amasino, R.M. Isolation of LUMINIDEPENDENS: A gene involved in the control of flowering time in Arabidopsis. Plant Cell 1994, 6, 75–83. [Google Scholar] [CrossRef]
- Chernoff, Y.O. Are there prions in plants? Proc. Natl. Acad. Sci. USA 2016, 113, 6097–6099. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.M.; Chapman, M.R. Curli biogenesis and function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef]
- Liebman, S.W.; Chernoff, Y.O. Prions in yeast. Genetics 2012, 191, 1041–1072. [Google Scholar] [CrossRef]
- Chernova, T.A.; Wilkinson, K.D.; Chernoff, Y.O. Prions, Chaperones, and Proteostasis in Yeast. Cold Spring Harb. Perspect. Biol. 2017, 9, a023663. [Google Scholar] [CrossRef]
- Wickner, R.B.; Edskes, H.K.; Kryndushkin, D.; McGlinchey, R.; Bateman, D.; Kelly, A. Prion diseases of yeast: Amyloid structure and biology. Semin. Cell Dev. Biol. 2011, 22, 469–475. [Google Scholar] [CrossRef]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef]
- Saupe, S.J.; Daskalov, A. The [Het-s] prion, an amyloid fold as a cell death activation trigger. PLoS Pathog. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Grizel, A.V.; Rubel, A.A.; Zelinsky, A.A.; Chandramowlishwaran, P.; Chernova, T.A. Application of yeast to studying amyloid and prion diseases. Adv. Genet. 2020, 105, 293–380. [Google Scholar] [CrossRef]
- Michelitsch, M.D.; Weissman, J.S. A census of glutamine/asparagine-rich regions: Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA 2000, 97, 11910–11915. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Harrison, P.M. The evolutionary scope and neurological disease linkage of yeast-prion-like proteins in humans. Biol. Direct. 2016, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, V.; Paladin, L.; Juan-Blanco, T.; Pallarès, I.; Aloy, P.; Tosatto, S.C.E.; Ventura, S. In silico Characterization of Human Prion-Like Proteins: Beyond Neurological Diseases. Front. Physiol. 2019, 10, 314. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.F.; Cunningham, D.; Spigelman, L.; Aten, B. Insulin biosynthesis: Evidence for a precursor. Science 1967, 157, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Tesar, J.T.; Koenig, H.; Hughes, C. Hormone storage granules in the beef anterior pituitary. I. Isolation, ultrastructure, and some biochemical properties. J. Cell Biol. 1969, 40, 225–235. [Google Scholar] [CrossRef]
- Dannies, P.S. Mechanisms for storage of prolactin and growth hormone in secretory granules. Mol. Genet. Metab. 2002, 76, 6–13. [Google Scholar] [CrossRef]
- Perdue, J.F.; Mcshan, W.H. Association of Adrenocorticotropic Hormone Activity with Small Secretory Granules from Rat Anterior Pituitary Glands. Endocrinology 1966, 78, 406–408. [Google Scholar] [CrossRef]
- Habener, J.F.; Amherdt, M.; Ravazzola, M.; Orci, L. Parathyroid hormone biosynthesis. Correlation of conversion of biosynthetic precursors with intracellular protein migration as determined by electron microscope autoradiography. J. Cell Biol. 1979, 80, 715–731. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.R.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef]
- Fernandez, C.J.; Haugwitz, M.; Eaton, B.; Moore, H.-P.H. Distinct Molecular Events during Secretory Granule Biogenesis Revealed by Sensitivities to Brefeldin A. Mol. Biol. Cell 1997, 8, 2171–2185. [Google Scholar] [CrossRef]
- Dannies, P.S. Protein Hormone Storage in Secretory Granules: Mechanisms for Concentration and Sorting. Endocr. Rev. 1999, 20, 3–21. [Google Scholar] [CrossRef]
- Zanini, A.; Giannattasio, G.; Nussdorfer, G.; Margolis, R.K.; Margolis, R.U.; Meldolesi, J. Molecular organization of prolactin granules. II. Characterization of glycosaminoglycans and glycoproteins of the bovine prolactin matrix. J. Cell Biol. 1980, 86, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Sankoorikal, B.-J.; Zhu, Y.L.; Hodsdon, M.E.; Lolis, E.; Dannies, P.S. Aggregation of Human Wild-Type and H27A-Prolactin in Cells and in Solution: Roles of Zn2+, Cu2+, and pH. Endocrinology 2002, 143, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.S.; Das, S.; Ghosh, S.; Anoop, A.; Jha, N.N.; Khan, T.; Singru, P.; Kumar, A.; Maji, S.K. Amyloid formation of growth hormone in presence of zinc: Relevance to its storage in secretory granules. Sci. Rep. 2016, 6, 23370. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Schubert, D.; Rivier, C.; Lee, S.; Rivier, J.E.; Riek, R. Amyloid as a depot for the formulation of long-acting drugs. PLoS Biol. 2008, 6, e17. [Google Scholar] [CrossRef]
- Lorenzo, A.; Yankner, B.A. Amyloid Fibril Toxicity in Alzheimer’s Disease and Diabetesa. Ann. N. Y. Acad. Sci. 1996, 777, 89–95. [Google Scholar] [CrossRef]
- Amico-Ruvio, S.A.; Paganelli, M.A.; Myers, J.M.; Popescu, G.K. Ifenprodil effects on GluN2B-containing glutamate receptors. Mol. Pharmacol. 2012, 82, 1074–1081. [Google Scholar] [CrossRef]
- Haider, S.; Cho, D.; Amelard, R.; Wong, A.; Clausi, D.A. Enhanced classification of malignant melanoma lesions via the integration of physiological features from dermatological photographs. In Proceedings of the 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Chicago, IL, USA, 26–30 September 2014; pp. 6455–6458. [Google Scholar]
- Fedorow, H.; Tribl, F.; Halliday, G.; Gerlach, M.; Riederer, P.; Double, K.L. Neuromelanin in human dopamine neurons: Comparison with peripheral melanins and relevance to Parkinson’s disease. Prog. Neurobiol. 2005, 75, 109–124. [Google Scholar] [CrossRef]
- Lillie, R.D. The basophilia of melanins. J. Histochem. Cytochem. 1955, 3, 453–454. [Google Scholar] [CrossRef]
- Lillie, R.D. Metal reduction reactions of the melanins: histochemical studies. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1957, 5, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.G. On the origin and significance of neuromelanin. Arch. Pathol. Lab. Med. 1979, 103, 359–362. [Google Scholar] [PubMed]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef]
