On the Potential of Silicon as a Building Block for Life




Abstract
1. Introduction
2. General Requirements for the Chemistry of Life
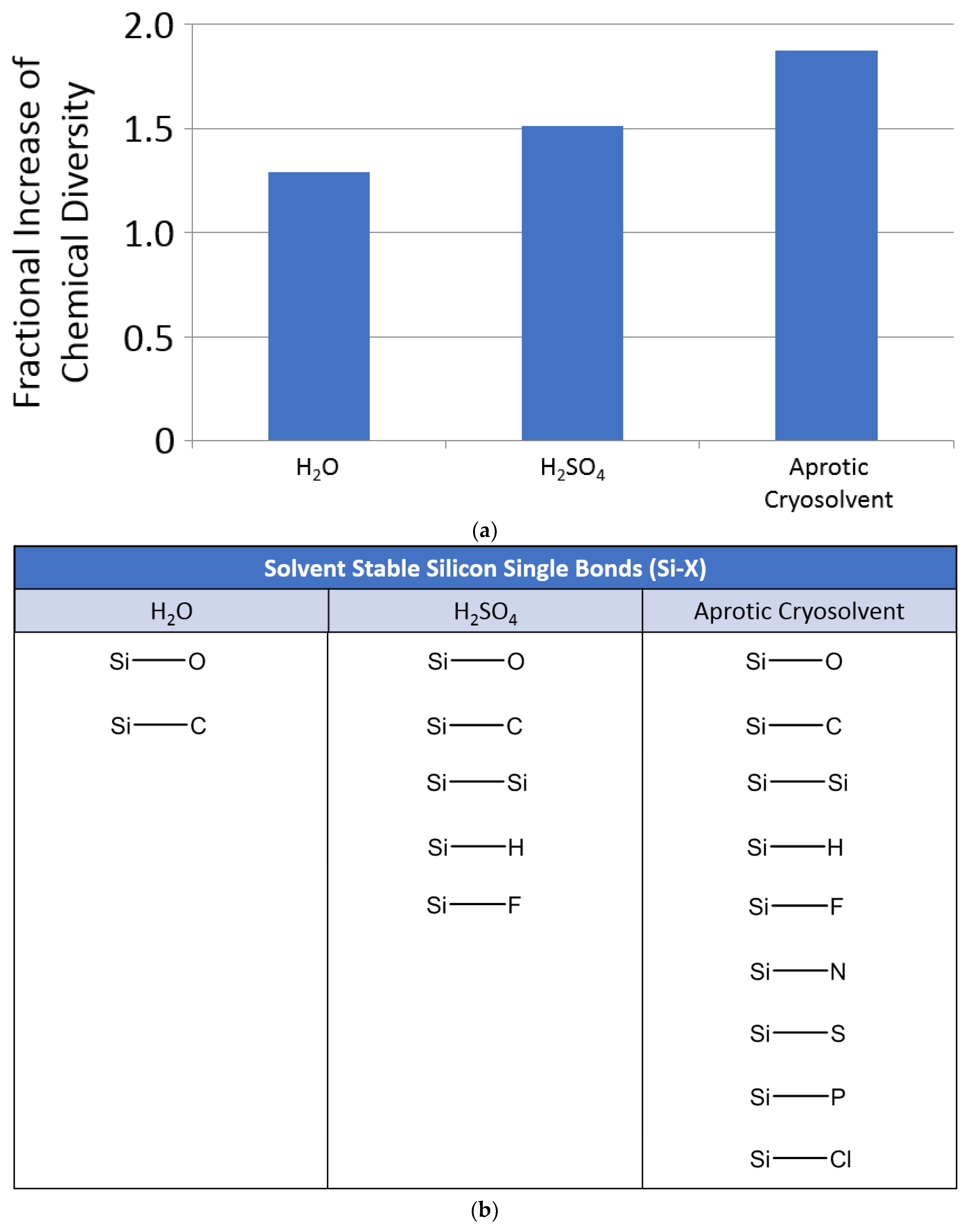
2.1. Chemical Diversity
2.2. Chemical Stability and Reactivity
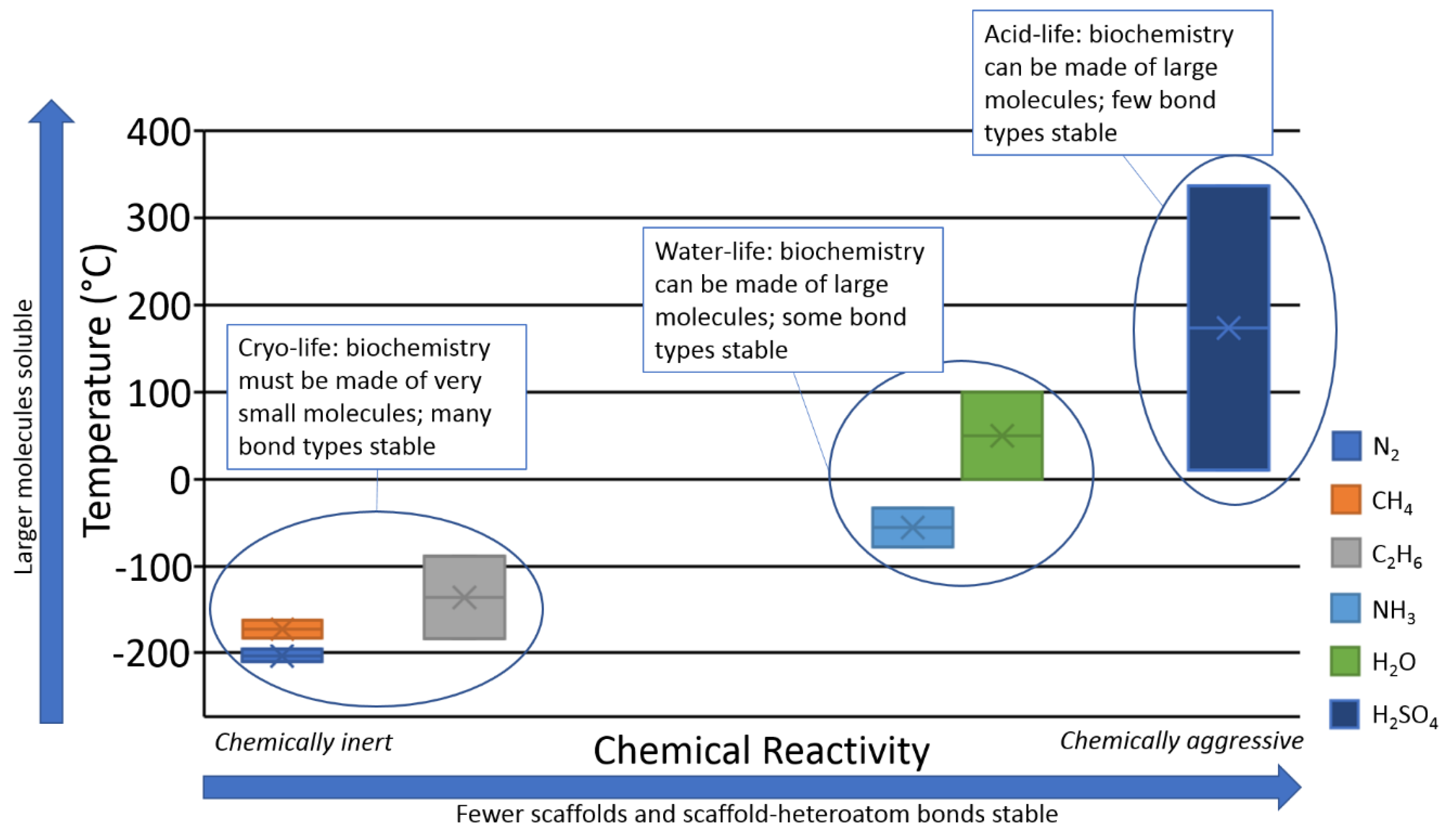
2.3. Solvent
3. Overview of Silicon Chemistry
3.1. Silicon Chemistry Overview in Comparison to Carbon
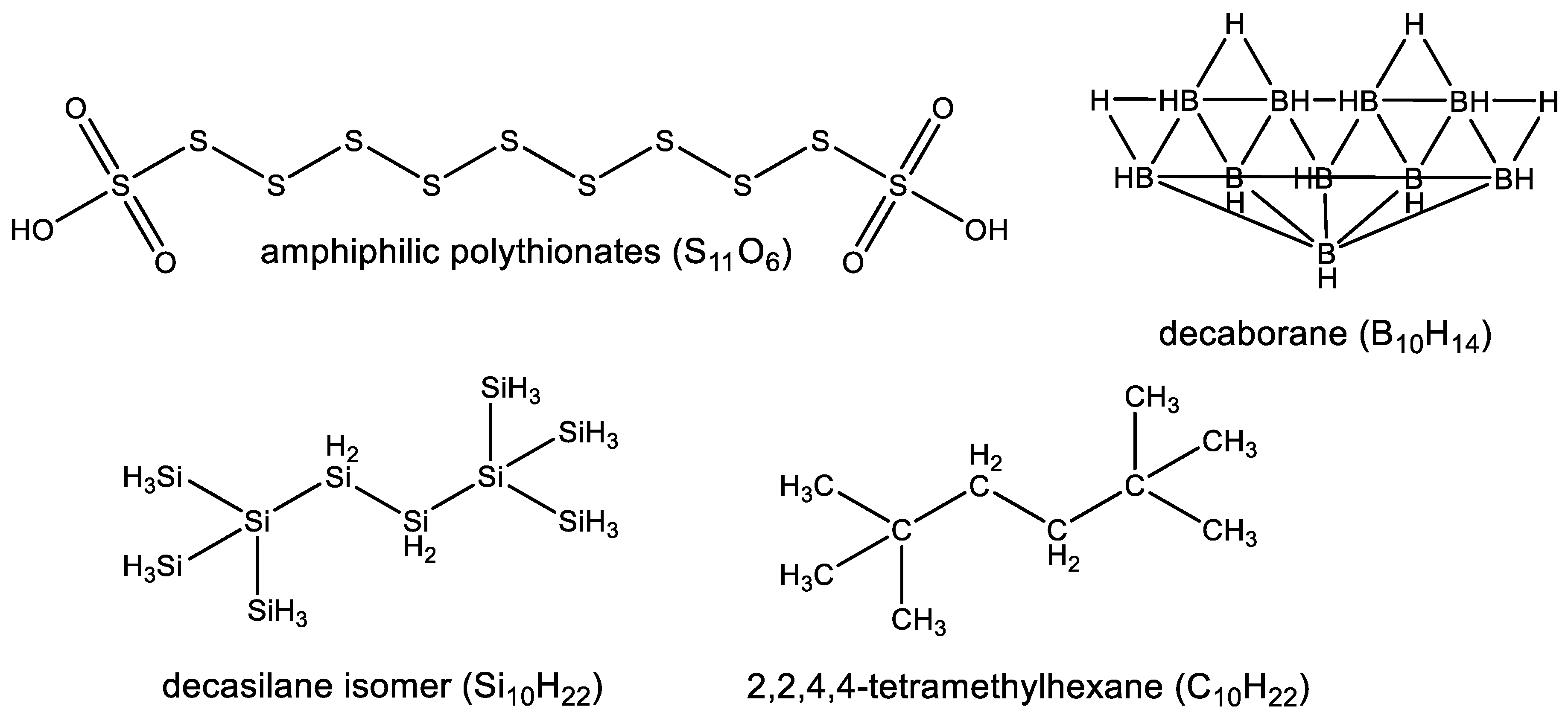
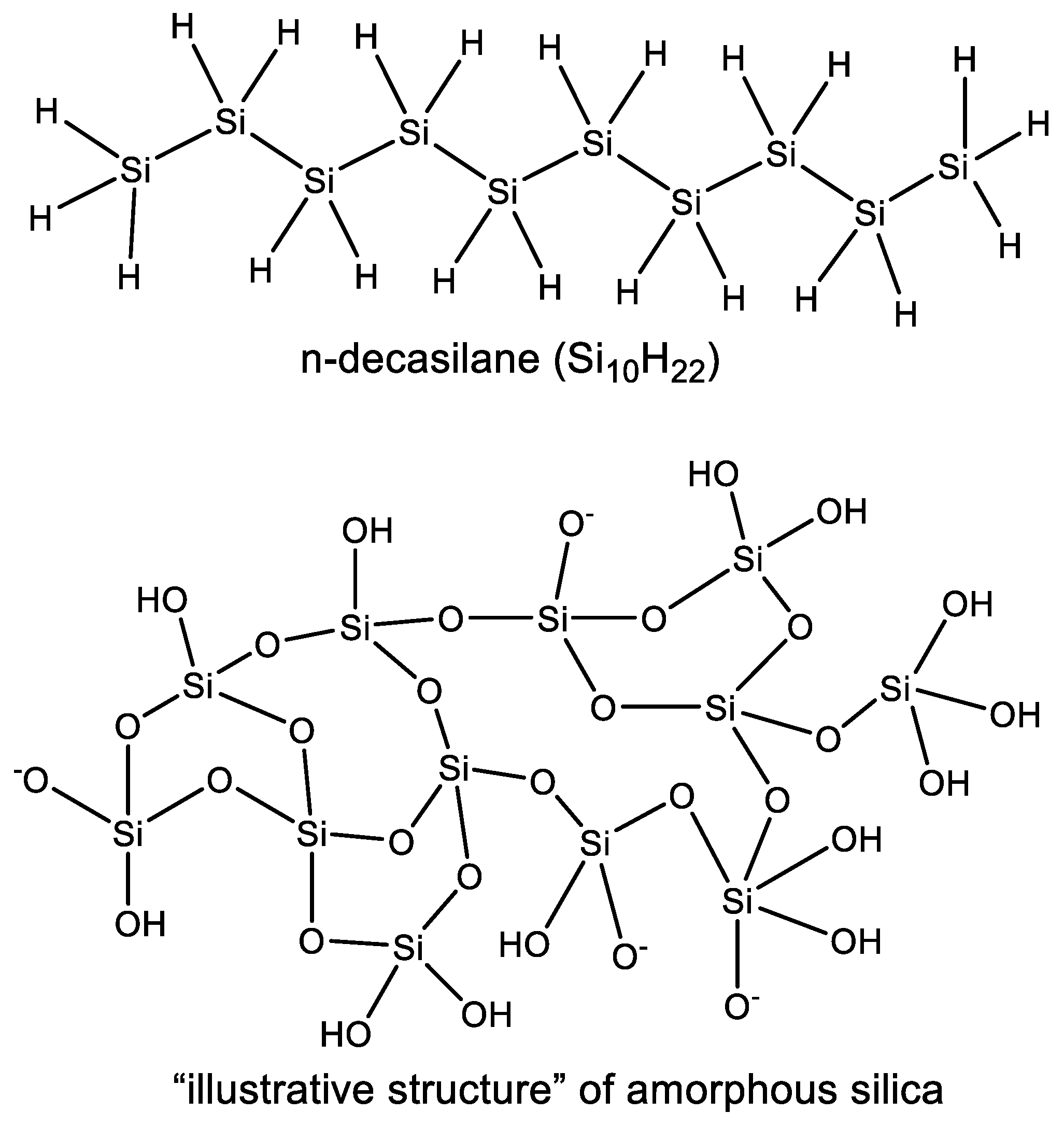
3.2. Diversity of Silicon Chemistry
3.2.1. Theoretical Diversity of Silicon Chemistry
3.2.2. Observed Functional Diversity of Silicon Chemistry
3.2.3. Observed Structural Diversity of Complex Silicon Polymer Chemistry

3.3. Thermodynamic Aspects of Formation of Silicon Compounds
4. Potential for Silicon Biochemistry in Different Solvents
4.1. Silicon Biochemistry in Water



4.2. Silicon Biochemistry in Non-Aqueous Acid/Base Solvents
4.3. Silicon Biochemistry in Sulfuric Acid
4.4. Silicon Biochemistry in Cold Aprotic Solvents
5. Synthesis and Conclusions
- Silicon and carbon are “false twins”. Their similarities are superficial and insufficient to mitigate their crucial differences. Chemistry that is stable and normal for carbon is unstable and exotic for silicon, and, similarly, chemistry that is unstable and impossible for carbon is stable and routine for silicon. Silicon’s distinct chemical characteristics and reactivity make it a challenging choice for life (Section 3.1).
- Silicon-based life that uses Si exclusively as a scaffold element is often portrayed in science fiction. An iconic example of a fictional silicon-based life form is Horta from the twenty-fifth episode—“The Devil in the Dark”—of the first season of the popular science fiction television series Star Trek TOS, itself possibly based on the “Siliconey” in Asimov’s short story “The Talking Stone” (1955). However, silicon-based life that uses Si exclusively as a scaffold element is almost certainly impossible.
- −
- Despite the potentially rich chemistry of silicon, direct substitution of C for Si in organic molecules is often impossible (Section 3.1; Section 3.2).
- −
- Formation of many biologically crucial functional groups is much less favorable for silicon than for their carbon counterparts (e.g., unsaturated silicon structures are generally only stable at cryogenic temperatures, if at all) (Section 3.2.1).
- −
- Due to the very high affinity of silicon to oxygen the most common stable polymers of silicon are built from a meshwork of Si–O chains instead of linear Si–Si silane polymers (Section 3.1).
- −
