Bird Eye View of Protein Subcellular Localization Prediction



Abstract
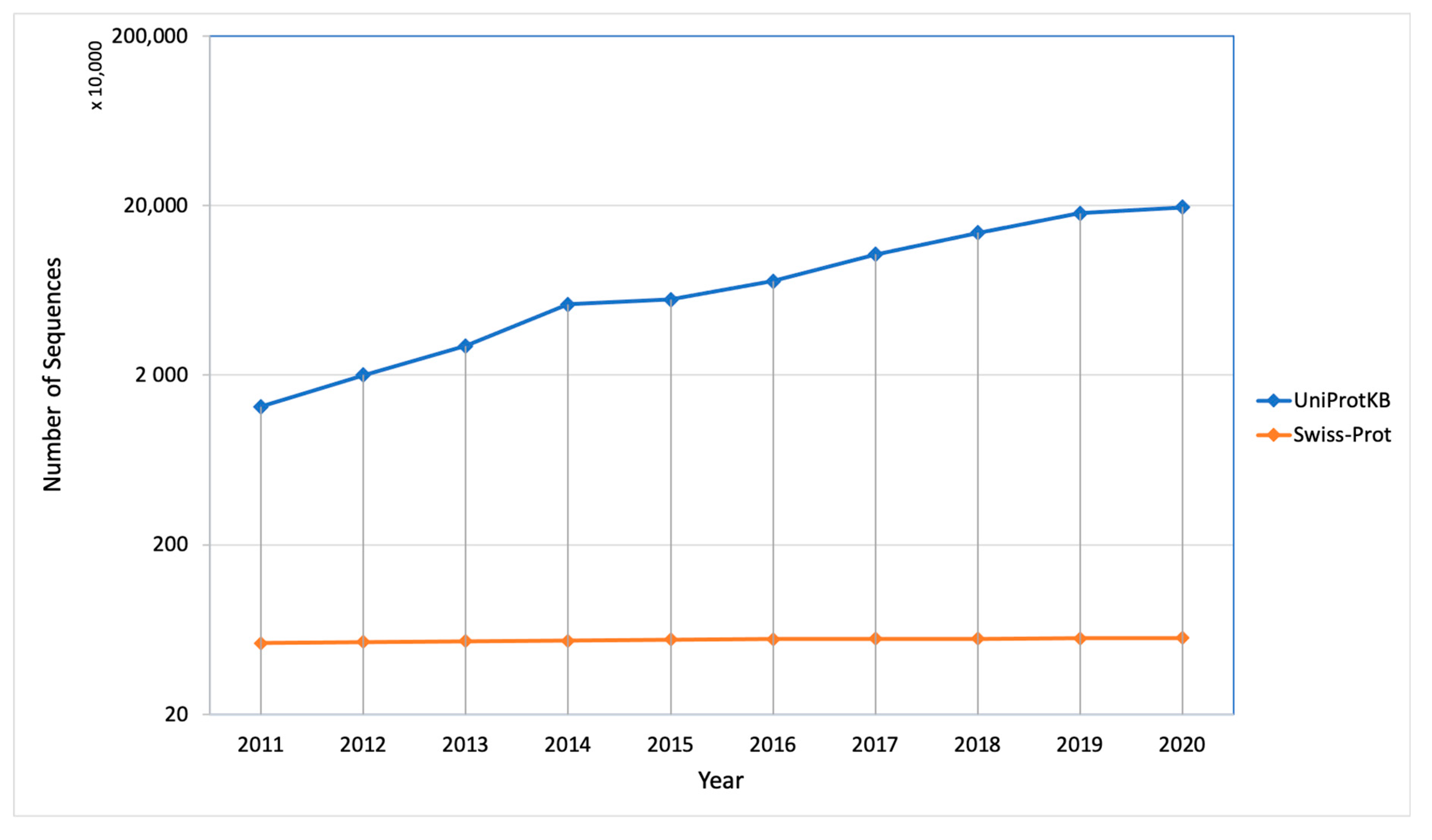
1. Introduction
2. Experimental Approaches for Protein Localization
3. Computational Approaches for Protein Localization
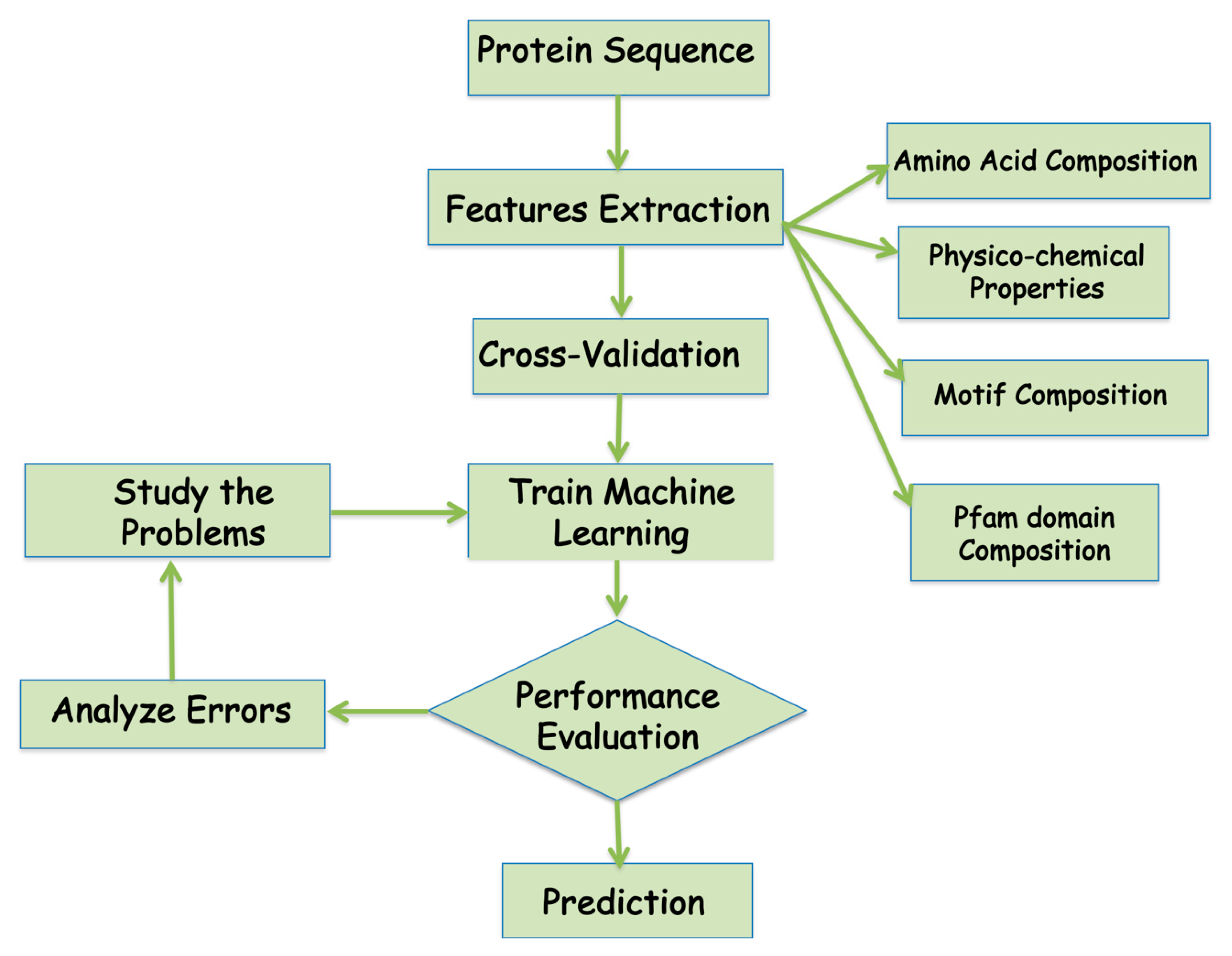
3.1. Sequence Feature-Based Method
3.2. Homology Based Method
3.3. Functional Motifs, Domains, and other Signatures Based Method
3.4. Signal Peptide Based Method
3.5. Non-Sequence Derived Features
3.6. Integrated Method
4. Machine Learning Tools Used in Protein Prediction
4.1. Support Vector Machine
4.2. Random Forest
4.3. Neural Network and Deep Learning
5. Importance and Future Aspects of Protein Subcellular Localization
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harper, J.W.; Bennett, E.J. Proteome complexity and the forces that drive proteome imbalance. Nature 2016, 537, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, J.; Nabil, M.M.; Zhang, J. Deep Forest-based Prediction of Protein Subcellular Localization. Curr. Gene Ther. 2018, 18, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.-Z.; Wu, Y.; Gao, Q.-Z.; Zhao, L.; Xu, Y.-Y. Automated classification of protein subcellular localization in immunohistochemistry images to reveal biomarkers in colon cancer. BMC Bioinform. 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cai, L.; Liao, B.; Fu, X.; Bing, P.; Yang, J. Prediction of Protein Subcellular Localization Based on Fusion of Multi-view Features. Molecules 2019, 24, 919. [Google Scholar] [CrossRef]
- Mooney, C.; Wang, Y.; Pollastri, G. SCLpred: Protein subcellular localization prediction by N-to-1 neural networks. Bioinformatics 2011, 27, 2812–2819. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Nielsen, H.; Von Heijne, G. ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci. 1999, 8, 978–984. [Google Scholar] [CrossRef]
- Kumar, R.; Jain, S.; Kumari, B.; Kumar, M. Protein Sub-Nuclear Localization Prediction Using SVM and Pfam Domain Information. PLoS ONE 2014, 9, e98345. [Google Scholar] [CrossRef]
- Kumar, M.; Raghava, G. Prediction of nuclear proteins using SVM and HMM models. BMC Bioinform. 2009, 10, 22. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, P.; Luo, J.; Jiang, Y. Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm. Genome 2003, 14, 859–865. [Google Scholar] [CrossRef]
- Li, G.-P.; Du, P.-F.; Shen, Z.-A.; Liu, H.-Y.; Luo, T. DPPN-SVM: Computational Identification of Mis-Localized Proteins in Cancers by Integrating Differential Gene Expressions With Dynamic Protein-Protein Interaction Networks. Front. Genet. 2020, 11, 600454. [Google Scholar] [CrossRef]
- Kumar, R.; Kumari, B.; Kumar, M. Proteome-wide prediction and annotation of mitochondrial and sub-mitochondrial proteins by incorporating domain information. Mitochondrion 2018, 42, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Singhal, N.; Kumar, R.; Kumar, M. mRNALoc: A novel machine-learning based in-silico tool to predict mRNA subcellular localization. Nucleic Acids Res. 2020, 48, W239–W243. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Kaleel, M.; Zheng, Y.; Chen, J.; Feng, X.; Simpson, J.C.; Pollastri, G.; Mooney, C. SCLpred-EMS: Subcellular localization prediction of endomembrane system and secretory pathway proteins by Deep N-to-1 Convolutional Neural Networks. Bioinformatics 2020, 36, 3343–3349. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Bruciaferri, N.; Tartari, G.; Martelli, P.L.; Casadio, R. DeepMito: Accurate prediction of protein sub-mitochondrial localization using convolutional neural networks. Bioinformatics 2020, 36, 56–64. [Google Scholar] [CrossRef]