- Herrero, M.T.; Hirsch, E.C.; Kastner, A.; Luquin, M.R.; Javoy-Agid, F.; Gonzalo, L.M.; Obeso, J.A.; Agid, Y. Neuromelanin Accumulation with Age in Catecholaminergic Neurons from Macaca fascicularis Brainstem. Dev. Neurosci. 1993, 15, 37–48. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Knörle, R. Neuromelanin in Parkinson’s Disease: from Fenton Reaction to Calcium Signaling. Neurotox. Res. 2018, 33, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Fujikawa, K.; Zucca, F.A.; Zecca, L.; Ito, S. The structure of neuromelanin as studied by chemical degradative methods. J. Neurochem. 2003, 86, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Vanna, R.; Bellei, C.; Zucca, F.A.; Wakamatsu, K.; Monzani, E.; Ito, S.; Casella, L.; Zecca, L. Neuromelanins of human brain have soluble and insoluble components with dolichols attached to the melanic structure. PLoS ONE 2012, 7, e48490. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef]
- Pawelek, J.M.; Lerner, A.B. 5,6-Dihydroxyindole is a melanin precursor showing potent cytotoxicity. Nature 1978, 276, 627–628. [Google Scholar] [CrossRef]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Rochin, L.; van Niel, G. PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation. Int. J. Mol. Sci. 2016, 17, 1438. [Google Scholar] [CrossRef] [PubMed]
- Hellström, A.R.; Watt, B.; Fard, S.S.; Tenza, D.; Mannström, P.; Narfström, K.; Ekesten, B.; Ito, S.; Wakamatsu, K.; Larsson, J.; et al. Inactivation of Pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet. 2011, 7, e1002285. [Google Scholar] [CrossRef]
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson’s disease. NPJ Park. Dis. 2018, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Bellei, C.; Costi, P.; Albertini, A.; Monzani, E.; Casella, L.; Gallorini, M.; Bergamaschi, L.; Moscatelli, A.; Turro, N.J.; et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc. Natl. Acad. Sci. USA 2008, 105, 17567–17572. [Google Scholar] [CrossRef]
- Stanka, P.; Rathjen, P.; Sahlmann, B. Evidence of membrane transformation during melanogenesis. Electron microscopic study on the retinal pigment epithelium of chick embryos. Cell Tissue Res. 1981, 214, 343–353. [Google Scholar] [CrossRef]
- Raposo, G.; Tenza, D.; Murphy, D.M.; Berson, J.F.; Marks, M.S. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell Biol. 2001, 152, 809–824. [Google Scholar] [CrossRef]
- Ancans, J.; Tobin, D.J.; Hoogduijn, M.J.; Smit, N.P.; Wakamatsu, K.; Thody, A.J. Melanosomal pH controls rate of melanogenesis, eumelanin/phaeomelanin ratio and melanosome maturation in melanocytes and melanoma cells. Exp. Cell Res. 2001, 268, 26–35. [Google Scholar] [CrossRef]
- Bycroft, M.; Bateman, A.; Clarke, J.; Hamill, S.J.; Sandford, R.; Thomas, R.L.; Chothia, C. The structure of a PKD domain from polycystin-1: implications for polycystic kidney disease. EMBO J. 1999, 18, 297–305. [Google Scholar] [CrossRef]
- Hoashi, T.; Tamaki, K.; Hearing, V.J. The secreted form of a melanocyte membrane-bound glycoprotein (Pmel17/gp100) is released by ectodomain shedding. FASEB J. 2010, 24, 916–930. [Google Scholar] [CrossRef]
- Berson, J.F.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 2001, 12, 3451–3464. [Google Scholar] [CrossRef] [PubMed]
- Maresh, G.A.; Marken, J.S.; Neubauer, M.; Aruffo, A.; Hellström, I.; Hellström, K.E.; Marquardt, H. Cloning and Expression of the Gene for the Melanoma-Associated ME20 Antigen. DNA Cell Biol. 1994, 13, 87–95. [Google Scholar] [CrossRef]
- Maresh, G.A.; Wang, W.C.; Beam, K.S.; Malacko, A.R.; Hellstrom, I.; Hellstrom, K.E.; Marquardt, H. Differential Processing and Secretion of the Melanoma-Associated Me20 Antigen. Arch. Biochem. Biophys. 1994, 311, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Nufer, O.; Guldbrandsen, S.; Degen, M.; Kappeler, F.; Paccaud, J.-P.; Tani, K.; Hauri, H.-P. Role of cytoplasmic C-terminal amino acids of membrane proteins in ER export. J. Cell Sci. 2002, 115, 619–628. [Google Scholar] [PubMed]
- Theos, A.C.; Berson, J.F.; Theos, S.C.; Herman, K.E.; Harper, D.C.; Tenza, D.; Sviderskaya, E.V.; Lamoreux, M.L.; Bennett, D.C.; Raposo, G.; et al. Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol. Biol. Cell 2006, 17, 3598–3612. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.C.; Theos, A.C.; Herman, K.E.; Tenza, D.; Raposo, G.; Marks, M.S. Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of Pmel17 that have been proteolytically processed in endosomes. J. Biol. Chem. 2008, 283, 2307–2322. [Google Scholar] [CrossRef] [PubMed]
- Valencia, J.C.; Rouzaud, F.; Julien, S.; Chen, K.G.; Passeron, T.; Yamaguchi, Y.; Abu-Asab, M.; Tsokos, M.; Costin, G.E.; Yamaguchi, H.; et al. Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils. J. Biol. Chem. 2007, 282, 11266–11280. [Google Scholar] [CrossRef]
- Theos, A.C.; Truschel, S.T.; Tenza, D.; Hurbain, I.; Harper, D.C.; Berson, J.F.; Thomas, P.C.; Raposo, G.; Marks, M.S. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 2006, 10, 343–354. [Google Scholar] [CrossRef]
- Berson, J.F.; Theos, A.C.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 2003, 161, 521–533. [Google Scholar] [CrossRef]
- Leonhardt, R.M.; Vigneron, N.; Rahner, C.; Cresswell, P. Proprotein convertases process Pmel17 during secretion. J. Biol. Chem. 2011, 286, 9321–9337. [Google Scholar] [CrossRef]
- Shimshek, D.R.; Jacobson, L.H.; Kolly, C.; Zamurovic, N.; Balavenkatraman, K.K.; Morawiec, L.; Kreutzer, R.; Schelle, J.; Jucker, M.; Bertschi, B.; et al. Pharmacological BACE1 and BACE2 inhibition induces hair depigmentation by inhibiting PMEL17 processing in mice. Sci. Rep. 2016, 6, 21917. [Google Scholar] [CrossRef] [PubMed]