- The excess cosmic abundance of elemental oxygen ensures that the great majority of the available silicon is almost exclusively, and stably, bonded to oxygen (in the form of unreactive silica). Therefore, while “carbon chemistry is the chemistry of life, silicon chemistry is the chemistry of rocks” (Section 4.1). The substantial energy needed to turn rocks into life, compared to that needed to turn CO2 into life, argues against silicon.
- The vast potential theoretical space of silicon chemistry is almost entirely unstable in water, and hence not available to a biochemistry based on water as a solvent (Section 3.2.1; Section 4.1).
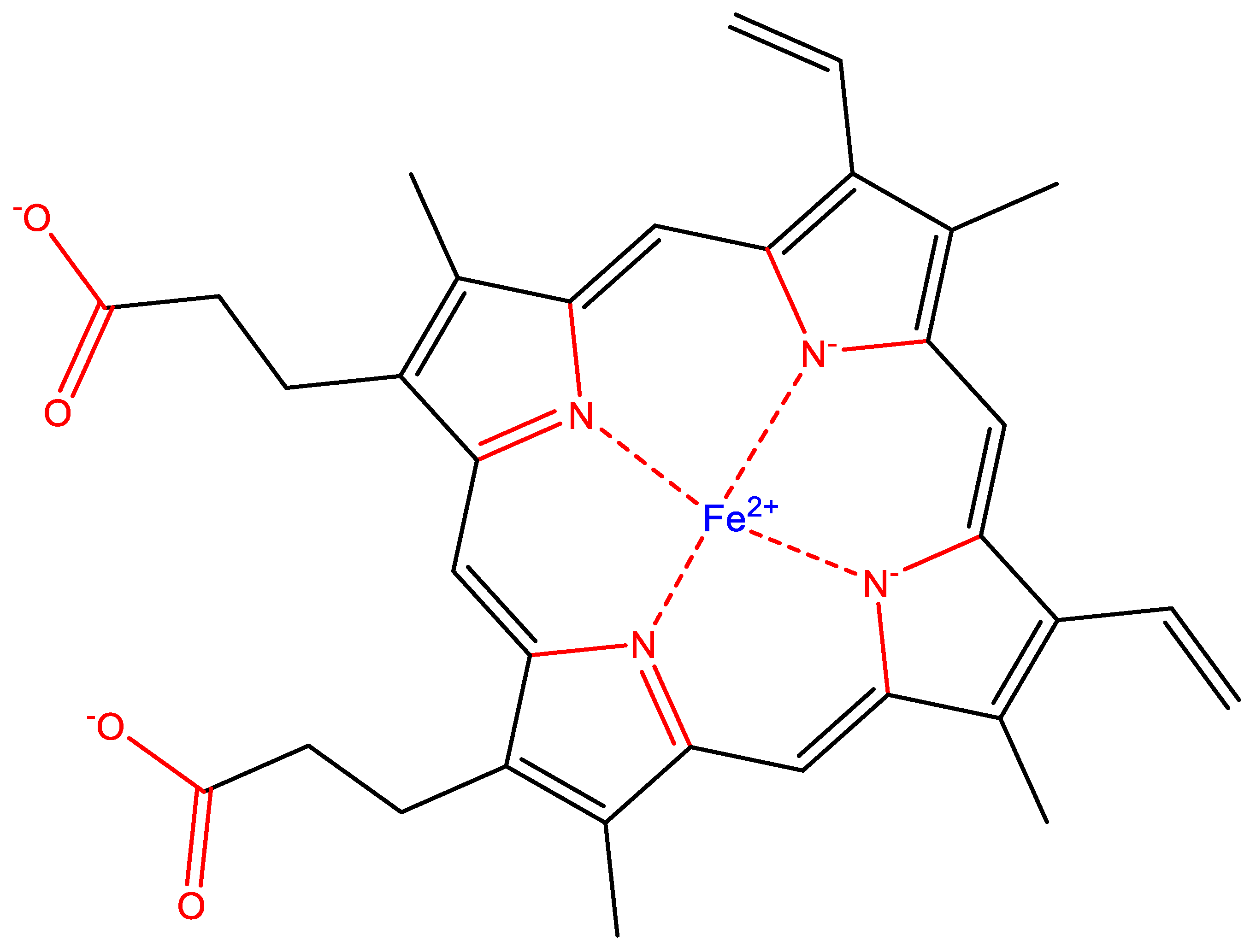
- Earth’s life silicon biochemistry is extremely chemically limited. In all of Earth’s life, the silicon atom is bonded exclusively to oxygen, forming a Si–O single bond. No naturally occurring life forms synthesize bonds between silicon and any other atom (Section 4.1).
- The usage of Si in the capacity of a rare heteroatom for water-based biochemistry can, in principle, be possible. The water stability of Si–C and Si–O bonds makes silicon attractive as a carrier of specialized biological functions. We postulate that some small number of organosilicon natural products with rare silicon heteroatoms await discovery (Section 4.1).
- The energies of formation of Si-containing compounds are generally much higher, and, therefore, less favorable, than their carbon counterparts. The thermodynamics puts additional, although not absolute, constraints on the potential of silicon-based life (Section 3.3).
- Any sort of biochemistry is implausible in cryogenic solvents, because of solubility limits (Section 4.4).
- Going forward, we should think about silicon as a contributor to biochemistry (as a common heteroatom in sulfuric acid and a rare heteroatom in water solvent) rather than a main building block of life.
- A larger fraction of the silicon chemical space is stable in concentrated sulfuric acid than in water. Such greater diversity of possible stable silicon molecules could be exploited by hypothetical sulfuric-acid-based life (e.g., a hypothetical, strictly aerial biosphere living in the sulfuric acid clouds of Venus). Even though carbon would still dominate, silicon could be widely used as a heteroatom component in sulfuric acid biochemistry (Section 4.3).
Author Contributions
Funding
Conflicts of Interest
Appendix A. Biochemistry of Silicon in Life on Earth
Appendix A.1. Silicon Biochemistry in Plants


Appendix A.2. Silicon Biochemistry in Diatoms
Appendix A.3. Silicon Biochemistry in Animals

Appendix B. Chemical Diversity of Silicon Compounds Observed in the Natural Environment
Appendix C. Formation of Unsaturated Organosilicon Compounds


Appendix D. Silicon Carbide and Carbon Planets as Potentially Suitable Environments for Silicon Biochemistry?
Appendix D.1. The Introduction to Carbon Planets
Appendix D.2. Can Carbon Planets Have Favourable Conditions for Complex Silicon Chemistry?