- Lv, Z.; Jin, S.; Ding, H.; Zou, Q. A Random Forest Sub-Golgi Protein Classifier Optimized via Dipeptide and Amino Acid Composition Features. Front. Bioeng. Biotechnol. 2019, 7, 215. [Google Scholar] [CrossRef]
- Yu, B.; Qiu, W.; Chen, C.; Ma, A.; Jiang, J.; Zhou, H.; Ma, Q. SubMito-XGBoost: Predicting protein submitochondrial localization by fusing multiple feature information and eXtreme gradient boosting. Bioinformatics 2020, 36, 1074–1081. [Google Scholar] [CrossRef]
- Nishikawa, K.; Ooi, T. Correlation of the Amino Acid Composition of a Protein to Its Structural and Biological Characters1. J. Biochem. 1982, 91, 1821–1824. [Google Scholar] [CrossRef]
- Nishikawa, K.; Kubota, Y.; Ooi, T. Classification of Proteins into Groups Based on Amino Acid Composition and Other Characters. II. Grouping into Four Types. J. Biochem. 1983, 94, 997–1007. [Google Scholar] [CrossRef]
- Behbahani, M.; Nosrati, M.; Moradi, M.; Mohabatkar, H. Using Chou’s General Pseudo Amino Acid Composition to Classify Laccases from Bacterial and Fungal Sources via Chou’s Five-Step Rule. Appl. Biochem. Biotechnol. 2020, 190, 1035–1048. [Google Scholar] [CrossRef]
- Kumar, R.; Kumari, B.; Kumar, M. Prediction of endoplasmic reticulum resident proteins using fragmented amino acid composition and support vector machine. PeerJ 2017, 5, e3561. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Cozzetto, D.; Jones, D.T. Computational Methods for Annotation Transfers from Sequence. In The Gene Ontology Handbook Methods in Molecular Biology; Dessimoz, C.Š.N., Ed.; Humana Press: New York, NY, USA, 2017. [Google Scholar]
- Nair, R.; Rost, B. Sequence conserved for subcellular localization. Protein Sci. 2002, 11, 2836–2847. [Google Scholar] [CrossRef] [PubMed]
- Silver, P.A.; Chiang, A.; Sadler, I. Mutations that alter both localization and production of a yeast nuclear protein. Genes Dev. 1988, 2, 707–717. [Google Scholar] [CrossRef]
- Freeman, B.T.; Sokolowski, M.; Roy-Engel, A.M.; Smither, M.E.; Belancio, V.P. Identification of charged amino acids required for nuclear localization of human L1 ORF1 protein. Mob. DNA 2019, 10, 20. [Google Scholar] [CrossRef]
- Laurila, K.; Vihinen, M. Prediction of disease-related mutations affecting protein localization. BMC Genom. 2009, 10, 122. [Google Scholar] [CrossRef]
- Nakai, K.; Horton, P. Computational Prediction of Subcellular Localization. Methods Mol. Biol. 2007, 390, 429–466. [Google Scholar] [CrossRef]
- Loewenstein, Y.; Raimondo, D.; Redfern, O.C.; Watson, J.; Frishman, D.; Linial, M.; Orengo, C.; Thornton, J.; Tramontano, A. Protein function annotation by homology-based inference. Genome Biol. 2009, 10, 1–8. [Google Scholar] [CrossRef]
- Mott, R.; Schultz, J.; Bork, P.; Ponting, C.P. Predicting Protein Cellular Localization Using a Domain Projection Method. Genome Res. 2002, 12, 1168–1174. [Google Scholar] [CrossRef]
- Guda, C.; Subramaniam, S. TARGET: A new method for predicting protein subcellular localization in eukaryotes. Bioinformatics 2005, 21, 3963–3969. [Google Scholar] [CrossRef]