- Rochin, L.; Hurbain, I.; Serneels, L.; Fort, C.; Watt, B.; Leblanc, P.; Marks, M.S.; De Strooper, B.; Raposo, G.; van Niel, G. BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10658–10663. [Google Scholar] [CrossRef]
- Kummer, M.P.; Maruyama, H.; Huelsmann, C.; Baches, S.; Weggen, S.; Koo, E.H. Formation of Pmel17 amyloid is regulated by juxtamembrane metalloproteinase cleavage, and the resulting C-terminal fragment is a substrate for gamma-secretase. J. Biol. Chem. 2009, 284, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, T.; Muller, J.; Vieira, W.D.; Rouzaud, F.; Kikuchi, K.; Tamaki, K.; Hearing, V.J. The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers. J. Biol. Chem. 2006, 281, 21198–21208. [Google Scholar] [CrossRef]
- Watt, B.; van Niel, G.; Fowler, D.M.; Hurbain, I.; Luk, K.C.; Stayrook, S.E.; Lemmon, M.A.; Raposo, G.; Shorter, J.; Kelly, J.W.; et al. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem. 2009, 284, 35543–35555. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Shewmaker, F.; McPhie, P.; Monterroso, B.; Thurber, K.; Wickner, R.B. The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis. Proc. Natl. Acad. Sci. USA 2009, 106, 13731–13736. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Lee, J.C. Why Study Functional Amyloids? Lessons from the Repeat Domain of Pmel17. J. Mol. Biol. 2018, 430, 3696–3706. [Google Scholar] [CrossRef]
- Dean, D.N.; Lee, J.C. pH-Dependent fibril maturation of a Pmel17 repeat domain isoform revealed by tryptophan fluorescence. Biochim. Biophys. Acta Proteins Proteomics 2019, 1867, 961–969. [Google Scholar] [CrossRef]
- Hee, J.S.; Mitchell, S.M.; Liu, X.; Leonhardt, R.M. Melanosomal formation of PMEL core amyloid is driven by aromatic residues. Sci. Rep. 2017, 7, 44064. [Google Scholar] [CrossRef]
- Graham, M.; Tzika, A.C.; Mitchell, S.M.; Liu, X.; Leonhardt, R.M. Repeat domain-associated O-glycans govern PMEL fibrillar sheet architecture. Sci. Rep. 2019, 9, 6101. [Google Scholar] [CrossRef]
- Si, K.; Giustetto, M.; Etkin, A.; Hsu, R.; Janisiewicz, A.M.; Miniaci, M.C.; Kim, J.-H.; Zhu, H.; Kandel, E.R. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell 2003, 115, 893–904. [Google Scholar] [CrossRef]
- Charlesworth, A.; Meijer, H.A.; de Moor, C.H. Specificity factors in cytoplasmic polyadenylation. WIREs RNA 2013, 4, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D. CPEB: a life in translation. Trends Biochem. Sci. 2007, 32, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Fiumara, F.; Rajasethupathy, P.; Antonov, I.; Kosmidis, S.; Sossin, W.S.; Kandel, E.R. MicroRNA-22 Gates Long-Term Heterosynaptic Plasticity in Aplysia through Presynaptic Regulation of CPEB and Downstream Targets. Cell Rep. 2015, 11, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Krüttner, S.; Traunmüller, L.; Dag, U.; Jandrasits, K.; Stepien, B.; Iyer, N.; Fradkin, L.G.; Noordermeer, J.N.; Mensh, B.D.; Keleman, K. Synaptic Orb2A Bridges Memory Acquisition and Late Memory Consolidation in Drosophila. Cell Rep. 2015, 11, 1953–1965. [Google Scholar] [CrossRef] [PubMed]
- Si, K.; Lindquist, S.; Kandel, E.R. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003, 115, 879–891. [Google Scholar] [CrossRef]
- Theis, M.; Si, K.; Kandel, E.R. Two previously undescribed members of the mouse CPEB family of genes and their inducible expression in the principal cell layers of the hippocampus. Proc. Natl. Acad. Sci. USA 2003, 100, 9602–9607. [Google Scholar] [CrossRef]
- Stephan, J.S.; Fioriti, L.; Lamba, N.; Colnaghi, L.; Karl, K.; Derkatch, I.L.; Kandel, E.R. The CPEB3 Protein Is a Functional Prion that Interacts with the Actin Cytoskeleton. Cell Rep. 2015, 11, 1772–1785. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Kan, M.-C.; Lin, C.-L.; Richter, J.D. CPEB3 and CPEB4 in neurons: analysis of RNA-binding specificity and translational control of AMPA receptor GluR2 mRNA. EMBO J. 2006, 25, 4865–4876. [Google Scholar] [CrossRef]
- Pavlopoulos, E.; Trifilieff, P.; Chevaleyre, V.; Fioriti, L.; Zairis, S.; Pagano, A.; Malleret, G.; Kandel, E.R. Neuralized1 activates CPEB3: a function for nonproteolytic ubiquitin in synaptic plasticity and memory storage. Cell 2011, 147, 1369–1383. [Google Scholar] [CrossRef]
- Si, K.; Kandel, E.R. The Role of Functional Prion-Like Proteins in the Persistence of Memory. Cold Spring Harb. Perspect. Biol. 2016, 8, a021774. [Google Scholar] [CrossRef] [PubMed]
- Drisaldi, B.; Colnaghi, L.; Fioriti, L.; Rao, N.; Myers, C.; Snyder, A.M.; Metzger, D.J.; Tarasoff, J.; Konstantinov, E.; Fraser, P.E.; et al. SUMOylation Is an Inhibitory Constraint that Regulates the Prion-like Aggregation and Activity of CPEB3. Cell Rep. 2015, 11, 1694–1702. [Google Scholar] [CrossRef]
- Ford, L.; Ling, E.; Kandel, E.R.; Fioriti, L. CPEB3 inhibits translation of mRNA targets by localizing them to P bodies. Proc. Natl. Acad. Sci. USA 2019, 116, 18078–18087. [Google Scholar] [CrossRef]
- Cougot, N.; Bhattacharyya, S.N.; Tapia-Arancibia, L.; Bordonné, R.; Filipowicz, W.; Bertrand, E.; Rage, F. Dendrites of mammalian neurons contain specialized P-body-like structures that respond to neuronal activation. J. Neurosci. 2008, 28, 13793–13804. [Google Scholar] [CrossRef] [PubMed]
- Sudhakaran, I.P.; Ramaswami, M. Long-term memory consolidation: The role of RNA-binding proteins with prion-like domains. RNA Biol. 2017, 14, 568–586. [Google Scholar] [CrossRef] [PubMed]
- Miniaci, M.C.; Kim, J.-H.; Puthanveettil, S.V.; Si, K.; Zhu, H.; Kandel, E.R.; Bailey, C.H. Sustained CPEB-dependent local protein synthesis is required to stabilize synaptic growth for persistence of long-term facilitation in Aplysia. Neuron 2008, 59, 1024–1036. [Google Scholar] [CrossRef]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM structure of a neuronal functional amyloid implicated in memory persistence in Drosophila. Science 2020, 367, 1230–1234. [Google Scholar] [CrossRef]