Appendix E. Chemical Diversity, Thermodynamics and Solubility Calculations
Appendix E.1. Chemical Diversity Calculations with Combimol-B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | O | P | S | F | Cl | S=O | S=S | S(=O)=O | S(=S)=O | P=O | P=S | Si | |
| N | + | + | + | + | + | + | + | + | + | + | + | + | |
| O | + | + | + | + | + | + | + | + | + | ||||
| P | + | + | + | + | + | + | + | ||||||
| S | + | + | + | + | + | + | + | + | + | + | + | + | |
| F | + | + | + | + | + | + | + | + | + | + | |||
| Cl | + | + | + | + | + | + | + | + | + | ||||
| S=O | + | + | + | + | + | + | + | + | + | ||||
| S=S | + | + | + | + | + | + | + | + | |||||
| S(=O)=O | + | + | + | + | + | + | + | + | + | ||||
| S(=S)=O | + | + | + | + | + | + | + | + | + | ||||
| P=O | + | + | + | + | + | + | + | + | |||||
| P=S | + | + | + | + | + | + | + | + | |||||
| Si | + | + | + | + | + | + | + | + | + | + | + | + | + |
| (b) | |||||||||||||
| N | O | P | S | F | Cl | S=O | S=S | S(=O)=O | S(=S)=O | P=O | P=S | Si | |
| N | + | + | + | + | + | + | + | + | + | ||||
| O | + | + | + | + | + | + | + | + | |||||
| P | + | + | |||||||||||
| S | + | + | + | + | + | + | + | + | |||||
| F | + | ||||||||||||
| Cl | |||||||||||||
| S=O | + | + | + | + | + | ||||||||
| S=S | + | + | + | + | |||||||||
| S(=O)=O | + | + | + | + | + | + | |||||||
| S(=S)=O | + | + | + | + | + | + | |||||||
| P=O | + | + | + | ||||||||||
| P=S | + | + | + | ||||||||||
| Si | |||||||||||||
| (c) | |||||||||||||
| N | O | P+ | S | F | Cl | S=O | S=S | S(=O)=O | S(=S)=O | Si | |||
| N | + | + | |||||||||||
| O | + | + | + | + | + | ||||||||
| P+ | |||||||||||||
| S | + | + | + | + | |||||||||
| F | + | ||||||||||||
| Cl | + | ||||||||||||
| S=O | + | + | + | ||||||||||
| S=S | + | ||||||||||||
| S(=O)=O | + | + | + | + | + | ||||||||
| S(=S)=O | + | + | + | + | + | ||||||||
| Si | + | + | |||||||||||
Appendix E.2. Thermodynamics of Formation of Silicon Compounds
- (1)
- XO2 + 4H2 -> XH4 + 2H2O
- (2)
- 2XO2 +7H2 -> X2H6 + 4H2O
- (3)
- 3XO2 + 10H2 -> X3H8 + 6H2O
- (4)
- XO2 + 4CO2 + 16H2 -> XC4H12 + 10H2O
- (5)
- XO2 + 4CO2 + 16H2 -> XC4H12 + 10H2O
- (6)
- XO2 + 3CO2 + 12H2 -> XC3H10O + 7H2O
- (7)
- XO2 + HCl + 3H2 -> XH3Cl + 2H2O
- (8)
- 2XO2 + 6HCl + H2 -> X2Cl6 + 4H2O
- (9)
- XO2 + 4HF -> XF4 + 2H2O
- (10)
- XO2 + 3CO2 + ½ N2 + 13½H2 -> XC3H11N + 8H2O
- (11)
- H3PO4 + XO2 + 7H2 -> XPH5 + 6H2O
Appendix E.3. Estimation of the Solubility of Silicon Chemicals in Liquid Nitrogen
References
- Bains, W. Many chemistries could be used to build living systems. Astrobiology 2004, 4, 137–167. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Ricardo, A.; Carrigan, M.A. Is there a common chemical model for life in the universe? Curr. Opin. Chem. Biol. 2004, 8, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Makuch, D.; Irwin, L.N. The prospect of alien life in exotic forms on other worlds. Naturwissenschaften 2006, 93, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Baross, J.; Benner, S.A.; Cody, G.D.; Copley, S.D.; Pace, N.R.; Scott, J.H.; Shapiro, R.; Sogin, M.L.; Stein, J.L.; Summons, R.; et al. The Limits of Organic Life in Planetary Systems; National Academies Press: Washington, DC, USA, 2007; ISBN 0309179564. [Google Scholar]
- Peng, S. Silicon-Based Life in the Solar System. Silicon 2015, 7, 1–3. [Google Scholar] [CrossRef]
- Schulze-Makuch, D.; Irwin, L.N. Life in the Universe: Expectations and Constraints; Springer: Cham, Switzerland, 2018; ISBN 978-3-319-97657-0. [Google Scholar]
- Darling, D.; Schulze-Makuch, D. The Extraterrestrial Encyclopedia; First Edition Design eBook Publishing: Sarasota, FL, USA, 2016; ISBN 9781506901442. [Google Scholar]
- Asimov, I. The elementary composition of the earth’s crust. J. Chem. Educ. 1954, 31, 70. [Google Scholar] [CrossRef]
- Ferris, T. RollingStone; Penske Media Corporation: New York City, NY, USA, June 1973. [Google Scholar]
- Hoehler, T.; Bains, W.; Davila, A.; Parenteau, M.; Pohorille, A. Life’s requirements, habitability, and biological potential. In Planetary Astrobiology; Meadows, V., Des Marais, D.J., Arney, G., Schmidt, B., Eds.; University of Arizona Press: Tucson, AZ, USA, 2020; in press. [Google Scholar]
- Griebel, J.J.; Glass, R.S.; Char, K.; Pyun, J. Polymerizations with elemental sulfur: A novel route to high sulfur content polymers for sustainability, energy and defense. Prog. Polym. Sci. 2016, 58, 90–125. [Google Scholar] [CrossRef]
- Adams, R.M. Boron, Metallo-Boron Compounds, and Boranes; Interscience Publishers: New York City, NY, USA, 1964. [Google Scholar]
- Benner, S.A. Defining life. Astrobiology 2010, 10, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Naturally occurring organofluorines. In Organofluorines; Springer: Berlin/Heidelberg, Germany, 2002; pp. 121–136. [Google Scholar]
- Christopoulos, V.; Rotzinger, M.; Gerwig, M.; Seidel, J.; Kroke, E.; Holthausen, M.; Wunnicke, O.; Torvisco, A.; Fischer, R.; Haas, M. Synthesis and Properties of Branched Hydrogenated Nonasilanes and Decasilanes. Inorg. Chem. 2019, 58, 8820–8828. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Kubinec, M.; Martin, L.; Schainker, J.; Vento, S. Sugar and Sulfuric Acid. Available online: https://www.youtube.com/watch?v=ZOedJgqTT9E (accessed on 1 May 2020).
- Pohorille, A.; Pratt, L.R. Is water the universal solvent for life? Orig. Life Evol. Biosph. 2012, 42, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Szostak, J.W.; Bartel, D.P.; Luisi, P.L. Synthesizing life. Nature 2001, 409, 387–390. [Google Scholar] [CrossRef] [PubMed]
- McKay, P.C. Titan as the Abode of Life. Life 2016, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- McLendon, C.; Opalko, F.J.; Illangkoon, H.I.; Benner, S.A. Solubility of polyethers in hydrocarbons at low temperatures. A model for potential genetic backbones on warm titans. Astrobiology 2015, 15, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, F.J.; Fernandez-Soto, A.; Martínez, V.J. Diving into Exoplanets: Are Water Seas the Most Common? Astrobiology 2019, 19, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Budisa, N.; Schulze-Makuch, D. Supercritical carbon dioxide and its potential as a life-sustaining solvent in a planetary environment. Life 2014, 4, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.; Burschka, C.; Platte, D.; Tacke, R. Pentacoordination of Silicon by Five Different Ligand Atoms: Neutral Silicon (IV) Complexes with SiClSONC and SiISONC Skeletons. Angew. Chem. 2007, 119, 7136–7139. [Google Scholar] [CrossRef]
- Junold, K.; Baus, J.A.; Burschka, C.; Vent-Schmidt, T.; Riedel, S.; Tacke, R. Five-coordinate silicon (II) compounds with Si–M bonds (M = Cr, Mo, W, Fe): Bis [N, N′-diisopropylbenzamidinato (−)] silicon (II) as a ligand in transition-metal complexes. Inorg. Chem. 2013, 52, 11593–11599. [Google Scholar] [CrossRef] [PubMed]
- Stock, A. Hydrides of Boron and Silicon; Cornell University Press: Tucson, AZ, USA, 1933. [Google Scholar]
- Barron, A.R. Chemistry of the Main Group Elements; Rice University Press: Houston, TX, USA, 2014. [Google Scholar]
- Benner, S.A. Detecting Darwinism from Molecules in the Enceladus Plumes, Jupiter’s Moons, and Other Planetary Water Lagoons. Astrobiology 2017, 17, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Spivack, J.L.; Pohl, E.R.; Kochs, P. Organoalkoxysilanes, organosilanols, and organosiloxanols. In Organosilicon Materials; Springer: Berlin/Heidelberg, Germany, 1997; pp. 105–135. [Google Scholar]
- Patai, S.; Rappoport, Z. The Chemistry of Organic Silicon Compounds; Wiley: Hoboken, NJ, USA, 1989. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Walsh, R. Bond dissociation energy values in silicon-containing compounds and some of their implications. Acc. Chem. Res. 1981, 14, 246–252. [Google Scholar] [CrossRef]
- Muller, T.; Zilche, W.; Auner, N. Recent advances in the chemistry of Si-heteroatom multiple bonds. Chem. Org. silicon Compd. 1998, 2, 857–1062. [Google Scholar]
- Bains, W.; Seager, S. A combinatorial approach to biochemical space: Description and application to the redox distribution of metabolism. Astrobiology 2012, 12, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Seager, S.; Bains, W.; Petkowski, J.J. Toward a List of Molecules as Potential Biosignature Gases for the Search for Life on Exoplanets and Applications to Terrestrial Biochemistry. Astrobiology 2016, 16, 465–485. [Google Scholar] [CrossRef] [PubMed]
- Kipping, F.S. The bakerian lecture organic derivatives of silicon. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1937, 159, 139–148. [Google Scholar]
- Hengge, E.; Janoschek, R. Homocyclic silanes. Chem. Rev. 1995, 95, 1495–1526. [Google Scholar] [CrossRef]