- Nair, R.; Rost, B. Mimicking Cellular Sorting Improves Prediction of Subcellular Localization. J. Mol. Biol. 2005, 348, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.A.; Cerutti, L.; Hulo, N.; Gattiker, A.; Falquet, L.; Pagni, M.; Bairoch, A.; Bucher, P. PROSITE: A documented database using patterns and profiles as motif descriptors. Brief. Bioinform. 2002, 3, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Tsirigos, K.D.; Brunak, S.; Von Heijne, G. A Brief History of Protein Sorting Prediction. Protein J. 2019, 38, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Casadio, R. DeepSig: Deep learning improves signal peptide detection in proteins. Bioinformatics 2018, 34, 1690–1696. [Google Scholar] [CrossRef]
- Nielsen, H.; Engelbrecht, J.; Brunak, S.; Von Heijne, G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997, 10, 1–6. [Google Scholar] [CrossRef]
- Nielsen, H.; Krogh, A. Prediction of signal peptides and signal anchors by a hidden Markov model. Proc. Int. Conf. Intell. Syst. Mol. Boil. 1998, 6, 122–130. [Google Scholar]
- Bendtsen, J.D.; Nielsen, H.; Von Heijne, G.; Brunak, S. Improved Prediction of Signal Peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Thoms, S. Import of proteins into peroxisomes: Piggybacking to a new home away from home. Open Biol. 2015, 5, 150148. [Google Scholar] [CrossRef] [PubMed]
- Tessier, T.M.; MacNeil, K.M.; Mymryk, J.S. Piggybacking on Classical Import and Other Non-Classical Mechanisms of Nuclear Import Appear Highly Prevalent within the Human Proteome. Biology 2020, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Nair, R. LOC3D: Annotate sub-cellular localization for protein structures. Nucleic Acids Res. 2003, 31, 3337–3340. [Google Scholar] [CrossRef][Green Version]
- Kumar, A.; Rao, A.; Bhavani, S.; Newberg, J.Y.; Murphy, R.F. Automated analysis of immunohistochemistry images identifies candidate location biomarkers for cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18249–18254. [Google Scholar] [CrossRef]
- Xu, Y.-Y.; Shen, H.-B.; Murphy, R.F. Learning complex subcellular distribution patterns of proteins via analysis of immunohistochemistry images. Bioinformatics 2020, 36, 1908–1914. [Google Scholar] [CrossRef]
- Tahir, M.; Khan, A.; Kaya, H. Protein subcellular localization in human and hamster cell lines: Employing local ternary patterns of fluorescence microscopy images. J. Theor. Biol. 2014, 340, 85–95. [Google Scholar] [CrossRef]
- Xiao, X.; Shao, S.; Ding, Y.; Huang, Z.; Chou, K.-C. Using cellular automata images and pseudo amino acid composition to predict protein subcellular location. Amino Acids 2006, 30, 49–54. [Google Scholar] [CrossRef]
- Garapati, H.S.; Male, G.; Mishra, K. Predicting subcellular localization of proteins using protein-protein interaction data. Genomics 2020, 112, 2361–2368. [Google Scholar] [CrossRef]
- Ryngajłło, M.; Childs, L.H.; Lohse, M.; Giorgi, F.M.; Elude, A.; Selbig, J.; Usadel, B. SLocX: Predicting subcellular localization of Arabidopsis proteins leveraging gene expression data. Front. Plant Sci. 2011, 2, 43. [Google Scholar] [CrossRef]
- Mehrabad, E.M.; Hassanzadeh, R.; Eslahchi, C. PMLPR: A novel method for predicting subcellular localization based on recommender systems. Sci. Rep. 2018, 8, 12006. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Kanehisa, M. Expert system for predicting protein localization sites in gram-negative bacteria. Proteins 1991, 11, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Nakai, K. Better prediction of protein cellular localization sites with the k nearest neighbors classifier. Proc. Int. Conf. Intell. Syst. Mol. Boil. 1997, 5, 147–152. [Google Scholar]