- Fan, Y.; Yue, J.; Xiao, M.; Han-Zhang, H.; Wang, Y.V.; Ma, C.; Deng, Z.; Li, Y.; Yu, Y.; Wang, X.; et al. FXR1 regulates transcription and is required for growth of human cancer cells with TP53/FXR2 homozygous deletion. Elife 2017, 6, e26129. [Google Scholar] [CrossRef]
- Brown, V.; Jin, P.; Ceman, S.; Darnell, J.C.; O’Donnell, W.T.; Tenenbaum, S.A.; Jin, X.; Feng, Y.; Wilkinson, K.D.; Keene, J.D.; et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001, 107, 477–487. [Google Scholar] [CrossRef]
- Le Tonqueze, O.; Kollu, S.; Lee, S.; Al-Salah, M.; Truesdell, S.S.; Vasudevan, S. Regulation of monocyte induced cell migration by the RNA binding protein, FXR1. Cell Cycle 2016, 15, 1874–1882. [Google Scholar] [CrossRef]
- Siomi, M.C.; Zhang, Y.; Siomi, H.; Dreyfuss, G. Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol. Cell. Biol. 1996, 16, 3825–3832. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Zarnescu, D.C.; Ceman, S.; Nakamoto, M.; Mowrey, J.; Jongens, T.A.; Nelson, D.L.; Moses, K.; Warren, S.T. Biochemical and genetic interaction between the fragile X mental retardation protein 3nd the microRNA pathway. Nat. Neurosci. 2004, 7, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Bogert, B.A.; Li, W.; Su, K.; Lee, A.; Gao, F.-B. The fragile X-related gene affects the crawling behavior of Drosophila larvae by regulating the mRNA level of the DEG/ENaC protein pickpocket1. Curr. Biol. 2004, 14, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Steitz, J.A. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 2007, 128, 1105–1118. [Google Scholar] [CrossRef]
- Caudy, A.A.; Myers, M.; Hannon, G.J.; Hammond, S.M. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 2002, 16, 2491–2496. [Google Scholar] [CrossRef]
- Mazroui, R.; Huot, M.-E.; Tremblay, S.; Filion, C.; Labelle, Y.; Khandjian, E.W. Trapping of messenger RNA by Fragile X Mental Retardation protein into cytoplasmic granules induces translation repression. Hum. Mol. Genet. 2002, 11, 3007–3017. [Google Scholar] [CrossRef]
- Hofmann, I.; Casella, M.; Schnölzer, M.; Schlechter, T.; Spring, H.; Franke, W.W. Identification of the junctional plaque protein plakophilin 3 in cytoplasmic particles containing RNA-binding proteins and the recruitment of plakophilins 1 and 3 to stress granules. Mol. Biol. Cell 2006, 17, 1388–1398. [Google Scholar] [CrossRef]
- Sjekloća, L.; Pauwels, K.; Pastore, A. On the aggregation properties of FMRP—a link with the FXTAS syndrome? FEBS J. 2011, 278, 1912–1921. [Google Scholar] [CrossRef]
- Sopova, J.V.; Koshel, E.I.; Belashova, T.A.; Zadorsky, S.P.; Sergeeva, A.V.; Siniukova, V.A.; Shenfeld, A.A.; Velizhanina, M.E.; Volkov, K.V.; Nizhnikov, A.A.; et al. RNA-binding protein FXR1 is presented in rat brain in amyloid form. Sci. Rep. 2019, 9, 18983. [Google Scholar] [CrossRef]
- Spargo, S.C.; Hope, R.M. Evolution and nomenclature of the zona pellucida gene family. Biol. Reprod. 2003, 68, 358–362. [Google Scholar] [CrossRef]
- Lefièvre, L.; Conner, S.J.; Salpekar, A.; Olufowobi, O.; Ashton, P.; Pavlovic, B.; Lenton, W.; Afnan, M.; Brewis, I.A.; Monk, M.; et al. Four zona pellucida glycoproteins are expressed in the human. Hum. Reprod. 2004, 19, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Sander, C. A large domain common to sperm receptors (Zp2 and Zp3) and TGF-beta type III receptor. FEBS Lett. 1992, 300, 237–240. [Google Scholar] [CrossRef]
- Greve, J.M.; Wassarman, P.M. Mouse egg extracellular coat is a matrix of interconnected filaments possessing a structural repeat. J. Mol. Biol. 1985, 181, 253–264. [Google Scholar] [CrossRef]
- Rankin, T.L.; O’Brien, M.; Lee, E.; Wigglesworth, K.; Eppig, J.; Dean, J. Defective zonae pellucidae in Zp2-null mice disrupt folliculogenesis, fertility and development. Development 2001, 128, 1119–1126. [Google Scholar] [PubMed]
- Monné, M.; Han, L.; Jovine, L. Tracking down the ZP domain: From the mammalian zona pellucida to the molluscan vitelline envelope. Semin. Reprod. Med. 2006, 24, 204–216. [Google Scholar] [CrossRef]
- Egge, N.; Muthusubramanian, A.; Cornwall, G.A. Amyloid properties of the mouse egg zona pellucida. PLoS ONE 2015, 10, e0129907. [Google Scholar] [CrossRef]
- Han, L.; Monné, M.; Okumura, H.; Schwend, T.; Cherry, A.L.; Flot, D.; Matsuda, T.; Jovine, L. Insights into egg coat assembly and egg-sperm interaction from the X-ray structure of full-length ZP3. Cell 2010, 143, 404–415. [Google Scholar] [CrossRef]
- Tsubamoto, H.; Hasegawa, A.; Nakata, Y.; Naito, S.; Yamasaki, N.; Koyama, K. Expression of recombinant human zona pellucida protein 2 and its binding capacity to spermatozoa. Biol. Reprod. 1999, 61, 1649–1654. [Google Scholar] [CrossRef]
- Gahlay, G.; Gauthier, L.; Baibakov, B.; Epifano, O.; Dean, J. Gamete recognition in mice depends on the cleavage status of an egg’s zona pellucida protein. Science 2010, 329, 216–219. [Google Scholar] [CrossRef]
- Avella, M.A.; Xiong, B.; Dean, J. The molecular basis of gamete recognition in mice and humans. Mol. Hum. Reprod. 2013, 19, 279–289. [Google Scholar] [CrossRef]