- Dettlaff-Weglikowska, U.; Hönle, W.; Molassioti-Dohms, A.; Finkbeiner, S.; Weber, J. Structure and optical properties of the planar silicon compounds polysilane and Wöhler siloxene. Phys. Rev. B 1997, 56, 13132. [Google Scholar] [CrossRef]
- Baney, R.H.; Itoh, M.; Sakakibara, A.; Suzuki, T. Silsesquioxanes. Chem. Rev. 1995, 95, 1409–1430. [Google Scholar] [CrossRef]
- Harrison, P.G. Silicate cages: Precursors to new materials. J. Organomet. Chem. 1997, 542, 141–183. [Google Scholar] [CrossRef]
- Jennings, A.R.; Iacono, S.T.; Mabry, J.M. Polyhedral Silsesquioxanes. In Handbook of Sol-Gel Science and Technology; Klein, L., Aparicio, M., Jitianu, A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–24. ISBN 978-3-319-19454-7. [Google Scholar]
- Feher, F.J. Polyhedral oligosilsesquioxanes and heterosilsesquioxanes. Silicon Ger. Tin Compd. Met. Alkoxides Met. Diketons Silicones. Gelest Tullytown, PA 2000, 43–59. [Google Scholar]
- Haas, A. The chemistry of silicon-sulfur compounds. Angew. Chem. Int. Ed. English 1965, 4, 1014–1023. [Google Scholar] [CrossRef]
- Chung, M.-K.; Schlaf, M. A catalytic synthesis of thiosilanes and silthianes: Palladium nanoparticle-mediated cross-coupling of silanes with thio phenyl and thio vinyl ethers through selective carbon− sulfur bond activation. J. Am. Chem. Soc. 2004, 126, 7386–7392. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tokitoh, N.; Nagase, S.; Okazaki, R. The First Genuine Silicon-Sulfur Double-Bond Compound: Synthesis and Crystal Structure of a Kinetically Stabilized Silanethione. J. Am. Chem. Soc. 1994, 116, 11578–11579. [Google Scholar] [CrossRef]
- Minkovich, B.; Ruderfer, I.; Kaushansky, A.; Bravo-Zhivotovskii, D.; Apeloig, Y. α-Sila-Dipeptides: Synthesis and Characterization. Angew. Chem. 2018, 130, 13445–13449. [Google Scholar] [CrossRef]
- Sharma, H.K.; Pannell, K.H. Activation of the Si–Si bond by transition metal complexes. Chem. Rev. 1995, 95, 1351–1374. [Google Scholar] [CrossRef]
- Tokito, N.; Okazaki, R. Polysilanes: Conformation, chromotropism and conductivity. Chem. Org. silicon 1998, 2, 1063–1104. [Google Scholar]
- West, R. Polysilanes: Conformations, chromotropism and conductivity. PATAI’S Chem. Funct. Groups 2009. [Google Scholar] [CrossRef]
- Tacke, R.; Puelm, M.; Wagner, B. Zwitterionic pentacoordinate silicon compounds. Adv. Organomet. Chem. 1999, 44, 221–275. [Google Scholar]
- Laine, R.M.; Blohowiak, K.Y.; Robinson, T.R.; Hoppe, M.L.; Nardi, P.; Kampf, J.; Uhm, J. Synthesis of pentacoordinate silicon complexes from SiO2. Nature 1991, 353, 642–644. [Google Scholar] [CrossRef]
- Tacke, R.; Burschka, C.; Richter, I.; Wagner, B.; Willeke, R. Pentacoordinate Silicon Compounds with Si O5 Skeletons Containing SiOH or SiOSi Groups: Derivatives of the Pentahydroxosilicate (1−) Anion [Si (OH) 5]-and Its Anhydride [(HO) 4Si− O− Si (OH) 4] 2. J. Am. Chem. Soc. 2000, 122, 8480–8485. [Google Scholar] [CrossRef]
- Stevenson III, W.H.; Wilson, S.; Martin, J.C.; Farnham, W.B. Pseudorotational mechanism for the inversion of 10-Si-5 siliconates: Ligand structure and reactivity. J. Am. Chem. Soc. 1985, 107, 6340–6352. [Google Scholar] [CrossRef]
- Muller, T.; West, R. Cations of group 14 organometallics. Adv. Organomet. Chem. 2005, 53, 155–216. [Google Scholar]
- Kost, D.; Kingston, V.; Gostevskii, B.; Ellern, A.; Stalke, D.; Walfort, B.; Kalikhman, I. Donor-Stabilized Silyl Cations. 3. Ionic Dissociation of Hexacoordinate Silicon Complexes to Pentacoordinate Siliconium Salts Driven by Ion Solvation1. Organometallics 2002, 21, 2293–2305. [Google Scholar] [CrossRef]
- Kalikhman, I.; Gostevskii, B.; Girshberg, O.; Krivonos, S.; Kost, D. Donor-Stabilized Silyl Cations 4: N-Isopropylidene Hydrazides, Novel Bidentate Ligands for Penta-and Hexacoordinate Silicon Chelates1. Organometallics 2002, 21, 2551–2554. [Google Scholar] [CrossRef]
- Gostevskii, B.; Pestunovich, V.; Kalikhman, I.; Sivaramakrishna, A.; Kocher, N.; Deuerlein, S.; Leusser, D.; Stalke, D.; Kost, D. Donor-Stabilized Silyl Cations. 8. Carbon− Carbon Bond Formation through a Novel Interchelate Molecular Rearrangement in Pentacoordinate Siliconium-Ion Salts1. Organometallics 2004, 23, 4346–4348. [Google Scholar] [CrossRef]
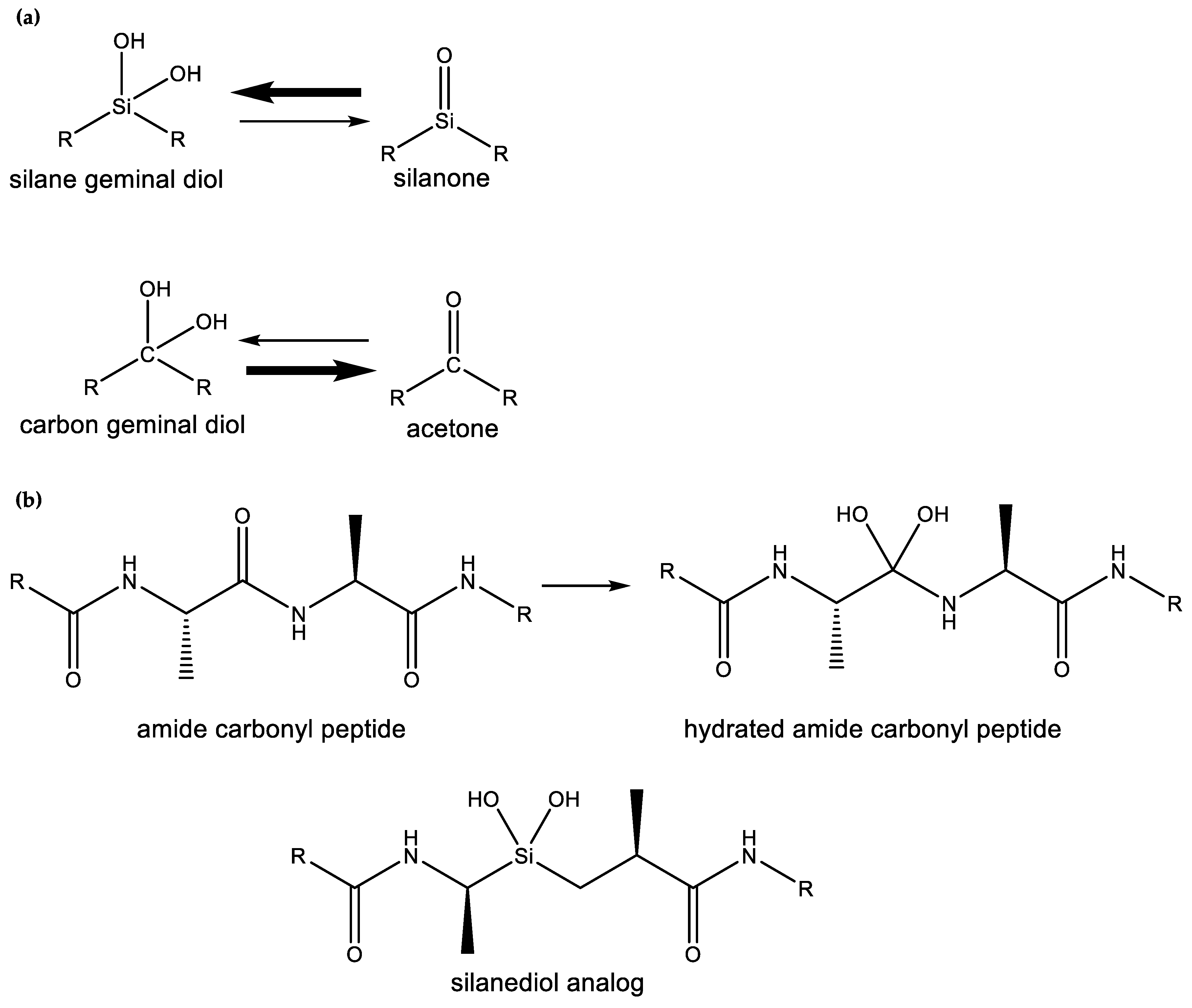
- Sieburth, S.M.; Nittoli, T.; Mutahi, A.M.; Guo, L. Silanediols: A new class of potent protease inhibitors. Angew. Chem. Int. Ed. 1998, 37, 812–814. [Google Scholar] [CrossRef]
- Franz, A.K.; Wilson, S.O. Organosilicon molecules with medicinal applications. J. Med. Chem. 2013, 56, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Henker, J.; Wirmer-Bartoschek, J.; Bendel, L.E.; Xiang, Y.; Fu, C.; Harms, K.; Schwalbe, H.; Meggers, E. Progress in the synthesis and bioactivity of hexacoordinate silicon (IV) complexes. Eur. J. Inorg. Chem. 2016, 2016, 5161–5170. [Google Scholar] [CrossRef]
- Pace, N.R. The universal nature of biochemistry. Proc. Natl. Acad. Sci. USA 2001, 98, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Yilgör, E.; Yilgör, I. Silicone containing copolymers: Synthesis, properties and applications. Prog. Polym. Sci. 2014, 39, 1165–1195. [Google Scholar] [CrossRef]
- Ito, H.; Taenaka, A.; Nagasaki, Y.; Kataoka, K.; Kato, M.; Tsuruta, T. Silicon-containing block copolymer membranes. Polymer (Guildford) 1996, 37, 633–637. [Google Scholar] [CrossRef]
- Czarnecki, S.; Bertin, A. Hybrid Silicon-Based Organic/Inorganic Block Copolymers with Sol–Gel Active Moieties: Synthetic Advances, Self-Assembly and Applications in Biomedicine and Materials Science. Chem. Eur. J. 2018, 24, 3354–3373. [Google Scholar] [CrossRef] [PubMed]
- Indulekha, K.; Roy, R.E.; Vishnu, A.G.; Rajeev, R.S.; Ninan, K.N.; Gouri, C. Silicone copolymers bearing reactive vinyl and hydride functionalities: Synthesis, characterisation and particulate composite thereof for specialty applications. Mater. Chem. Phys. 2018, 206, 213–223. [Google Scholar] [CrossRef]
- Corden, C.; Tyrer, D.; Menadue, H.; Calreo, J.; Dade, J.; Leferink, R. Socio-economic evaluation of the global silicones industry. Final Rep. 2016, 1, 1–115. [Google Scholar]
- Kamata, N.; Terunuma, D.; Ishii, R.; Satoh, H.; Aihara, S.; Yaoita, Y.; Tonsyo, S. Efficient energy transfer from polysilane molecules and its application to electroluminescence. J. Organomet. Chem. 2003, 685, 235–242. [Google Scholar] [CrossRef]
- Lickiss, P.D.; Litster, S.A.; Redhouse, A.D.; Wisener, C.J. Isolation of a tetrahydroxydisiloxane formed during hydrolysis of an alkyltrichlorosilane: Crystal and molecular structure of [But (OH) 2Si] 2O. J. Chem. Soc. Chem. Commun. 1991, 173–174. [Google Scholar] [CrossRef]
- Lickiss, P.D.; Redhouse, A.D.; Thompson, R.J.; Stańczyk, W.A.; Rózga, K. The crystal structure of (HOMe2Si) 2O. J. Organomet. Chem. 1993, 453, 13–16. [Google Scholar] [CrossRef]
- Unno, M.; Takada, K.; Matsumoto, H. Formation of supermolecule by assembling of two different silanols. Chem. Lett. 2000, 29, 242–243. [Google Scholar] [CrossRef]
- Bährle-Rapp, M. Methylsilanol Acetyltyrosine. In Springer Lexikon Kosmetik und Körperpflege; Springer: Berlin/Heidelberg, Germany, 2007; p. 354. [Google Scholar]
- Mori, T.; Sato, M.; Shimoike, Y.; Notsu, K. High SiF 4/HF ratio detected in Satsuma-Iwojima volcano’s plume by remote FT-IR observation. Earth Planets Sp. 2002, 54, 249–256. [Google Scholar] [CrossRef]
- Gurvich, L.V.; Veyts, I. Thermodynamic Properties of Individual Substances: Elements and Compounds; CRC Press: Boca Raton, FL, USA, 1990; Volume 2, ISBN 0891165339. [Google Scholar]
- Becerra, R.; Walsh, R. Thermochemistry of organosilicon compounds. In Organosilicon Compounds; Elsevier: Amsterdam, The Netherlands, 2017; pp. 79–113. [Google Scholar]
- Chase, M.W., Jr. NIST-JANAF thermochemical tables. J. Phys. Chem. Ref. data. 1998. Monograph. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Semiempirical Molecular Orbital Methods. In Reviews in Computational Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 45–81. ISBN 9780470125786. [Google Scholar]
- Winget, P.; Clark, T. Enthalpies of formation from B3LYP calculations. J. Comput. Chem. 2004, 25, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, G.; Shapiro, R. Life beyond Earth: The Intelligent Earthling’s Guide to Life in the Universe; William Morrow &Company: New York City, NY, USA, 1980; ISBN 0688036422. [Google Scholar]
- Léger, A.; Rouan, D.; Schneider, J.; Barge, P.; Fridlund, M.; Samuel, B.; Ollivier, M.; Guenther, E.; Deleuil, M.; Deeg, H.J. Transiting exoplanets from the CoRoT space mission-VIII. CoRoT-7b: The first super-Earth with measured radius. Astron. Astrophys. 2009, 506, 287–302. [Google Scholar] [CrossRef]
- Kuchner, M.J.; Seager, S. Extrasolar carbon planets. arXiv Prepr. 2005, arXiv:astro-ph/0504214, 1–17. Available online: https://arxiv.org/abs/astro-ph/0504214 (accessed on 1 May 2020).