- Bannai, H.; Tamada, Y.; Maruyama, O.; Nakai, K.; Miyano, S. Extensive feature detection of N-terminal protein sorting signals. Bioinformatics 2002, 18, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Spencer, C.; Wang, K.; Ester, M.; Tusnády, G.E.; Simon, I.; Hua, S.; Defays, K.; Lambert, C.; Nakai, K.; et al. PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 2003, 31, 3613–3617. [Google Scholar] [CrossRef]
- Gardy, J.L.; Laird, M.R.; Brinkman, F.S.L.; Chen, F.; Rey, S.; Walsh, C.J.; Ester, M. PSORTb v.2.0: Expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 2005, 21, 617–623. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef]
- Langlois, R.; Frank, J. A clarification of the terms used in comparing semi-automated particle selection algorithms in Cryo-EM. J. Struct. Biol. 2011, 175, 348–352. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, H. Model Cross-Validation. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, H. Negative Predictive Value. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Vapnik, V. The Nature of Statical Learning Theory; Springer: New York, NY, USA, 1995. [Google Scholar]
- Yadav, A.K.; Singla, D. VacPred: Sequence-based prediction of plant vacuole proteins using machine-learning techniques. J. Biosci. 2020, 45, 1–9. [Google Scholar] [CrossRef]
- Kong, Y.; Yu, T. A Deep Neural Network Model using Random Forest to Extract Feature Representation for Gene Expression Data Classification. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.A.; Pujol, T.A.; McDonald, J.; Qiu, P. Leveraging TCGA gene expression data to build predictive models for cancer drug response. BMC Bioinform. 2020, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Adnan, N.; Lei, C.; Ruan, J. Robust edge-based biomarker discovery improves prediction of breast cancer metastasis. BMC Bioinform. 2020, 21, 1–18. [Google Scholar] [CrossRef]
- Guan, X.; Runger, G.; Liu, L. Dynamic incorporation of prior knowledge from multiple domains in biomarker discovery. BMC Bioinform. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- Guo, L.; Wang, Z.; Du, Y.; Mao, J.; Zhang, J.; Yu, Z.; Guo, J.; Zhao, J.; Zhou, H.; Wang, H.; et al. Random-forest algorithm based biomarkers in predicting prognosis in the patients with hepatocellular carcinoma. Cancer Cell Int. 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Tang, J.; Mou, M.; Wang, Y.; Luo, Y.; Zhu, F. MetaFS: Performance assessment of biomarker discovery in metaproteomics. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Yu, X.; Lai, S.; Chen, H.; Chen, M. Protein–protein interaction network with machine learning models and multiomics data reveal potential neurodegenerative disease-related proteins. Hum. Mol. Genet. 2020, 29, 1378–1387. [Google Scholar] [CrossRef]
- Niu, B.; Liang, C.; Lu, Y.; Zhao, M.; Chen, Q.; Zhang, Y.; Zheng, L.; Chou, K.-C. Glioma stages prediction based on machine learning algorithm combined with protein-protein interaction networks. Genomics 2020, 112, 837–847. [Google Scholar] [CrossRef]
- Su, R.; Liu, X.; Wei, L.; Zou, Q. Deep-Resp-Forest: A deep forest model to predict anti-cancer drug response. Methods 2019, 166, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Liu, S.; Chen, J.; Li, X.; Ma, Q.; Yu, B. Predicting drug-target interactions using Lasso with random forest based on evolutionary information and chemical structure. Genomics 2019, 111, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- Lind, A.P.; Anderson, P.C. Predicting drug activity against cancer cells by random forest models based on minimal genomic information and chemical properties. PLoS ONE 2019, 14, e0219774. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, D.; Cao, B.; Carvajal, R.; Kim, M. PDXGEM: Patient-derived tumor xenograft-based gene expression model for predicting clinical response to anticancer therapy in cancer patients. BMC Bioinform. 2020, 21, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Yang, Y.; Shen, H.-B. ImPLoc: A multi-instance deep learning model for the prediction of protein subcellular localization based on immunohistochemistry images. Bioinformatics 2020, 36, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Zhang, Z.; Pan, Z.; Ying, Y.; Xie, Z.; Adhikari, S.; Phillips, J.; Carstens, R.P.; Black, D.L.; Wu, Y.; Xing, Y. Deep-learning augmented RNA-seq analysis of transcript splicing. Nat. Methods 2019, 16, 307–310. [Google Scholar] [CrossRef]
- Guo, Y.; Li, W.; Wang, B.; Liu, H.; Zhou, D. DeepACLSTM: Deep asymmetric convolutional long short-term memory neural models for protein secondary structure prediction. BMC Bioinform. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Wardah, W.; Khan, M.; Sharma, A.; Rashid, M.A. Protein secondary structure prediction using neural networks and deep learning: A review. Comput. Biol. Chem. 2019, 81, 1–8. [Google Scholar] [CrossRef]
- Baptista, D.; Ferreira, P.G.; Rocha, M. Deep learning for drug response prediction in cancer. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- You, J.; McLeod, R.D.; Hu, P. Predicting drug-target interaction network using deep learning model. Comput. Biol. Chem. 2019, 80, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kuenzi, B.M.; Park, J.; Fong, S.H.; Sanchez, K.S.; Lee, J.; Kreisberg, J.F.; Ma, J.; Ideker, T. Predicting Drug Response and Synergy Using a Deep Learning Model of Human Cancer Cells. Cancer Cell 2020, 38, 672–684.e6. [Google Scholar] [CrossRef] [PubMed]
- González, G.; Evans, C.L. Biomedical Image Processing with Containers and Deep Learning: An Automated Analysis Pipeline: Data architecture, artificial intelligence, automated processing, containerization, and clusters orchestration ease the transition from data acquisition to insights in medium-to-large datasets. BioEssays 2019, 41, 1900004. [Google Scholar] [CrossRef]
- Jurtz, V.I.; Johansen, A.R.; Nielsen, M.; Armenteros, J.J.A.; Nielsen, H.; Sønderby, C.K.; Winther, O.; Sønderby, S.K. An introduction to deep learning on biological sequence data: Examples and solutions. Bioinformatics 2017, 33, 3685–3690. [Google Scholar] [CrossRef]
- Ananda, M.M.; Hu, J. NetLoc: Network based protein localization prediction using protein-protein interaction and co-expression networks. In Proceedings of the 2010 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Hong Kong, China, 18–21 December 2010; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2010; pp. 142–148. [Google Scholar]
- Mondal, A.M.; Lin, J.-R.; Hu, J. Network based subcellular localization prediction for multi-label proteins. In Proceedings of the 2011 IEEE International Conference on Bioinformatics and Biomedicine Workshops (BIBMW), Atlanta, GA, USA, 12–15 November 2011; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2011; pp. 473–480. [Google Scholar]
- Lee, K.; Chuang, H.-Y.; Beyer, A.; Sung, M.-K.; Huh, W.-K.; Lee, B.; Ideker, T. Protein networks markedly improve prediction of subcellular localization in multiple eukaryotic species. Nucleic Acids Res. 2008, 36, e136. [Google Scholar] [CrossRef]