- Hewetson, A.; Do, H.Q.; Myers, C.; Muthusubramanian, A.; Sutton, R.B.; Wylie, B.J.; Cornwall, G.A. Functional Amyloids in Reproduction. Biomolecules 2017, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Javed, Q.; Collins, J.; Hay, M. Biogenesis of structural intercellular junctions during cleavage in the mouse embryo. J. Cell Sci. Suppl. 1993, 17, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Kurotaki, Y.; Hatta, K.; Nakao, K.; Nabeshima, Y.-I.; Fujimori, T. Blastocyst axis is specified independently of early cell lineage but aligns with the ZP shape. Science 2007, 316, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Welz, P.-S.; Wullaert, A.; Vlantis, K.; Kondylis, V.; Fernández-Majada, V.; Ermolaeva, M.; Kirsch, P.; Sterner-Kock, A.; van Loo, G.; Pasparakis, M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011, 477, 330–334. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Teng, X.; Degterev, A.; Jagtap, P.; Xing, X.; Choi, S.; Denu, R.; Yuan, J.; Cuny, G.D. Structure–activity relationship study of novel necroptosis inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5039–5044. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Stanger, B.Z.; Leder, P.; Lee, T.H.; Kim, E.; Seed, B. RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 1995, 81, 513–523. [Google Scholar] [CrossRef]
- Sun, X.; Lee, J.; Navas, T.; Baldwin, D.T.; Stewart, T.A.; Dixit, V.M. RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem. 1999, 274, 16871–16875. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.W.; Huang, B.C.; Shen, M.; Quast, J.; Chan, E.; Xu, X.; Nolan, G.P.; Payan, D.G.; Luo, Y. Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkappaB. Curr. Biol. 1999, 9, 539–542. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Ali, M.; Roback, L.; Mocarski, E.S. Herpes simplex virus 1 ICP6 impedes TNF receptor 1-induced necrosome assembly during compartmentalization to detergent-resistant membrane vesicles. J. Biol. Chem. 2019, 294, 991–1004. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Peng, S.; Yu, X.; Li, W.; Shi, F.; Luo, X.; Tang, M.; Tan, Z.; Bode, A.M.; et al. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Mompeán, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253.e10. [Google Scholar] [CrossRef]
- Daskalov, A. On the evolutionary trajectories of signal-transducing amyloids in fungi and beyond. Prion 2016, 10, 362–368. [Google Scholar] [CrossRef]
- Riek, R.; Saupe, S.J. The HET-S/s Prion Motif in the Control of Programmed Cell Death. Cold Spring Harb. Perspect. Biol. 2016, 8, a023515. [Google Scholar] [CrossRef] [PubMed]
- Daskalov, A.; Habenstein, B.; Sabaté, R.; Berbon, M.; Martinez, D.; Chaignepain, S.; Coulary-Salin, B.; Hofmann, K.; Loquet, A.; Saupe, S.J. Identification of a novel cell death-inducing domain reveals that fungal amyloid-controlled programmed cell death is related to necroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Kita, H. Eosinophils: multifaceted biological properties and roles in health and disease. Immunol. Rev. 2011, 242, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.R.; Ackerman, S.J. Eosinophil granule proteins: form and function. J. Biol. Chem. 2014, 289, 17406–17415. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Yousefi, S.; Stoeckle, C.; Soriaga, A.B.; Sawaya, M.R.; Kozlowski, E.; Schmid, I.; Radonjic-Hoesli, S.; Boutet, S.; Williams, G.J.; et al. Toxicity of eosinophil MBP is repressed by intracellular crystallization and promoted by extracellular aggregation. Mol. Cell 2015, 57, 1011–1021. [Google Scholar] [CrossRef]
- Bankers-Fulbright, J.L.; Kephart, G.M.; Bartemes, K.R.; Kita, H.; O’Grady, S.M. Platelet-activating factor stimulates cytoplasmic alkalinization and granule acidification in human eosinophils. J. Cell Sci. 2004, 117, 5749–5757. [Google Scholar] [CrossRef][Green Version]
- Thomas, L.L.; Kubo, H.; Loegering, D.J.; Spillard, K.; Weaver, A.J.; McCormick, D.J.; Weiler, C.; Gleich, G.J. Peptide-based analysis of amino acid sequences important to the biological activity of eosinophil granule major basic protein. Immunol. Lett. 2001, 78, 175–181. [Google Scholar] [CrossRef]
- Werfel, T.; Allam, J.-P.; Biedermann, T.; Eyerich, K.; Gilles, S.; Guttman-Yassky, E.; Hoetzenecker, W.; Knol, E.; Simon, H.-U.; Wollenberg, A.; et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2016, 138, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Frigas, E.; Gleich, G.J. The eosinophil and the pathophysiology of asthma. J. Allergy Clin. Immunol. 1986, 77, 527–537. [Google Scholar] [CrossRef]
- Swaminathan, G.J.; Weaver, A.J.; Loegering, D.A.; Checkel, J.L.; Leonidas, D.D.; Gleich, G.J.; Acharya, K.R. Crystal structure of the eosinophil major basic protein at 1.8 A. An atypical lectin with a paradigm shift in specificity. J. Biol. Chem. 2001, 276, 26197–26203. [Google Scholar] [CrossRef] [PubMed]
- Leiferman, K.M.; Ackerman, S.J.; Sampson, H.A.; Haugen, H.S.; Venencie, P.Y.; Gleich, G.J. Dermal deposition of eosinophil-granule major basic protein in atopic dermatitis. Comparison with onchocerciasis. N. Engl. J. Med. 1985, 313, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.L.; Leiferman, K.M.; Gleich, G.J. Eosinophil granule protein localization in eosinophilic endomyocardial disease. N. Engl. J. Med. 2011, 365, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, A.M.; Beaven, M.A. Regulation of mast cell responses in health and disease. Crit. Rev. Immunol. 2011, 31, 475–529. [Google Scholar] [CrossRef]
- Cornwall, G.A. New insights into epididymal biology and function. Hum. Reprod. Update 2009, 15, 213–227. [Google Scholar] [CrossRef]
- Whelly, S.; Muthusubramanian, A.; Powell, J.; Johnson, S.; Hastert, M.C.; Cornwall, G.A. Cystatin-related epididymal spermatogenic subgroup members are part of an amyloid matrix and associated with extracellular vesicles in the mouse epididymal lumen. Mol. Hum. Reprod. 2016, 22, 729–744. [Google Scholar] [CrossRef]
- Whelly, S.; Serobian, G.; Borchardt, C.; Powell, J.; Johnson, S.; Hakansson, K.; Lindstrom, V.; Abrahamson, M.; Grubb, A.; Cornwall, G.A. Fertility defects in mice expressing the L68Q variant of human cystatin C: a role for amyloid in male infertility. J. Biol. Chem. 2014, 289, 7718–7729. [Google Scholar] [CrossRef]
- Parent, A.D.; Cornwall, G.A.; Liu, L.Y.; Smith, C.E.; Hermo, L. Alterations in the testis and epididymis associated with loss of function of the cystatin-related epididymal spermatogenic (CRES) protein. J. Androl. 2011, 32, 444–463. [Google Scholar] [CrossRef]