- Tacke, R.; Linoh, H. Bioorganosilicon chemistry. Org. Silicon Compd. 1989, 1, 1143–1206. [Google Scholar]
- Zani, P. Biotransformations of organosilicon compounds: Enantioselective reduction of acyl silanes by means of baker’s yeast. J. Mol. Catal. B Enzym. 2001, 11, 279–285. [Google Scholar] [CrossRef]
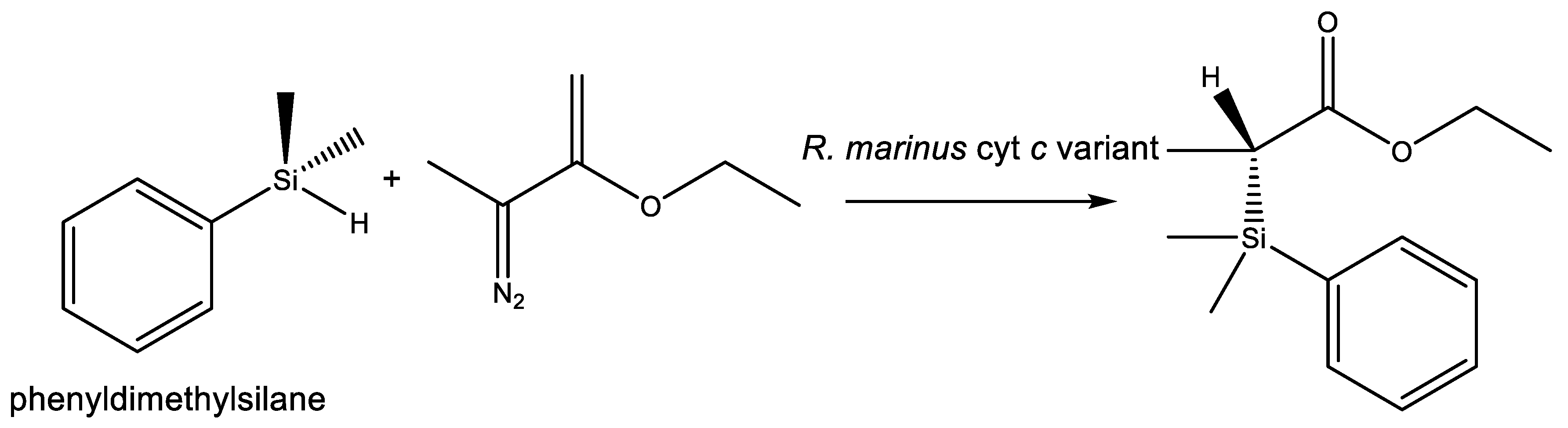
- Kan, S.B.J.; Lewis, R.D.; Chen, K.; Arnold, F.H. Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life. Science (80-) 2016, 354, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.H. Directed evolution: Bringing new chemistry to life. Angew. Chem. Int. Ed. 2018, 57, 4143–4148. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Srebnik, M. Synthesis and biological activity of α-aminoboronic acids, amine-carboxyboranes and their derivatives. Tetrahedron 2003, 59, 579–593. [Google Scholar] [CrossRef]
- Dembitsky, V.M.; Quntar, A.A.A.; Srebnik, M. Recent advances in the medicinal chemistry of α-Aminoboronic acids, amine-carboxyboranes and their derivatives. Mini Rev. Med. Chem. 2004, 4, 1001–1018. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhu, M.; Zhang, J.; Zhou, H. Synthesis of biologically active boron-containing compounds. Medchemcomm 2018, 9, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.T.; MacKinnon, S.L.; Yan, X.; Hammond, G.B.; Vaisberg, A.J.; Neto, C.C. Identification of triterpene hydroxycinnamates with in vitro antitumor activity from whole cranberry fruit (Vaccinium macrocarpon). J. Agric. Food Chem. 2003, 51, 3541–3545. [Google Scholar] [CrossRef] [PubMed]
- Murugavel, R.; Chandrasekhar, V.; Voigt, A.; Roesky, H.W.; Schmidt, H.-G.; Noltemeyer, M. New lipophilic air-stable silanetriols: First example of an x-ray crystal structure of a silanetriol with Si-N bonds. Organometallics 1995, 14, 5298–5301. [Google Scholar] [CrossRef]
- Pietschnig, R.; Spirk, S. The chemistry of organo silanetriols. Coord. Chem. Rev. 2016, 323, 87–106. [Google Scholar] [CrossRef]
- Lickiss, P.D. The synthesis and structure of organosilanols. Adv. Inorg. Chem. 1995, 42, 147–262. [Google Scholar]
- Lickiss, P.D. Polysilanols. Chem. Org. Silicon Compd. 2001, 3, 695–744. [Google Scholar]
- Chandrasekhar, V.; Nagendran, S.; Kingsley, S.; Krishnan, V.; Boomishankar, R. Si–O and P–O motifs in inorganic rings and clusters. J. Chem. Sci. 2000, 112, 171–178. [Google Scholar] [CrossRef]
- Bunning, J.D.; Lydon, J.E.; Eaborn, C.; Jackson, P.M.; Goodby, J.W.; Gray, G.W. Classification of the mesophase of di-isobutylsilanediol. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1982, 78, 713–724. [Google Scholar] [CrossRef]
- Mutahi, M.W.; Nittoli, T.; Guo, L.; Sieburth, S.M. Silicon-Based Metalloprotease Inhibitors: Synthesis and Evaluation of Silanol and Silanediol Peptide Analogues as Inhibitors of Angiotensin-Converting Enzyme1. J. Am. Chem. Soc. 2002, 124, 7363–7375. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Glekas, A.; Sieburth, S.M. Silanediol-based inhibitor of thermolysin. Bioorg. Med. Chem. Lett. 2002, 12, 3625–3627. [Google Scholar] [CrossRef]
- Chen, C.-A.; Sieburth, S.M.; Glekas, A.; Hewitt, G.W.; Trainor, G.L.; Erickson-Viitanen, S.; Garber, S.S.; Cordova, B.; Jeffry, S.; Klabe, R.M. Drug design with a new transition state analog of the hydrated carbonyl: Silicon-based inhibitors of the HIV protease. Chem. Biol. 2001, 8, 1161–1166. [Google Scholar] [CrossRef]
- Singh, S.; Sieburth, S.M. Serine protease inhibition by a silanediol peptidomimetic. Org. Lett. 2012, 14, 4422–4425. [Google Scholar] [CrossRef] [PubMed]
- Sieburth, S.M.; Chen, C. Silanediol protease inhibitors: From conception to validation. Eur. J. Org. Chem. 2006, 2006, 311–322. [Google Scholar] [CrossRef]
- Emeleus, H.J.; Wilkins, C.J. 122. Some new ethyl and phenyl silicon fluorides. J. Chem. Soc. 1944, 454–456. [Google Scholar] [CrossRef]
- Schmidt, M.; Schmidbaur, H. Über Silanolester anorganischer Säuren, III. Schwefelsäureester von Methylsilanolen. Chem. Ber. 1960, 93, 878–882. [Google Scholar] [CrossRef]
- Kipping, F.S. LXIX.—Organic derivatives of silicon. Part XXIV. dl-Derivatives of silicoethane. J. Chem. Soc. Trans. 1921, 119, 647–653. [Google Scholar] [CrossRef][Green Version]
- Kumada, M.; Yamaguchi, M.; Yamamoto, Y.; Nakajima, J.-I.; Shiina, K. Synthesis of some methyldisilanes containing functional groups. J. Org. Chem. 1956, 21, 1264–1268. [Google Scholar] [CrossRef]
- Plate, A.F.; Belikova, N.A.; Egorov, Y.P. Reaction of silacyclopentanes and silacyclohexanes with concentrated sulfuric acid. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1956, 5, 1101–1105. [Google Scholar] [CrossRef]
- Kipping, F.S.; Martin, G. LXII.—The action of fuming sulphuric acid on triphenylsilicol. J. Chem. Soc. Trans. 1909, 95, 489–494. [Google Scholar] [CrossRef]
- Sommer, L.H.; Bailey, D.L.; Goldberg, G.M.; Buck, C.E.; Bye, T.S.; Evans, F.J.; Whitmore, F.C. Vinylsilanes, chlorovinylsilanes and β-styryltrimethylsilane. Further studies on the α-silicon effect and β-eliminations involving silicon1. J. Am. Chem. Soc. 1954, 76, 1613–1618. [Google Scholar] [CrossRef]
- Patnode, W.; Wilcock, D.F. Methylpolysiloxanes1. J. Am. Chem. Soc. 1946, 68, 358–363. [Google Scholar] [CrossRef]
- Kollman, P.A.; Allen, L.C. Hydrogen bonded dimers and polymers involving hydrogen fluoride, water, and ammonia. J. Am. Chem. Soc. 1970, 92, 753–759. [Google Scholar] [CrossRef]
- Jensen, J.L.; Uaprasert, V. Acid-catalyzed hydration of dienes. III. Effects of ring strain on rate, enthalpy, and entropy for hydration of 1, 3-cycloalkadienes. J. Org. Chem. 1976, 41, 649–654. [Google Scholar] [CrossRef]
- Connelly, B.M.; Tolbert, M.A. Reaction of isoprene on thin sulfuric acid films: Kinetics, uptake, and product analysis. Environ. Sci. Technol. 2010, 44, 4603–4608. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.J.T. Organo Siloxanes and Their Production. U.S. Patent 2,592,682, 15 April 1952. [Google Scholar]
- Limaye, S.S.; Mogul, R.; Smith, D.J.; Ansari, A.H.; Słowik, G.P.; Vaishampayan, P. Venus’ Spectral Signatures and the Potential for Life in the Clouds. Astrobiology 2018, 18, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Makuch, D.; Grinspoon, D.H.; Abbas, O.; Irwin, L.N.; Bullock, M.A. A sulfur-based survival strategy for putative phototrophic life in the Venusian atmosphere. Astrobiology 2004, 4, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Seager, S.; Petkowski, J.J.; Gao, P.; Bains, W.; Bryan, N.C.; Ranjan, S.; Greaves, J. The Venusian Lower Atmosphere Haze as a Depot for Desiccated Microbial Life: A Proposed Life Cycle for Persistence of the Venusian Aerial Biosphere. Astrobiology 2020. accepted. [Google Scholar]
- Soderblom, L.A.; Kieffer, S.W.; Becker, T.L.; Brown, R.H.; Cook, A.F.; Hansen, C.J.; Johnson, T.V.; Kirk, R.L.; Shoemaker, E.M. Triton’s geyser-like plumes: Discovery and basic characterization. Science (80-) 1990, 250, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Connors, K.A. Chemical Kinetics: The Study of Reaction Rates in Solution; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1990; ISBN 156081053X. [Google Scholar]
- Kovalev, D.; Timoshenko, V.Y.; Künzner, N.; Gross, E.; Koch, F. Strong explosive interaction of hydrogenated porous silicon with oxygen at cryogenic temperatures. Phys. Rev. Lett. 2001, 87, 68301. [Google Scholar] [CrossRef] [PubMed]
- West, R. Multiple bonds to silicon: 20 years later. Polyhedron 2002, 21, 467–472. [Google Scholar] [CrossRef]
- Milnes, K.K.; Pavelka, L.C.; Baines, K.M. Cycloaddition of carbonyl compounds and alkynes to (di) silenes and (di) germenes: Reactivity and mechanism. Chem. Soc. Rev. 2016, 45, 1019–1035. [Google Scholar] [CrossRef] [PubMed]
- Rest, A.J.; Scurlock, R.G.; Wu, M.F. The solubilities of nitrous oxide, carbon dioxide, aliphatic ethers and alcohols, and water in cryogenic liquids. Chem. Eng. J. 1990, 43, 25–31. [Google Scholar] [CrossRef]
- Abraham, M.H.; Smith, R.E.; Luchtefeld, R.; Boorem, A.J.; Luo, R.; Acree Jr, W.E. Prediction of solubility of drugs and other compounds in organic solvents. J. Pharm. Sci. 2010, 99, 1500–1515. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L.; De Vries, T. The The Solubility of Solid Ethane, Ethylene, and Propylene in Liquid Nitrogen and Oxygen. J. Phys. Chem. 1950, 54, 665–670. [Google Scholar] [CrossRef]
- Preston, G.T.; Funk, E.W.; Prausnitz, J.M. Solubilities of hydrocarbons and carbon dioxide in liquid methane and in liquid argon. J. Phys. Chem. 1971, 75, 2345–2352. [Google Scholar] [CrossRef]
- Wojtaszek, Z.; Szczepaniec-Cieciak, E. The solubility of solid chlorine, hydrogen sulphide, sulphur dioxide, and ammonia in liquid nitrogen in the temperature range 77.4–63.5 K. Cryogenics (Guildford) 1975, 15, 257–260. [Google Scholar] [CrossRef]
- Wojtaszek, Z.; Szczepaniec-Cieciak, E.; Morzyniec, A. The solubility of solid nitrous oxide in liquid nitrogen in the 77.4–63.5 K temperature range. Cryogenics (Guildford) 1975, 15, 351–353. [Google Scholar] [CrossRef]
- Szczepaniec-Cieciak, E.; Nagraba, K. The solubility of solidified toluene in liquid nitrogen. Cryogenics (Guildford) 1978, 18, 601–603. [Google Scholar] [CrossRef]
- Szczepaniec-Cieciak, E.; Kondaurov, V.A.; Melikova, S.M. The solubility of light olefins in liquid nitrogen: Part 2. Cryogenics (Guildford) 1979, 19, 649–651. [Google Scholar] [CrossRef]
- Wojtaszek, Z.; Dabrowska, B.; Skwarczyńska, M. The solubility of solidified dichloromethane in liquid nitrogen at 77.4 K. Cryogenics (Guildford) 1979, 19, 399–400. [Google Scholar] [CrossRef]
- Chen, R.J.J.; Liaw, V.W.; Elliot, D.G. Prediction of CO2 Solubility in Light Hydrocarbon Mixtures at Low Temperatures. In Advances in Cryogenic Engineering; Springer: Boston, MA, USA, 1980; pp. 620–628. [Google Scholar]
- Kuebler, G.P.; McKinley, C. Solubility of Solid tert-Butyl Mercaptan in Liquid Methane and an LNG Mixture. In Advances in Cryogenic Engineering; Springer: Boston, MA, USA, 1980; pp. 616–619. [Google Scholar]
- Szczepaniec-Cieciak, E.; Kondaurov, V.A.; Melikova, S.M. Study on the solubility light alkanes in liquid nitrogen. Cryogenics (Guildford) 1980, 20, 48–51. [Google Scholar] [CrossRef]