- Mintz-Oron, S.; Aharoni, A.; Ruppin, E.; Shlomi, T. Network-based prediction of metabolic enzymes’ subcellular localization. Bioinformatics 2009, 25, i247–i1252. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Method | Tools Used | Performance Matrix | Locations/Organism | Availability | Year |
|---|---|---|---|---|---|
| SignalP-5.0 * | convolutional and recurrent (LSTM) neural networks | MCC, precision and recall | Archaea, Gram-positive Bacteria, Gram-negative Bacteria and Eukarya | http://www.cbs.dtu.dk/services/SignalP/ | 2019 |
| TargetP 2.0 * | recurrent neural networks (RNNs) network | Precision, recall, F1-score, MCC | mitochondrial, chloroplastic, secretory pathway | http://www.cbs.dtu.dk/services/TargetP/ | 2019 |
| SigUNet | Convolutional neural network | MCC, precision, recall, F1 measure | Eukaryotes, Gram-positive and Gram-negative bacteria | https://github.com/mbilab/SigUNet | 2019 |
| DeepSig | Convolutional Neural Networks | MCC, False Positive Rate, precision and recall | Eukaryotes, Gram-positive bacteria and Gram-negative bacteria | https://deepsig.biocomp.unibo.it | 2018 |
| SChloro | SVM | Accuracy, Recall, Precision, F1-score, and MCC | six chloroplastic sub-compartments | http://schloro.biocomp.unibo.it | 2017 |
| PredSL | combination of neural networks, Markov chains, scoring matrices (PrediSi), and HMMs, | Accuracy | Eukaryotic subcellular location | http://bioinformatics.biol.uoa.gr/PredSL/ | 2006 |
| TatP | HMM/artificial neural networks. | S-score and the C-score, Y-score, D-score | bacteria | http://www.cbs.dtu.dk/services/TatP/ | 2005 |
| ChloroP | Neural network | MCC, sensitivity, specificity | chloroplast transit peptides | http://www.cbs.dtu.dk/services/ChloroP/ | 1999 |
| Method | Tools Used | Performance Matrix | Feature Based | Locations/Organism | Availability | Year |
|---|---|---|---|---|---|---|
| DeepPred-SubMito | Convolutional neural network | Accuracy, MCC | Sequence information | Mitochondrial and submitochondrial proteins | https://github.com/jinyinping/DeepPred-SubMito.git | 2020 |
| SubMito-XGBoost | Extreme gradient boosting (XGBoost) | Sensitivity, Specificity, False positive rate, MCC, F1-measure, precision | Sequence information | Submitochondrial proteins | https://github.com/QUST-AIBBDRC/SubMito-XGBoost/ | 2020 |
| mRNALoc | SVM | Sensitivity, Specificity, Accuracy, MCC | Sequence information | eukaryotic | http://proteininformatics.org/mkumar/mrnaloc | 2020 |
| SCLpred-EMS | Convolutional neural network | Sensitivity, Specificity, False positive rate, MCC | Sequence information | endomembrane system and secretory pathway | http://distilldeep.ucd.ie/SCLpred2/ | 2020 |
| BUSCA | Integrated method of DeepSig, TPpred3, PredGPI, BetAware and ENSEMBLE3.0 | Precision, recall, F1-score, MCC | Sequence information, signal and transit peptides, glycophosphatidylinositol (GPI) anchors and transmembrane domains | Gram-positive, gram-negative, fungi, plant, animal | http://busca.biocomp.unibo.it | 2018 |
| SubMitoPred | SVM | Sensitivity, Specificity, Accuracy, MCC | Sequence and domain information | Mitochondrial and submitochondrial proteins | http://proteininformatics.org/mkumar/submitopred/ | 2018 |
| pLoc-mEuk | ML-GKR (multi-label Gaussian kernel regression) classifier | Coverage, Accuracy, Absolute true, Absolute false | Gene Ontology and Chou’s general PseAAC | 22 different subcellular localizations of eukaryotic proteins | http://www.jci-bioinfo.cn/pLoc-mEuk/ | 2018 |