- Chau, K.M.; Cornwall, G.A. Reduced fertility in vitro in mice lacking the cystatin CRES (cystatin-related epididymal spermatogenic): Rescue by exposure of spermatozoa to dibutyryl cAMP and isobutylmethylxanthine. Biol. Reprod. 2011, 84, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Guyonnet, B.; Egge, N.; Cornwall, G.A. Functional Amyloids in the Mouse Sperm Acrosome. Mol. Cell. Biol. 2014, 34, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Wassler, M.; Syntin, P.; Sutton-Walsh, H.G.; Hsia, N.; Hardy, D.M.; Cornwall, G.A. Identification and characterization of cystatin-related epididymal spermatogenic protein in human spermatozoa: localization in the equatorial segment. Biol. Reprod. 2002, 67, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; French, K.C.; Popova, L.A.; Lednev, I.K.; Lopez, M.M.; Makhatadze, G.I. Mechanism of fibril formation by a 39-residue peptide (PAPf39) from human prostatic acidic phosphatase. Biochemistry 2009, 48, 11582–11591. [Google Scholar] [CrossRef] [PubMed]
- French, K.C.; Makhatadze, G.I. Core sequence of PAPf39 amyloid fibrils and mechanism of pH-dependent fibril formation: the role of monomer conformation. Biochemistry 2012, 51, 10127–10136. [Google Scholar] [CrossRef] [PubMed]
- Andrich, K.; Bieschke, J. The Effect of (-)-Epigallo-catechin-(3)-gallate on Amyloidogenic Proteins Suggests a Common Mechanism. Adv. Exp. Med. Biol. 2015, 863, 139–161. [Google Scholar] [CrossRef]
- Castellano, L.M.; Bart, S.M.; Holmes, V.M.; Weissman, D.; Shorter, J. Repurposing Hsp104 to Antagonize Seminal Amyloid and Counter HIV Infection. Chem. Biol. 2015, 22, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef]
- Easterhoff, D.; Ontiveros, F.; Brooks, L.R.; Kim, Y.; Ross, B.; Silva, J.N.; Olsen, J.S.; Feng, C.; Hardy, D.J.; Dunman, P.M.; et al. Semen-derived enhancer of viral infection (SEVI) binds bacteria, enhances bacterial phagocytosis by macrophages, and can protect against vaginal infection by a sexually transmitted bacterial pathogen. Antimicrob. Agents Chemother. 2013, 57, 2443–2450. [Google Scholar] [CrossRef]
- Münch, J.; Rücker, E.; Ständker, L.; Adermann, K.; Goffinet, C.; Schindler, M.; Wildum, S.; Chinnadurai, R.; Rajan, D.; Specht, A.; et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell 2007, 131, 1059–1071. [Google Scholar] [CrossRef]
- Easterhoff, D.; DiMaio, J.T.M.; Doran, T.M.; Dewhurst, S.; Nilsson, B.L. Enhancement of HIV-1 infectivity by simple, self-assembling modular peptides. Biophys. J. 2011, 100, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2018, 50, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef]
- Berke, I.C.; Yu, X.; Modis, Y.; Egelman, E.H. MDA5 assembles into a polar helical filament on dsRNA. Proc. Natl. Acad. Sci. USA 2012, 109, 18437–18441. [Google Scholar] [CrossRef]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef]
- Ye, J.; Maniatis, T. A prion-like trigger of antiviral signaling. Cell 2011, 146, 348–350. [Google Scholar] [CrossRef][Green Version]
- Moresco, E.M.Y.; La Vine, D.; Beutler, B. Prion-like behavior of MAVS in RIG-I signaling. Cell Res. 2011, 21, 1643–1645. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Gauthier, A.E.; Mills, E.W.; Ingolia, N.T.; Kagan, J.C. A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell 2014, 156, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.W.; Rixon, M.W.; Que, B.G.; Davie, E.W. Cloning of fibrinogen genes and their cDNA. Ann. N. Y. Acad. Sci. 1983, 408, 449–456. [Google Scholar] [CrossRef]
- Chung, D.W.; Harris, J.E.; Davie, E.W. Nucleotide sequences of the three genes coding for human fibrinogen. Adv. Exp. Med. Biol. 1990, 281, 39–48. [Google Scholar] [CrossRef]
- Kranenburg, O.; Bouma, B.; Kroon-Batenburg, L.M.J.; Reijerkerk, A.; Wu, Y.-P.; Voest, E.E.; Gebbink, M.F.B.G. Tissue-type plasminogen activator is a multiligand cross-beta structure receptor. Curr. Biol. 2002, 12, 1833–1839. [Google Scholar] [CrossRef]
- Averett, L.E.; Geer, C.B.; Fuierer, R.R.; Akhremitchev, B.B.; Gorkun, O.V.; Schoenfisch, M.H. Complexity of “A-a” knob-hole fibrin interaction revealed by atomic force spectroscopy. Langmuir 2008, 24, 4979–4988. [Google Scholar] [CrossRef]
- Yermolenko, I.S.; Lishko, V.K.; Ugarova, T.P.; Magonov, S.N. High-resolution visualization of fibrinogen molecules and fibrin fibers with atomic force microscopy. Biomacromolecules 2011, 12, 370–379. [Google Scholar] [CrossRef]
- Protopopova, A.D.; Barinov, N.A.; Zavyalova, E.G.; Kopylov, A.M.; Sergienko, V.I.; Klinov, D.V. Visualization of fibrinogen αC regions and their arrangement during fibrin network formation by high-resolution AFM. J. Thromb. Haemost. 2015, 13, 570–579. [Google Scholar] [CrossRef]
- Pretorius, E.; Page, M.J.; Mbotwe, S.; Kell, D.B. Lipopolysaccharide-binding protein (LBP) can reverse the amyloid state of fibrin seen or induced in Parkinson’s disease. PLoS ONE 2018, 13, e0192121. [Google Scholar] [CrossRef]
- Zhmurov, A.; Kononova, O.; Litvinov, R.I.; Dima, R.I.; Barsegov, V.; Weisel, J.W. Mechanical transition from α-helical coiled coils to β-sheets in fibrin(ogen). J. Am. Chem. Soc. 2012, 134, 20396–20402. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, R.I.; Faizullin, D.A.; Zuev, Y.F.; Weisel, J.W. The α-helix to β-sheet transition in stretched and compressed hydrated fibrin clots. Biophys. J. 2012, 103, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Bester, J.; Soma, P.; Kell, D.B.; Pretorius, E. Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget 2015, 6, 35284–35303. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Bester, J.; Vermeulen, N.; Alummoottil, S.; Soma, P.; Buys, A.V.; Kell, D.B. Poorly controlled type 2 diabetes is accompanied by significant morphological and ultrastructural changes in both erythrocytes and in thrombin-generated fibrin: implications for diagnostics. Cardiovasc. Diabetol. 2015, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Bester, J.; Vermeulen, N.; Lipinski, B.; Gericke, G.S.; Kell, D.B. Profound morphological changes in the erythrocytes and fibrin networks of patients with hemochromatosis or with hyperferritinemia, and their normalization by iron chelators and other agents. PLoS ONE 2014, 9, e85271. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Mbotwe, S.; Bester, J.; Robinson, C.J.; Kell, D.B. Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J. R. Soc. Interface 2016, 13, 20160539. [Google Scholar] [CrossRef]