- Zelikina, G.Y.; Meister, T.G.; Mamchenko, T.B. Spectroscopic determination of the solubility of several substances in liquefied gases according to electronic absorption bands. J. Appl. Spectrosc. 1980, 32, 348–352. [Google Scholar] [CrossRef]
- Dabrowska, B. The solubility of selected halogen hydrocarbons in liquid nitrogen at 77.4 K. Cryogenics (Guildford) 1984, 24, 276–277. [Google Scholar] [CrossRef]
- Dabrowska, B. Solubility of CFCl3, CHCl3, CCl4 and C2HCl3 in liquid nitrogen at 77.4 K. Cryogenics (Guildford) 1991, 31, 896–899. [Google Scholar] [CrossRef]
- Kuebler, G.P.; McKinley, C. Solubility of solid benzene, toluene, n-hexane, and n-heptane in liquid methane. In Advances in Cryogenic Engineering; Springer: Boston, MA, USA, 1995; pp. 320–326. [Google Scholar]
- Dabrowska, B. The solubility of solidified bromoethane C2H5Br in liquid nitrogen at 77.4 K. Cryogenics (Guildford) 1996, 36, 985–988. [Google Scholar] [CrossRef]
- Szczepaniec-Cieciak, E.; Krzeczkowska, M. Solubility of 1-pentene ice in liquid nitrogen and argon at the standard boiling points of the solvents. J. Solut. Chem. 1998, 27, 485–494. [Google Scholar] [CrossRef]
- Kurdziel, M.; Szczepaniec-Cięciak, E.; Żarnowska, E.; Stach, J.; Nitek, W.; Dąbrowska, B. Solubility of solid 1-hexene and 2-methylpentane in liquid argon and nitrogen at the standard boiling points of the solvents. J. Solut. Chem. 2001, 30, 781–794. [Google Scholar] [CrossRef]
- Kurdziel, M.; Szczepaniec-Cięciak, E.; Golonka, M.; Dąbrowska, B.; Nitek, W. Solubility of solid 1-hexyne in liquid argon and nitrogen at the standard boiling points of the solvents. J. Solut. Chem. 2002, 31, 253–260. [Google Scholar] [CrossRef]
- Kurdziel, M.; Szczepaniec-Cięciak, E.; Dąbrowska, B.; Nitek, W.; Paliś, K.; Ślusarska, E. Solubility of solid 2, 3-dimethylbutane and cyclopentane in liquid argon and nitrogen at the standard boiling points of the solvents. J. Solut. Chem. 2003, 32, 601–615. [Google Scholar] [CrossRef]
- Kurdziel, M.; Szczepaniec-Cieciak, E.; Watorczyk, M.; Dabrowska, B. Solubility of solid 2-methyl-1, 3-butadiene (isoprene) in liquid argon and nitrogen at the standard boiling points of the solvents. J. Solut. Chem. 2004, 33, 453–464. [Google Scholar] [CrossRef]
- Singh, S.; Combe, J.-P.; Cordier, D.; Wagner, A.; Chevrier, V.F.; McMahon, Z. Experimental determination of acetylene and ethylene solubility in liquid methane and ethane: Implications to Titan’s surface. Geochim. Cosmochim. Acta 2017, 208, 86–101. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Cairns-Smith, G.A.; Hartman, H. Clay Minerals and the Origin of Life; Cambridge University Press: Cambridge, UK, 1986; Volume 204. [Google Scholar]
- Wainwright, M. The neglected microbiology of silicon-from the origin of life to an explanation for what Henry Charlton Bastian saw. Soc. Gen. Microbiol. Q. 1997, 24, 83–85. [Google Scholar]
- Wainwright, M.; Al-Wajeeh, K.; Grayston, S.J. Effect of silicic acid and other silicon compounds on fungal growth in oligotrophic and nutrient-rich media. Mycol. Res. 1997, 101, 933–938. [Google Scholar] [CrossRef]
- Das, S.; Mandal, S.; Chakrabarty, A.N.; Dastidar, S.G. Metabolism of silicon as a probable pathogenicity factor for Mycobacterium & Nocardia spp. Indian J. Med. Res. 1992, 95, 59–65. [Google Scholar] [PubMed]
- Epstein, E. The anomaly of silicon in plant biology. Proc. Natl. Acad. Sci. USA 1994, 91, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Raven, J.A. The Transport and Function of Silicon in Plants. Biol. Rev. 1983, 58, 179–207. [Google Scholar] [CrossRef]
- Emadian, S.F.; Newton, R.J. Growth Enhancement of Loblolly Pine (Pinus taeda L.) Seedlings by Silicon. J. Plant Physiol. 1989, 134, 98–103. [Google Scholar] [CrossRef]
- Ma, J.F.; Yamaji, N. Silicon uptake and accumulation in higher plants. Trends Plant Sci. 2006, 11, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, R.R.; Benhamou, N.; Menzies, J.G. Cytological Evidence of an Active Role of Silicon in Wheat Resistance to Powdery Mildew (Blumeria graminis f. sp. tritici). Phytopathology 2003, 93, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Côté-Beaulieu, C.; Chain, F.; Menzies, J.G.; Kinrade, S.D.; Bélanger, R.R. Absorption of aqueous inorganic and organic silicon compounds by wheat and their effect on growth and powdery mildew control. Environ. Exp. Bot. 2009, 65, 155–161. [Google Scholar] [CrossRef]
- Epstein, E. Silicon: Its manifold roles in plants. Ann. Appl. Biol. 2009, 155, 155–160. [Google Scholar] [CrossRef]
- Adrees, M.; Ali, S.; Rizwan, M.; Zia-ur-Rehman, M.; Ibrahim, M.; Abbas, F.; Farid, M.; Qayyum, M.F.; Irshad, M.K. Mechanisms of silicon-mediated alleviation of heavy metal toxicity in plants: A review. Ecotoxicol. Environ. Saf. 2015, 119, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Exley, C. Silicon in life: A bioinorganic solution to bioorganic essentiality. J. Inorg. Biochem. 1998, 69, 139–144. [Google Scholar] [CrossRef]
- Epstein, E. Silicon. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 641–664. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.P.; Ma, G.R.; Shui, M.X.; Wan, D.F.; Li, R.H. Silicon isotope study on rice plants from the Zhejiang province, China. Chem. Geol. 2005, 218, 41–50. [Google Scholar] [CrossRef]
- Casey, W.H.; Kinrade, S.D.; Knight, C.T.G.; Rains, D.W.; Epstein, E. Aqueous silicate complexes in wheat, Triticum aestivum L. Plant. Cell Environ. 2004, 27, 51–54. [Google Scholar] [CrossRef]
- Ding, T.P.; Zhou, J.X.; Wan, D.F.; Chen, Z.Y.; Wang, C.Y.; Zhang, F. Silicon isotope fractionation in bamboo and its significance to the biogeochemical cycle of silicon. Geochim. Cosmochim. Acta 2008, 72, 1381–1395. [Google Scholar] [CrossRef]
- Ma, J.F.; Yamaji, N.; Mitani, N.; Tamai, K.; Konishi, S.; Fujiwara, T.; Katsuhara, M.; Yano, M. An efflux transporter of silicon in rice. Nature 2007, 448, 209. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.F.; Yamaji, N.; Mitani-Ueno, N. Transport of silicon from roots to panicles in plants. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2011, 87, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Mitani, N.; Yamaji, N.; Ma, J.F. HvLsi1 is a silicon influx transporter in barley. Plant J. 2009, 57, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Mitani, N.; Yamaji, N.; Ma, J.F. Identification of Maize Silicon Influx Transporters. Plant Cell Physiol. 2008, 50, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, N.; Mitatni, N.; Ma, J.F. A Transporter Regulating Silicon Distribution in Rice Shoots. Plant Cell 2008, 20, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, N.; Chiba, Y.; Mitani-Ueno, N.; Feng Ma, J. Functional Characterization of a Silicon Transporter Gene Implicated in Silicon Distribution in Barley. Plant Physiol. 2012, 160, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Currie, H.A.; Perry, C.C. Silica in plants: Biological, biochemical and chemical studies. Ann. Bot. 2007, 100, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.; Leishman, M.R. Is plant ecology more siliceous than we realise? Trends Plant Sci. 2011, 16, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lux, A.; Luxová, M.; Abe, J.; Tanimoto, E.; Hattori, T.; Inanaga, S. The dynamics of silicon deposition in the sorghum root endodermis. New Phytol. 2003, 158, 437–441. [Google Scholar] [CrossRef]
- Keller, C.; Rizwan, M.; Davidian, J.-C.; Pokrovsky, O.S.; Bovet, N.; Chaurand, P.; Meunier, J.-D. Effect of silicon on wheat seedlings (Triticum turgidum L.) grown in hydroponics and exposed to 0 to 30 µM Cu. Planta 2015, 241, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Sauer, D.; Saccone, L.; Conley, D.J.; Herrmann, L.; Sommer, M. Review of methodologies for extracting plant-available and amorphous Si from soils and aquatic sediments. Biogeochemistry 2006, 80, 89–108. [Google Scholar] [CrossRef]
- Shakoor, S.A.; Bhat, M.A.; Mir, S.H. Phytoliths in plants: A review. Res. Rev. J. Bot. Sci. 2014, 3, 10–24. [Google Scholar]
- Hodson, M.J.; Sangster, A.G. Techniques for the microanalysis of higher plants with particular reference to silicon in cryofixed wheat tissues. Scanning Microsc. 1990, 4, 407–418. [Google Scholar]
- Guntzer, F.; Keller, C.; Meunier, J.-D. Benefits of plant silicon for crops: A review. Agron. Sustain. Dev. 2012, 32, 201–213. [Google Scholar] [CrossRef]
- Li, Z.; Song, Z.; Cornelis, J.-T. Impact of rice cultivar and organ on elemental composition of phytoliths and the release of bio-available silicon. Front. Plant Sci. 2014, 5, 529. [Google Scholar] [CrossRef] [PubMed]
- Balec, R.; Belanger, R.; Chapman, D.M.; Epstein, E.; Guevel, M.H.; Kinrade, S.D.; Knight, C.T.G.; Rains, D.W.; Terill, M.; Wang, J. Organosilicate chemistry: Evidence for a crosslinking role in plants. In III Silicon in Agriculture Conference. Ed Uberlândia; Federal University of Uberlandia: Uberlandia, Brazil, 2005; Volume 76. [Google Scholar]
- Currie, H.A.; Perry, C.C. Chemical evidence for intrinsic “Si” within Equisetum cell walls. Phytochemistry 2009, 70, 2089–2095. [Google Scholar] [CrossRef] [PubMed]
- Inanaga, S.; Okasaka, A. Calcium and silicon binding compounds in cell walls of rice shoots. Soil Sci. Plant Nutr. 1995, 41, 103–110. [Google Scholar] [CrossRef]
- Fang, J.; Ma, X. In vitro simulation studies of silica deposition induced by lignin from rice. J. Zhejiang Univ. Sci. B 2006, 7, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Law, C.; Exley, C. New insight into silica deposition in horsetail (Equisetum arvense). BMC Plant Biol. 2011, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Fleck, A.T.; Schulze, S.; Hinrichs, M.; Specht, A.; Waßmann, F.; Schreiber, L.; Schenk, M.K. Silicon promotes exodermal Casparian band formation in Si-accumulating and Si-excluding species by forming phenol complexes. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, L.; Liu, J.; Liu, X.; Li, X.; Ma, J.; Lin, Y.; Xu, F. Evidence for ‘silicon’within the cell walls of suspension-cultured rice cells. New Phytol. 2013, 200, 700–709. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Ma, J.; Wang, L. A hemicellulose-bound form of silicon with potential to improve the mechanical properties and regeneration of the cell wall of rice. New Phytol. 2015, 206, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Funakawa, H.; Miwa, K. Synthesis of borate cross-linked rhamnogalacturonan II. Front. Plant Sci. 2015, 6, 223. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.; Lerch, S.J.L.; Shrestha, R.P. Understanding diatom cell wall silicification—Moving forward. Front. Mar. Sci. 2018, 5, 125. [Google Scholar] [CrossRef]