| ERPred | SVM | Sensitivity, Specificity, Accuracy, MCC | Sequence information, | ER Proteins | http://proteininformatics.org/mkumar/erpred/index.html | 2017 |
| DeepLoc | deep recurrent neural networks | Accuracy, MCC | Sequence information | 10 different location of eukaryotic proteins | http://www.cbs.dtu.dk/services/DeepLoc | 2017 |
| SubNucPred | SVM | Sensitivity, Specificity, Accuracy, MCC | Sequence and domain information | Nuclear and subnuclear protein | http://proteininformatics.org/mkumar/subnucpred/ | 2014 |
| LocTree3 | SVM and homology | Accuracy, recall, standard deviation, standard error | Homology-based, Gene Ontology | 18 classes for eukaryotes, in six for bacteria and in three for archaea | http://www.rostlab.org/services/loctree3 | 2014 |
| PlantLoc | localization motif search | accuracy | localization motif information | 11 different location of plant proteins | http://cal.tongji.edu.cn/PlantLoc/ | 2013 |
| iLoc-Cell, package of predictors for subcellular locations of proteins. It includes iLoc-Hum, iLoc-Animal, iLoc-Plant, iLoc-Euk, iLoc-Virus, iLoc-Gpos, iLoc-Gneg | multi-label learning, multi-label KNN | Accuracy, Precision, Recall, Absolute-true rate, Absolute-false rate, | Sequence information, gene ontology, PSSM, | Different subcellular location of Human, animals, plants, eukaryotic, Virus, gram-positive, gram-negatives | http://www.jci-bioinfo.cn/iLoc-Cell | 2011, 2012, 2013 |
| MARSpred | SVM | Sensitivity, specificity, Accuracy, MCC | Sequence information, PSSM | cytosolic and mitochondrial aminoacyl tRNA synthetase | http://www.imtech.res.in/raghava/marspred/ | 2012 |
| SCLPred | Neural Network | Sensitivity, specificity, False positive rate, MCC | primary sequence and multiple sequence alignments | four classes for animals and fungi and five classes for plants | http://distill.ucd.ie/distill/ | 2011 |
| AtSubP | SVM | Sensitivity, specificity, error rate, MCC, ROC curve | Sequence information, PSSM | subcellular localization of Arabidopsis | http://bioinfo3.noble.org/AtSubP | 2010 |
| Euk-mPLoc 2.0 | OET-KNN (Optimized Evidence-Theoretic K-Nearest Neighbor) classifiers | accuracy | gene ontology information, functional domain information, and sequential evolutionary information | eukaryotic proteins among the following 22 locations | http://www.csbio.sjtu.edu.cn/bioinf/euk-multi-2/ | 2010 |
| PSORTb | SVM | Precision, recall, accuracy, MCC | Sequence information | Different subcellular location of Gram-negative, Gram-positive, archaea | http://www.psort.org/psortb | 2010 |
| YLoc | naïve Bayes alongside entropy-based discretization | overall accuracy, F1-score | Sequences information, GO-term and motif | animal, fungal and plant proteins | www.multiloc.org/YLoc | 2010 |
| SubChlo | evidence-theoretic K-nearest neighbor (ET-KNN) algorithm | overall accuracy, accuracy | Sequences information (PseAAC), | chloroplast proteins | http://bioinfo.au.tsinghua.edu.cn/subchlo | 2009 |
| MultiLoc2 | SVM | Sensitivity, specificity, Accuracy, MCC | phylogenetic profiles and gene ontology terms | Plant, Animal, Fungal | https://abi-services.informatik.uni-tuebingen.de/multiloc2/webloc.cgi | 2009 |
| AAIndexLoc | SVM | Sensitivity, specificity, Accuracy, MCC | Sequence information and physicochemical properties | Animal, Fungal and plants | http://aaindexloc.bii.a-star.edu.sg | 2008 |