- Page, M.J.; Thomson, G.J.A.; Nunes, J.M.; Engelbrecht, A.-M.; Nell, T.A.; de Villiers, W.J.S.; de Beer, M.C.; Engelbrecht, L.; Kell, D.B.; Pretorius, E. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci. Rep. 2019, 9, 3102. [Google Scholar] [CrossRef]
- Serpell, L.C.; Benson, M.; Liepnieks, J.J.; Fraser, P.E. Structural analyses of fibrinogen amyloid fibrils. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2007, 14, 199–203. [Google Scholar] [CrossRef]
- Stangou, A.J.; Banner, N.R.; Hendry, B.M.; Rela, M.; Portmann, B.; Wendon, J.; Monaghan, M.; Maccarthy, P.; Buxton-Thomas, M.; Mathias, C.J.; et al. Hereditary fibrinogen A alpha-chain amyloidosis: phenotypic characterization of a systemic disease and the role of liver transplantation. Blood 2010, 115, 2998–3007. [Google Scholar] [CrossRef]
- Haidinger, M.; Werzowa, J.; Kain, R.; Antlanger, M.; Hecking, M.; Pfaffenberger, S.; Mascherbauer, J.; Gremmel, T.; Gilbertson, J.A.; Rowczenio, D.; et al. Hereditary amyloidosis caused by R554L fibrinogen Aα-chain mutation in a Spanish family and review of the literature. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2013, 20, 72–79. [Google Scholar] [CrossRef]
- Kell, D.B.; Pretorius, E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Prog. Biophys. Mol. Biol. 2017, 123, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Zamolodchikov, D.; Berk-Rauch, H.E.; Oren, D.A.; Stor, D.S.; Singh, P.K.; Kawasaki, M.; Aso, K.; Strickland, S.; Ahn, H.J. Biochemical and structural analysis of the interaction between β-amyloid and fibrinogen. Blood 2016, 128, 1144–1151. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Zamolodchikov, D.; Strickland, S. Aβ delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood 2012, 119, 3342–3351. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: more than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Mateju, D.; Mediani, L.; Carra, S. Granulostasis: Protein Quality Control of RNP Granules. Front. Mol. Neurosci. 2017, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rayman, J.B.; Kandel, E.R.; Derkatch, I.L. Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol. Cell 2014, 55, 305–318. [Google Scholar] [CrossRef]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef]
- Riemschoss, K.; Arndt, V.; Bolognesi, B.; von Eisenhart-Rothe, P.; Liu, S.; Buravlova, O.; Duernberger, Y.; Paulsen, L.; Hornberger, A.; Hossinger, A.; et al. Fibril-induced glutamine-/asparagine-rich prions recruit stress granule proteins in mammalian cells. Life Sci. Alliance 2019, 2, e201800280. [Google Scholar] [CrossRef]
- Rayman, J.B.; Kandel, E.R. TIA-1 Is a Functional Prion-Like Protein. Cold Spring Harb. Perspect. Biol. 2017, 9, a030718. [Google Scholar] [CrossRef]
- Kedersha, N.; Anderson, P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Piecyk, M.; Wax, S.; Beck, A.R.; Kedersha, N.; Gupta, M.; Maritim, B.; Chen, S.; Gueydan, C.; Kruys, V.; Streuli, M.; et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J. 2000, 19, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Heck, M.V.; Azizov, M.; Stehning, T.; Walter, M.; Kedersha, N.; Auburger, G. Dysregulated expression of lipid storage and membrane dynamics factors in Tia1 knockout mouse nervous tissue. Neurogenetics 2014, 15, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Civas, A.; Lama, Z.; Lagaudrière-Gesbert, C.; Blondel, D. Rabies Virus Infection Induces the Formation of Stress Granules Closely Connected to the Viral Factories. PLoS Pathog. 2016, 12, e1005942. [Google Scholar] [CrossRef]
- Zhang, Q.; Sharma, N.R.; Zheng, Z.-M.; Chen, M. Viral Regulation of RNA Granules in Infected Cells. Virol. Sin. 2019, 34, 175–191. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Vanderweyde, T.E.; Youmans, K.L.; Apicco, D.J.; Wolozin, B. Pathological stress granules in Alzheimer’s disease. Brain Res. 2014, 1584, 52–58. [Google Scholar] [CrossRef]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; Lummertz da Rocha, E.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef]
- Ahmed, A.B.; Kajava, A.V. Breaking the amyloidogenicity code: methods to predict amyloids from amino acid sequence. FEBS Lett. 2013, 587, 1089–1095. [Google Scholar] [CrossRef]
- Wilson, C.J.; Bommarius, A.S.; Champion, J.A.; Chernoff, Y.O.; Lynn, D.G.; Paravastu, A.K.; Liang, C.; Hsieh, M.C.; Heemstra, J.M. Biomolecular Assemblies: Moving from Observation to Predictive Design. Chem. Rev. 2018, 118, 11519–11574. [Google Scholar] [CrossRef]
- Cremades, N.; Dobson, C.M. The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol. Dis. 2018, 109, 178–190. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Alexandrov, I.M.; Mitkevich, O.V.; Shkundina, I.S.; Ter-Avanesyan, M.D. Purification and analysis of prion and amyloid aggregates. Methods 2006, 39, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Wear, M.P.; Shewmaker, F. Amyloid cannot resist identification. Prion 2013, 7, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Alexandrov, A.I.; Ryzhova, T.A.; Mitkevich, O.V.; Dergalev, A.A.; Ter-Avanesyan, M.D.; Galkin, A.P. Proteomic screening for amyloid proteins. PLoS ONE 2014, 9, e116003. [Google Scholar] [CrossRef]