- Otzen, D. The role of proteins in biosilicification. Scientifica (Cairo) 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Seaborn, C.D.; Nielsen, F.H. Silicon deprivation decreases collagen formation in wounds and bone, and ornithine transaminase enzyme activity in liver. Biol. Trace Elem. Res. 2002, 89, 251–261. [Google Scholar] [CrossRef]
- Ratcliffe, S.; Jugdaohsingh, R.; Vivancos, J.; Marron, A.; Deshmukh, R.; Ma, J.F.; Mitani-Ueno, N.; Robertson, J.; Wills, J.; Boekschoten, M. V Identification of a mammalian silicon transporter. Am. J. Physiol. Physiol. 2017, 312, C550–C561. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K. A bound form of silicon in glycosaminoglycans and polyuronides. Proc. Natl. Acad. Sci. USA 1973, 70, 1608–1612. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, E.M. Proceedings: Silicon as an essential element. In Federation Proceedings; Federation of American Societies for Experimental Biology: Bethesda, MA, USA, 1974; Volume 33, p. 1758. [Google Scholar]
- Carlisle, E.M. In vivo requirement for silicon in articular cartilage and connective tissue formation in the chick. J. Nutr. 1976, 106, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, E.M. A silicon requirement for normal skull formation in chicks. J. Nutr. 1980, 110, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Schröer, H.C.; Krasko, A.; Le Pennec, G.; Adell, T.; Wiens, M.; Hassanein, H.; Müller, I.M.; Müller, W.E.G. Silicase, an enzyme which degrades biogenous amorphous silica: Contribution to the metabolism of silica deposition in the demosponge Suberites domuncula. In Silicon Biomineralization; Springer: Berlin/Heidelberg, Germany, 2003; pp. 249–268. [Google Scholar]
- Shimizu, K.; Morse, D.E. Silicatein: A unique silica-synthesizing catalytic triad hydrolase from marine sponge skeletons and its multiple applications. In Methods in Enzymology; Elsevier: Cambridge, MA, USA, 2018; Volume 605, pp. 429–455. ISBN 0076-6879. [Google Scholar]
- Fegley, B. Carbon chemistry and organic compound synthesis in the solar nebula. Meteoritics 1987, 22, 378. [Google Scholar]
- Gladstone, G.R.; Towe, K.M.; Kasting, J. Photochemistry in the primitive solar nebula. Science (80-) 1993, 261, 1058–1060. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hanon, P.; Chaussidon, M.; Robert, F. The redox state of the solar nebula: C and H concentrations in chondrules. Meteorit. Planet. Sci. Suppl. 1996, 31, A57. [Google Scholar]
- Llorca, J. Gas-grain chemistry of carbon in interplanetary dust particles- Kinetics and mechanism of hydrocarbon formation. Lunar Planet. Sci. XXIX 1998, 29, 1119. [Google Scholar]
- Varela, M.E.; Metrich, N. Carbon in olivines of chondritic meteorites. Geochim. Cosmochim. Acta 2000, 64, 3433–3438. [Google Scholar] [CrossRef]
- McElroy, D.; Walsh, C.; Markwick, A.J.; Cordiner, M.A.; Smith, K.; Millar, T.J. The UMIST database for astrochemistry 2012. Astron. Astrophys. 2013, 550, A36. [Google Scholar] [CrossRef]
- Umeki, H.; Nakajima, M.; Endo, Y. Laboratory detections of SiC2N and SiC3N by Fourier transform microwave spectroscopy. J. Chem. Phys. 2014, 141, 184303. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.C.; Gottlieb, C.A.; Thaddeus, P. Silicon molecules in space and in the laboratory. Mol. Phys. 2003, 101, 697–704. [Google Scholar] [CrossRef][Green Version]
- Cernicharo, J.; McCarthy, M.C.; Gottlieb, C.A.; Agúndez, M.; Prieto, L.V.; Baraban, J.H.; Changala, P.B.; Guélin, M.; Kahane, C.; Martin-Drumel, M.A. Discovery of SiCSi in IRC+ 10216: A missing link between gas and dust carriers of Si–C bonds. Astrophys. J. Lett. 2015, 806, L3. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; Fonti, S.; Orofino, V. The 4.6 micron feature of–SiH groups in silicate dust grains and infrared cometary spectra. Planet. Space Sci. 1999, 47, 781–785. [Google Scholar] [CrossRef]
- Cernicharo, J.; Waters, L.; Decin, L.; Encrenaz, P.; Tielens, A.; Agúndez, M.; De Beck, E.; Müller, H.S.P.; Goicoechea, J.R.; Barlow, M.J. A high-resolution line survey of IRC+ 10216 with Herschel/HIFI-First results: Detection of warm silicon dicarbide (SiC). Astron. Astrophys. 2010, 521, L8. [Google Scholar] [CrossRef]
- Velilla Prieto, L.; Cernicharo, J.; Quintana-Lacaci, G.; Agúndez, M.; Castro-Carrizo, A.; Fonfría, J.P.; Marcelino, N.; Zúñiga, J.; Requena, A.; Bastida, A. Si-bearing Molecules Toward IRC+ 10216: ALMA Unveils the Molecular Envelope of CWLeo. Astrophys. J. 2015, 805. [Google Scholar] [CrossRef]
- Fonfría, J.P.; Cernicharo, J.; Richter, M.J.; Fernández-López, M.; Velilla Prieto, L.; Lacy, J.H. The abundance of 28Si32S, 29Si32S, 28Si34S, and 30Si32S in the inner layers of the envelope of IRC+ 10216. Mon. Not. R. Astron. Soc. 2015, 453, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Agúndez, M.; Fonfría, J.P.; Cernicharo, J.; Kahane, C.; Daniel, F.; Guélin, M. Molecular abundances in the inner layers of IRC+ 10216. Astron. Astrophys. 2012, 543, A48. [Google Scholar] [CrossRef]
- Guélin, M.; Muller, S.; Cernicharo, J.; McCarthy, M.C.; Thaddeus, P. Detection of the SiNC radical in IRC+ 10216. Astron. Astrophys. 2004, 426, L49–L52. [Google Scholar] [CrossRef]
- Guélin, M.; Muller, S.; Cernicharo, J.; Apponi, A.J.; McCarthy, M.C.; Gottlieb, C.A.; Thaddeus, P. Astronomical detection of the free radical SiCN. Astron. Astrophys. 2000, 363, L9–L12. [Google Scholar]
- Cernicharo, J.; Agúndez, M.; Prieto, L.V.; Guélin, M.; Pardo, J.R.; Kahane, C.; Marka, C.; Kramer, C.; Navarro, S.; Quintana-Lacaci, G. Discovery of methyl silane and confirmation of silyl cyanide in IRC+ 10216. Astron. Astrophys. 2017, 606, L5. [Google Scholar] [CrossRef] [PubMed]
- Leung, I.; Guo, W.; Friedman, I.; Gleason, J. Natural occurrence of silicon carbide in a diamondiferous kimberlite from Fuxian. Nature 1990, 346, 352. [Google Scholar] [CrossRef]
- Mathez, E.A.; Fogel, R.A.; Hutcheon, I.D.; Marshintsev, V.K. Carbon isotopic composition and origin of SiC from kimberlites of Yakutia, Russia. Geochim. Cosmochim. Acta 1995, 59, 781–791. [Google Scholar] [CrossRef]
- Bernatowicz, T.J.; Amari, S.; Lewis, R.S. Refractory carbides in interstellar graphite. In Proceedings of the Lunar and Planetary Science Conference, Houston, TX, USA, 14–18 March 1994; Lunar and Planetary Institute: Houston, TX, USA, 1994; Volume 25, p. 103. [Google Scholar]
- Lee, M.R.; Russell, S.S.; Arden, J.W.; Pillinger, C.T. The isotopic composition and mineralogy of silicon nitride (Si3N4) within ordinary and enstatite chondrites. Meteoritics 1992, 27, 248–249. [Google Scholar]
- Travaglio, C.; Gallino, R.; Amari, S.; Zinner, E.; Woosley, S.; Lewis, R.S. Low-density graphite grains and mixing in type II supernovae. Astrophys. J. 1999, 510, 325. [Google Scholar] [CrossRef]
- Hoppe, P.; Strebel, R.; Eberhardt, P.; Amari, S.; Lewis, R.S. Small SiC grains and a nitride grain of circumstellar origin from the Murchison meteorite: Implications for stellar evolution and nucleosynthesis. Geochim. Cosmochim. Acta 1996, 60, 883–907. [Google Scholar] [CrossRef]
- Ebel, D.S.; Grossman, L. Condensation from supernova gas made of free atoms1. Geochim. Cosmochim. Acta 2001, 65, 469–477. [Google Scholar] [CrossRef]
- Zbik, M.; Jasieniak, M.; SMART, R.S.C. Organosilane occurrence in irghizite samples from the Zhamanshin impact crater, Kazakhstan. Meteorit. Planet. Sci. 2000, 35, 943–947. [Google Scholar] [CrossRef]
- Baus, J.A.; Burschka, C.; Bertermann, R.; Guerra, C.F.; Bickelhaupt, F.M.; Tacke, R. Neutral six-coordinate and cationic five-coordinate silicon (iv) complexes with two bidentate monoanionic n, s-pyridine-2-thiolato (−) ligands. Inorg. Chem. 2013, 52, 10664–10676. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C.; McEuen, P.; McEuen, P. Introduction to Solid State Physics; Wiley: New York, NY, USA, 1996; Volume 8. [Google Scholar]
- Ashcroft, N.W.; Mermin, N.D. Solid state physics. Saunders Coll. Phila. 1976, 120, 1–848. [Google Scholar]
- Muthukumaran, N.; Velappan, K.; Gour, K.; Prabusankar, G. N-heterocyclic carbene supported halosilylenes: New frontiers in an emerging field. Coord. Chem. Rev. 2018, 377, 1–43. [Google Scholar] [CrossRef]
- Abersfelder, K.; White, A.J.P.; Rzepa, H.S.; Scheschkewitz, D. A tricyclic aromatic isomer of hexasilabenzene. Science (80-) 2010, 327, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, A.; Kinjo, R.; Ichinohe, M. A stable compound containing a silicon-silicon triple bond. Science (80-) 2004, 305, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, T.; Guo, J.-D.; Hashizume, D.; Sasamori, T.; Nagase, S.; Tokitoh, N. The selective formation of a 1, 2-disilabenzene from the reaction of a disilyne with phenylacetylene. Dalt. Trans. 2018, 47, 13318–13322. [Google Scholar] [CrossRef] [PubMed]
- Han, J.S.; Sasamori, T.; Mizuhata, Y.; Tokitoh, N. Reactivity of an aryl-substituted silicon–silicon triple bond: 1, 2-disilabenzenes from the reactions of a 1, 2-diaryldisilyne with alkynes. Dalt. Trans. 2010, 39, 9238–9240. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, R.; Ichinohe, M.; Sekiguchi, A.; Takagi, N.; Sumimoto, M.; Nagase, S. Reactivity of a Disilyne RSi≍ SiR (R=Si i Pr [CH (SiMe3) 2] 2) toward π-Bonds: Stereospecific Addition and a New Route to an Isolable 1, 2-Disilabenzene. J. Am. Chem. Soc. 2007, 129, 7766–7767. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.; Seager, S.; Petkowski, J.J.; Sousa-Silva, C.; Ranjan, S.; Huang, J.; Bains, W. Assessment of Isoprene as a Possible Biosignature Gas in Exoplanets with Anoxic Atmospheres. Astrobiology 2020. in review. [Google Scholar]
- Blom, B.; Driess, M. Functional Molecular Silicon Compounds II; Springer: Berlin, Germany, 2014. [Google Scholar]