| Cell-PLoc package of predictors for subcellular locations of proteins. It includes Euk-mPLoc, Hum-mPLoc, Plant-PLoc, Gpos-PLoc, Gneg-PLoc, Virus-PLoc | KNN or OET-KN algorithm | Accuracy and F1 score | GO and functional domain information | 22 subcellular location of eukaryotic, human, plant, Gram-positive bacterial, Gram-negative bacterial and viral proteins | http://chou.med.harvard.edu/bioinf/Cell-PLoc | 2008 |
| ProLoc-GO | SVM-GO, k-NN-GO and fuzzy k-NN-GO | MCC | GO term information | eukaryotic, human, | http://iclab.life.nctu.edu.tw/prolocgo | 2008 |
| ProLoc | SVM | Accuracy | physicochemical composition | subnuclear localizations | http://iclab.life.nctu.edu.tw/proloc | 2007 |
| SherLoc | SVM | Sensitivity, specificity, MCC | Sequence information | eukaryotic proteins | http://www-bs.informatik.uni-tuebingen.de/Services/SherLoc/ | 2007 |
| MitPred | SVM | Sensitivity, specificity, Accuracy, MCC | Sequence information | Mitochondrial proteins | http://www.imtech.res.in/raghava/mitpred/ | 2006 |
| BaCelLo | SVM | Coverage, Normalized Accuracy, geometric average , overall accuracy, Generalized Correlation | Sequence information | Plant, Animal, Fungal | http://www.biocomp.unibo.it/bacell/ | 2006 |
| HSLpred | SVM | Accuracy, MCC, Reliability index | Sequence information | Human Protein | http://www.imtech.res.in/raghava/hslpred/ | 2005 |
| PSLpred | SVM | Accuracy, MCC, Reliability index | Sequence information | gram-negative bacterial proteins | http://www.imtech.res.in/raghava/pslpred/ | 2005 |
| ESLpred | SVM | Accuracy, MCC, Reliability index | Sequence information and PSSM | eukaryotic proteins | http://www.imtech.res.in/raghava/eslpred/ | 2004 |
| Metric | Explanation | Formula | References |
|---|---|---|---|
| Accuracy | It is the ratio of the number of correct predictions to the total number of input samples. It works well if there are an equal number of samples belonging to each class. | TP + TN/(TP + FP + TN + FN) | [61,62] |
| Specificity/True Negative Rate | Proportion of negatives that are correctly identified | TN/(TN + FP) | [61,62] |
| Precision/Positive Predictive Value | It tells us how many of the correctly predicted cases actually turned out to be positive. | TP/(TP + FP) | [61] |
| Recall/Sensitivity/True Positive Rate | It tells us how many of the actual positive cases we were able to predict correctly with our model. | TP/(TP + FN) | [61,62] |
| F1-Score | F1 score is the harmonic mean between precision and recall. Greater the F1 score, better the performance of the model. It tells how precise the classifier is and how robust it is. | F1 = 2 * precision * Recall/Precision + Recall | [63] |
| Negative Predictive Value | It is defined as the proportion of predicted negatives, which are real negatives. It reflects the probability that a predicted negative is a true negative. | TN/(TN + FN) | [64] |
| False Positive Rate | Number of incorrect positive predictions divided by the total number of negatives. | FP/(FP + TN) | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.; Dhanda, S.K. Bird Eye View of Protein Subcellular Localization Prediction. Life 2020, 10, 347. https://doi.org/10.3390/life10120347
Kumar R, Dhanda SK. Bird Eye View of Protein Subcellular Localization Prediction. Life. 2020; 10(12):347. https://doi.org/10.3390/life10120347
Chicago/Turabian StyleKumar, Ravindra, and Sandeep Kumar Dhanda. 2020. "Bird Eye View of Protein Subcellular Localization Prediction" Life 10, no. 12: 347. https://doi.org/10.3390/life10120347
APA StyleKumar, R., & Dhanda, S. K. (2020). Bird Eye View of Protein Subcellular Localization Prediction. Life, 10(12), 347. https://doi.org/10.3390/life10120347