- Nizhnikov, A.A.; Antonets, K.S.; Bondarev, S.A.; Inge-Vechtomov, S.G.; Derkatch, I.L. Prions, amyloids, and RNA: Pieces of a puzzle. Prion 2016, 10, 182–206. [Google Scholar] [CrossRef] [PubMed]
- Antonets, K.S.; Volkov, K.V.; Maltseva, A.L.; Arshakian, L.M.; Galkin, A.P.; Nizhnikov, A.A. Proteomic Analysis of Escherichia coli Protein Fractions Resistant to Solubilization by Ionic Detergents. Biochemistry 2016, 81, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Kosolapova, A.O.; Belousov, M.V.; Sulatskaya, A.I.; Belousova, M.E.; Sulatsky, M.I.; Antonets, K.S.; Volkov, K.V.; Lykholay, A.N.; Shtark, O.Y.; Vasileva, E.N.; et al. Two Novel Amyloid Proteins, RopA and RopB, from the Root Nodule Bacterium Rhizobium leguminosarum. Biomolecules 2019, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Ryzhova, T.A.; Sopova, J.V.; Zadorsky, S.P.; Siniukova, V.A.; Sergeeva, A.V.; Galkina, S.A.; Nizhnikov, A.A.; Shenfeld, A.A.; Volkov, K.V.; Galkin, A.P. Screening for amyloid proteins in the yeast proteome. Curr. Genet. 2018, 64, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Belousov, M.V.; Bondarev, S.A.; Kosolapova, A.O.; Antonets, K.S.; Sulatskaya, A.I.; Sulatsky, M.I.; Zhouravleva, G.A.; Kuznetsova, I.M.; Turoverov, K.K.; Nizhnikov, A.A. M60-like metalloprotease domain of the Escherichia coli YghJ protein forms amyloid fibrils. PLoS ONE 2018, 13, e0191317. [Google Scholar] [CrossRef]
- Sergeeva, A.V.; Sopova, J.V.; Belashova, T.A.; Siniukova, V.A.; Chirinskaite, A.V.; Galkin, A.P.; Zadorsky, S.P. Amyloid properties of the yeast cell wall protein Toh1 and its interaction with prion proteins Rnq1 and Sup35. Prion 2019, 13, 21–32. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Pripuzova, N.; Shewmaker, F.P. Isolation and Analysis of Prion and Amyloid Aggregates from Yeast Cells. Cold Spring Harb. Protoc. 2017, 2017. [Google Scholar] [CrossRef]
- Sivanathan, V.; Hochschild, A. Generating extracellular amyloid aggregates using E. coli cells. Genes Dev. 2012, 26, 2659–2667. [Google Scholar] [CrossRef]
- Sivanathan, V.; Hochschild, A. A bacterial export system for generating extracellular amyloid aggregates. Nat. Protoc. 2013, 8, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Taglialegna, A.; Matilla-Cuenca, L.; Dorado-Morales, P.; Navarro, S.; Ventura, S.; Garnett, J.A.; Lasa, I.; Valle, J. The biofilm-associated surface protein Esp of Enterococcus faecalis forms amyloid-like fibers. NPJ Biofilms Microbiomes 2020, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.F. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr. Opin. Biotechnol. 1995, 6, 494–500. [Google Scholar] [CrossRef]
- Zhouravleva, G.; Frolova, L.; Le Goff, X.; Le Guellec, R.; Inge-Vechtomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995, 14, 4065–4072. [Google Scholar] [CrossRef] [PubMed]
- Stansfield, I.; Jones, K.M.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Poznyakovski, A.I.; Paushkin, S.V.; Nierras, C.R.; Cox, B.S.; Ter-Avanesyan, M.D.; Tuite, M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373. [Google Scholar] [CrossRef]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nüske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 2018, 359, eaao5654. [Google Scholar] [CrossRef]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef]
- Paul, K.R.; Molliex, A.; Cascarina, S.; Boncella, A.E.; Taylor, J.P.; Ross, E.D. Effects of Mutations on the Aggregation Propensity of the Human Prion-Like Protein hnRNPA2B1. Mol. Cell. Biol. 2017, 37, e00652-16. [Google Scholar] [CrossRef]
- Chernova, T.A.; Chernoff, Y.O.; Wilkinson, K.D. Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery. Molecules 2019, 24, 3388. [Google Scholar] [CrossRef]
- Chandramowlishwaran, P.; Sun, M.; Casey, K.L.; Romanyuk, A.V.; Grizel, A.V.; Sopova, J.V.; Rubel, A.A.; Nussbaum-Krammer, C.; Vorberg, I.M.; Chernoff, Y.O. Mammalian amyloidogenic proteins promote prion nucleation in yeast. J. Biol. Chem. 2018, 293, 3436–3450. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN(+)]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef]
- Osherovich, L.Z.; Weissman, J.S. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell 2001, 106, 183–194. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Derkach, I.L.; Inge-Vechtomov, S.G. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr. Genet. 1993, 24, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Chernoff, Y.O.; Kushnirov, V.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996, 144, 1375–1386. [Google Scholar]
- Berchowitz, L.E.; Kabachinski, G.; Walker, M.R.; Carlile, T.M.; Gilbert, W.V.; Schwartz, T.U.; Amon, A. Regulated Formation of an Amyloid-like Translational Repressor Governs Gametogenesis. Cell 2015, 163, 406–418. [Google Scholar] [CrossRef]
- White-Grindley, E.; Li, L.; Mohammad Khan, R.; Ren, F.; Saraf, A.; Florens, L.; Si, K. Contribution of Orb2A stability in regulated amyloid-like oligomerization of Drosophila Orb2. PLoS Biol. 2014, 12, e1001786. [Google Scholar] [CrossRef]
- Wu, L.; Wells, D.; Tay, J.; Mendis, D.; Abbott, M.A.; Barnitt, A.; Quinlan, E.; Heynen, A.; Fallon, J.R.; Richter, J.D. CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron 1998, 21, 1129–1139. [Google Scholar] [CrossRef]
- Lathe, R.; Darlix, J.-L. Prion protein PrP nucleic acid binding and mobilization implicates retroelements as the replicative component of transmissible spongiform encephalopathy. Arch. Virol. 2020, 165, 535–556. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers. Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.P.; Hewitt, E.W. Why are Functional Amyloids Non-Toxic in Humans? Biomolecules 2017, 7. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).