- Maxka, J.; Huang, L.M.; West, R. Synthesis and NMR spectroscopy of permethylpolysilane oligomers Me (SiMe2) 10Me, Me (SiMe2) 16Me, and Me (Me2Si) 22Me. Organometallics 1991, 10, 656–659. [Google Scholar] [CrossRef]
- Fujino, M. Photoconductivity in organopolysilanes. Chem. Phys. Lett. 1987, 136, 451–453. [Google Scholar] [CrossRef]
- Koe, J.; Fujiki, M. Chapter 6—Polysilanes. In Organosilicon Compounds; Lee, V.Y., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 219–300. ISBN 978-0-12-814213-4. [Google Scholar]
- Dahn, J.R.; Way, B.M.; Fuller, E.; Tse, J.S. Structure of siloxene and layered polysilane (Si 6 H 6). Phys. Rev. B 1993, 48, 17872. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Ando, W.; Chojnowski, J. Silicon-Containing Polymers: The Science and Technology of Their Synthesis and Applications; Springer Science & Business Media: Berlin, Germany, 2013; ISBN 9401139393. [Google Scholar]
- Ranjan, S.; Wordsworth, R.; Sasselov, D.D. The surface UV environment on planets orbiting M dwarfs: Implications for prebiotic chemistry and the need for experimental follow-up. Astrophys. J. 2017, 843, 110. [Google Scholar] [CrossRef]
- Bond, J.C.; O’Brien, D.P.; Lauretta, D.S. The compositional diversity of extrasolar terrestrial planets. I. In situ simulations. Astrophys. J. 2010, 715, 1050. [Google Scholar] [CrossRef]
- Öberg, K.I.; Murray-Clay, R.; Bergin, E.A. The effects of snowlines on C/O in planetary atmospheres. Astrophys. J. Lett. 2011, 743, L16. [Google Scholar] [CrossRef]
- Booth, R.A.; Clarke, C.J.; Madhusudhan, N.; Ilee, J.D. Chemical enrichment of giant planets and discs due to pebble drift. Mon. Not. R. Astron. Soc. 2017, 469, 3994–4011. [Google Scholar] [CrossRef]
- Fortney, J.J. On the carbon-to-oxygen ratio measurement in nearby Sun-like stars: Implications for planet formation and the determination of stellar abundances. Astrophys. J. Lett. 2012, 747, L27. [Google Scholar] [CrossRef]
- Whitehouse, L.J.; Farihi, J.; Green, P.J.; Wilson, T.G.; Subasavage, J.P. Dwarf carbon stars are likely metal-poor binaries and unlikely hosts to carbon planets. Mon. Not. R. Astron. Soc. 2018, 479, 3873–3878. [Google Scholar] [CrossRef]
- Nissen, P.E. The carbon-to-oxygen ratio in stars with planets. Astron. Astrophys. 2013, 552, A73. [Google Scholar] [CrossRef]
- Bergfors, C.; Farihi, J. Do C/O> 1 main-sequence stars build carbon planets. In AAS/Division for Extreme Solar Systems Abstracts; American Astronomical Society: Washington, DC, USA, 2015; Volume 3. [Google Scholar]
- Wilson, D.J.; Gänsicke, B.T.; Farihi, J.; Koester, D. Carbon to oxygen ratios in extrasolar planetesimals. Mon. Not. R. Astron. Soc. 2016, 459, 3282–3286. [Google Scholar] [CrossRef]
- Madhusudhan, N.; Mousis, O.; Johnson, T.V.; Lunine, J.I. Carbon-rich giant planets: Atmospheric chemistry, thermal inversions, spectra, and formation conditions. Astrophys. J. 2011, 743, 191. [Google Scholar] [CrossRef]
- Kidokoro, Y.; Umemoto, K.; Hirose, K.; Ohishi, Y. Phase transition in SiC from zinc-blende to rock-salt structure and implications for carbon-rich extrasolar planets. Am. Mineral. 2017, 102, 2230–2234. [Google Scholar] [CrossRef]
- Miozzi, F.; Morard, G.; Antonangeli, D.; Clark, A.N.; Dorn, C.; Antoine, R.; Mezouar, M.; Baron, M.A.; Pakhomova, A.; Fiquet, G. An experimental approach to investigate carbon rich exoplanets interior. In Proceedings of the European Planetary Science Congress, Berlin, Germany, 16–21 September 2018; Volume 12. [Google Scholar]
- Miozzi, F.; Morard, G.; Antonangeli, D.; Clark, A.N.; Mezouar, M.; Dorn, C.; Rozel, A.; Fiquet, G. Equation of State of SiC at Extreme Conditions: New Insight Into the Interior of Carbon-Rich Exoplanets. J. Geophys. Res. Planets 2018, 123, 2295–2309. [Google Scholar] [CrossRef]
- Lodders, K.; Fegley Jr, B. Condensation chemistry of carbon stars. In AIP Conference Proceedings; American Institute of Physics: Melville, NY, USA, 1997; Volume 402, pp. 391–423. [Google Scholar]
- Wilson, H.F.; Militzer, B. Interior phase transformations and mass-radius relationships of silicon-carbon planets. Astrophys. J. 2014, 793, 34. [Google Scholar] [CrossRef]
- Futó, P.; Gucsik, A. Basic Mineralogical Models for Silicate-and Carbon-Rich Mega-Earths Considering Compositional and Geophysical Constraints. In Proceedings of the Lunar and Planetary Science Conference, The Woodlands, TX, USA, 9–23 March 2018; Lunar and Planetary Institute: Houston, TX, USA, 2018; Volume 49. [Google Scholar]
- Hakim, K.; Spaargaren, R.; Grewal, D.S.; Rohrbach, A.; Berndt, J.; Dominik, C.; Van Westrenen, W. Mineralogy, Structure, and Habitability of Carbon-Enriched Rocky Exoplanets: A Laboratory Approach. Astrobiology 2019, 19, 867–884. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, P.B.; Rugheimer, S. Hydrogen cyanide in nitrogen-rich atmospheres of rocky exoplanets. Icarus 2019, 329, 124–131. [Google Scholar] [CrossRef]
- Rase, H.F. Handbook of Commercial Catalysts: Heterogeneous Catalysts; CRC Press: Boca Raton, FL, USA, 2000; ISBN 1420036548. [Google Scholar]
- Singh, S.K.; Parida, K.M.; Mohanty, B.C.; Rao, S.B. High surface area silicon carbide from rice husk: A support material for catalysts. React. Kinet. Catal. Lett. 1995, 54, 29–34. [Google Scholar] [CrossRef]
- Bosque, R.; Sales, J. Polarizabilities of solvents from the chemical composition. J. Chem. Inf. Comput. Sci. 2002, 42, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.C. The estimation of solubility parameters and related properties of liquids. J. Chem. Technol. Biotechnol. Chem. Technol. 1984, 34, 38–42. [Google Scholar] [CrossRef]
- Abraham, M.H.; Platts, J.A. Hydrogen bond structural group constants. J. Org. Chem. 2001, 66, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology; International Union of Crystal: Oxford, UK, 2001; Volume 9, ISBN 0198509707. [Google Scholar]
- Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. CH/π hydrogen bonds in organic and organometallic chemistry. CrystEngComm 2009, 11, 1757–1788. [Google Scholar] [CrossRef]








| Feature | Carbon | Silicon |
|---|---|---|
| Electronic Configuration | C=1s2 2s2 2p2 | Si=1s2 2s2 2p6 3s2 3p2 |
| Covalent Radius | 77 pm | 117 pm |
| Coordination Numbers for Stable Compounds | 1, 2, 3, 4 | 3, 4, 5, 6 |
| Pauling Scale Electronegativity | 2.50 [Cδ-←Hδ+] | 1.8 [Siδ+→Hδ-] |
| Bond Energies (D) and Lengths of Biologically Important Bonds and Their Si Equivalents | C–C: 346 kJ/mol; 154 pm | Si–Si: 222 kJ/mol; 233 pm |
| C–O: 358 kJ/mol; 143 pm | Si–O: 452 kJ/mol; 163 pm | |
| C–N: 305 kJ/mol; 147 pm | Si–N: 355 kJ/mol; | |
| C–S: 272 kJ/mol; 182 pm | Si–S: 293 kJ/mol; 200 pm | |
| C–H: 411 kJ/mol; 109 pm | Si–H: 318 kJ/mol; 148 pm | |
| C–Si: 318 kJ/mol; 185 pm (longer and slightly weaker than C–C) | ||
| Bonding Geometry | e.g., N(CH3)3; pyramidal | e.g., N(SiH3)3; planar |
| Lipophilicity | Log P of PhCMe3: 4.0 | Log P of PhSiMe3: 4.7 |
| Chemical Properties of Selected Functional Groups | C–H; stable in aqueous solution | Si–H; susceptible to hydrolysis, especially under basic conditions (rate strongly dependent on substituents on Si and pH) |
| Si–C; stable in aqueous solution; useful pharmacological properties | ||
| C–O–C; stable in aqueous solution | Si–O–C; susceptible to hydrolysis (rate strongly dependent on substituents on Si and pH) | |
| C–OH; stable towards condensation, lower acidity | Si–OH; stable, but liable to condensation (rate strongly dependent on substituents on Si and pH), higher acidity | |
| C–N; stable in aqueous solution | Si–N; susceptible to hydrolysis, especially under acidic conditions (rate strongly dependent on substituents on Si and pH) | |
| C–S; stable in aqueous solution | Si–S; susceptible to hydrolysis | |
| Multiple Bonds * | C=C, C#C, C:C; stable in aqueous solution | Si=Si, Si#Si; few examples of Si=Si and Si#Si known, highly reactive and unstable in aqueous solution; Si:Si; only one hexasilicone system with dismutational aromaticity is known. Remarkably it is relatively air- and thermostable (<200 °C). |
| Si=C and Si#C bonds are known, although Si=C bonds are very unstable under almost all conditions; very few Si:C silabenzene systems are stable | ||
| Penta- and Hexacoordinate Systems | Unstable | Well known and stable (in some cases even in aqueous solution) as charged and uncharged species |
| Silicon Compound | Carbon Analogue | ΔG° Formation kJ/mol (298 K) | ΔG Formation from XO2, H2O, N2, HCl, HF, H3PO4 + H2 (Standard State) | ||
|---|---|---|---|---|---|
| Silicon | Carbon | Silicon | Carbon | ||
| Silane SiH4 | Methane CH4 | 57.2 | −51.12 | 434.17 | −141.62 |
| Disilane Si2H6 | Ethane C2H6 | 127.07 | −32.25 | 880.99 | −213.23 |
| Trisilane Si3H8 | Propane C3H8 | 185.18 | −23.84 | 1316.06 | −295.32 |
| Tetramethylsilane SiC4H10 | Neopentane C5H12 | −96.13 | −22.35 | −81.14 | −474.8 |
| Diethylsilane SiC4H10 | n-pentane C5H12 | −46.96 | −8.24 | −31.96 | −460.69 |
| Trimethylsilanol (CH3)3SiOH | Tert-butanol (CH3)3COH | −372.64 | −175.46 | −28.81 | −299.08 |
| Chlorosilane SiH3Cl | Chloromethane CH3Cl | −116.104 | −59.88 | −356.08 | −55.15 |
| Hexachlorodisilane Si2Cl6 | Hexachlorethane C2Cl6 | −970.08 | −57.61 | 355.16 | 332.73 |
| Silicon tetrafluoride SiF4 | Carbon tetrafluoride CF4 | −1572.71 | −888.51 | −444.42 | −227.67 |
| Trimethylsilanamine (CH3)3SiNH2 | Tert-butylamine (CH3)3CNH2 | −146.59 | 28.49 | −41.10 | −333.47 |
| Silylphosphane PSiH5 | Methylphosphane CH5P | 22.76 | −8.29 | 604.18 | 94.68 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petkowski, J.J.; Bains, W.; Seager, S. On the Potential of Silicon as a Building Block for Life. Life 2020, 10, 84. https://doi.org/10.3390/life10060084
Petkowski JJ, Bains W, Seager S. On the Potential of Silicon as a Building Block for Life. Life. 2020; 10(6):84. https://doi.org/10.3390/life10060084
Chicago/Turabian StylePetkowski, Janusz Jurand, William Bains, and Sara Seager. 2020. "On the Potential of Silicon as a Building Block for Life" Life 10, no. 6: 84. https://doi.org/10.3390/life10060084
APA StylePetkowski, J. J., Bains, W., & Seager, S. (2020). On the Potential of Silicon as a Building Block for Life. Life, 10(6), 84. https://doi.org/10.3390/life10060084