Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies

Abstract
1. Introduction
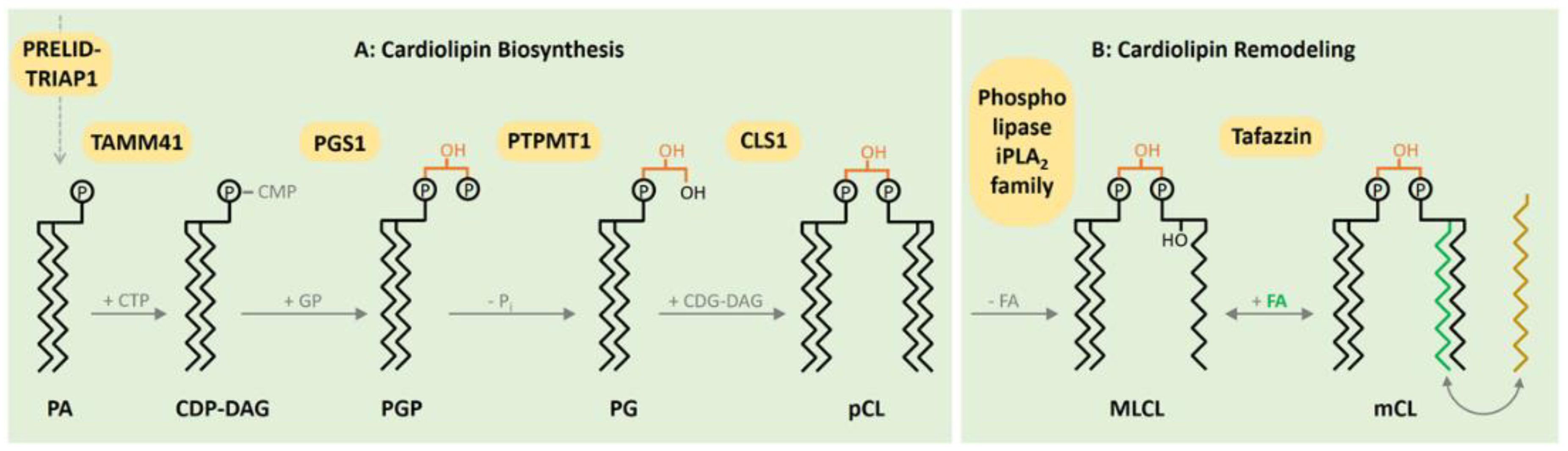
2. CL Biosynthesis
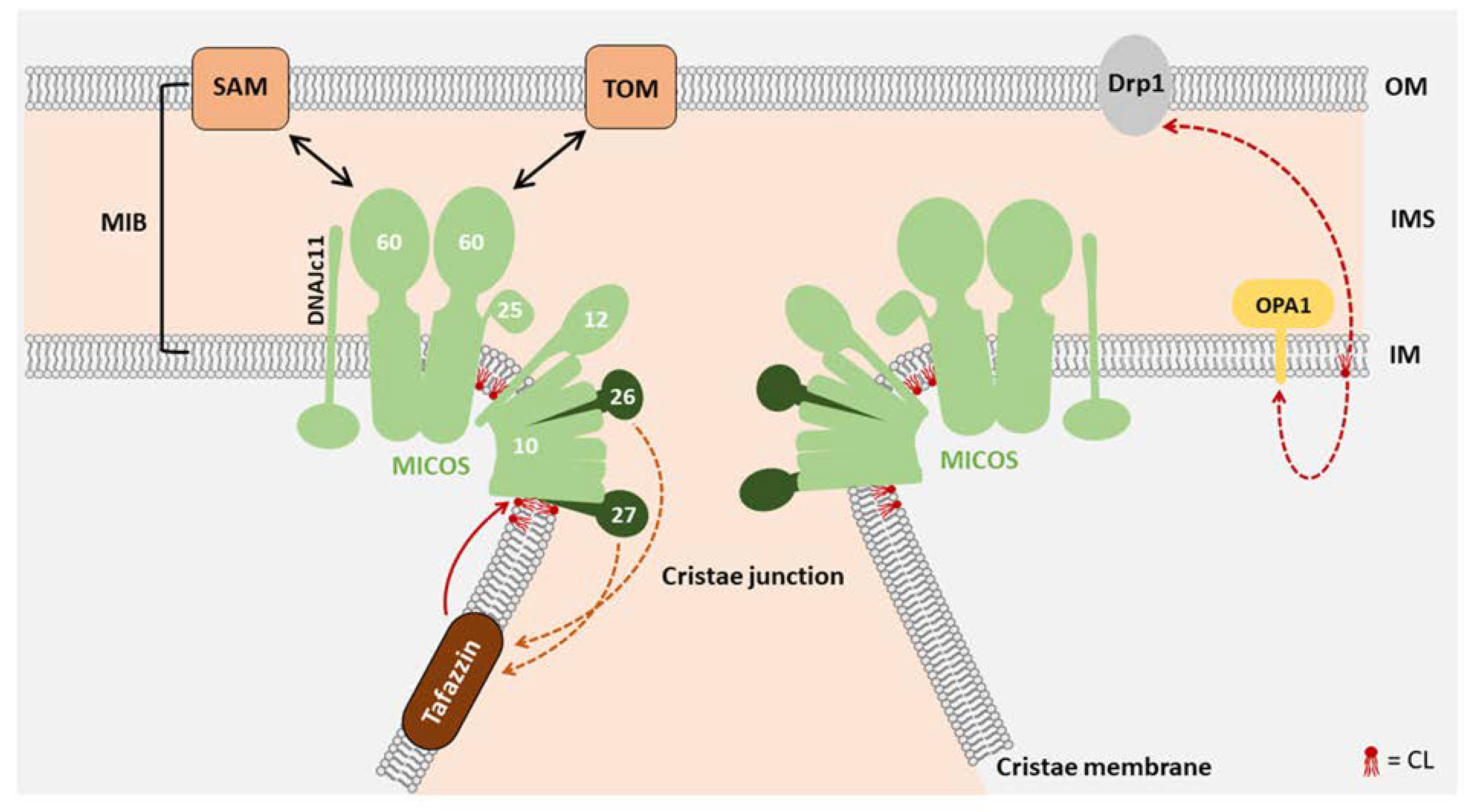
3. Function of CL in Mitochondrial Morphology
4. Function of CL in Energy Metabolism
5. Function of CL in Intermediate Metabolism
6. CL Function in Calcium Homeostasis
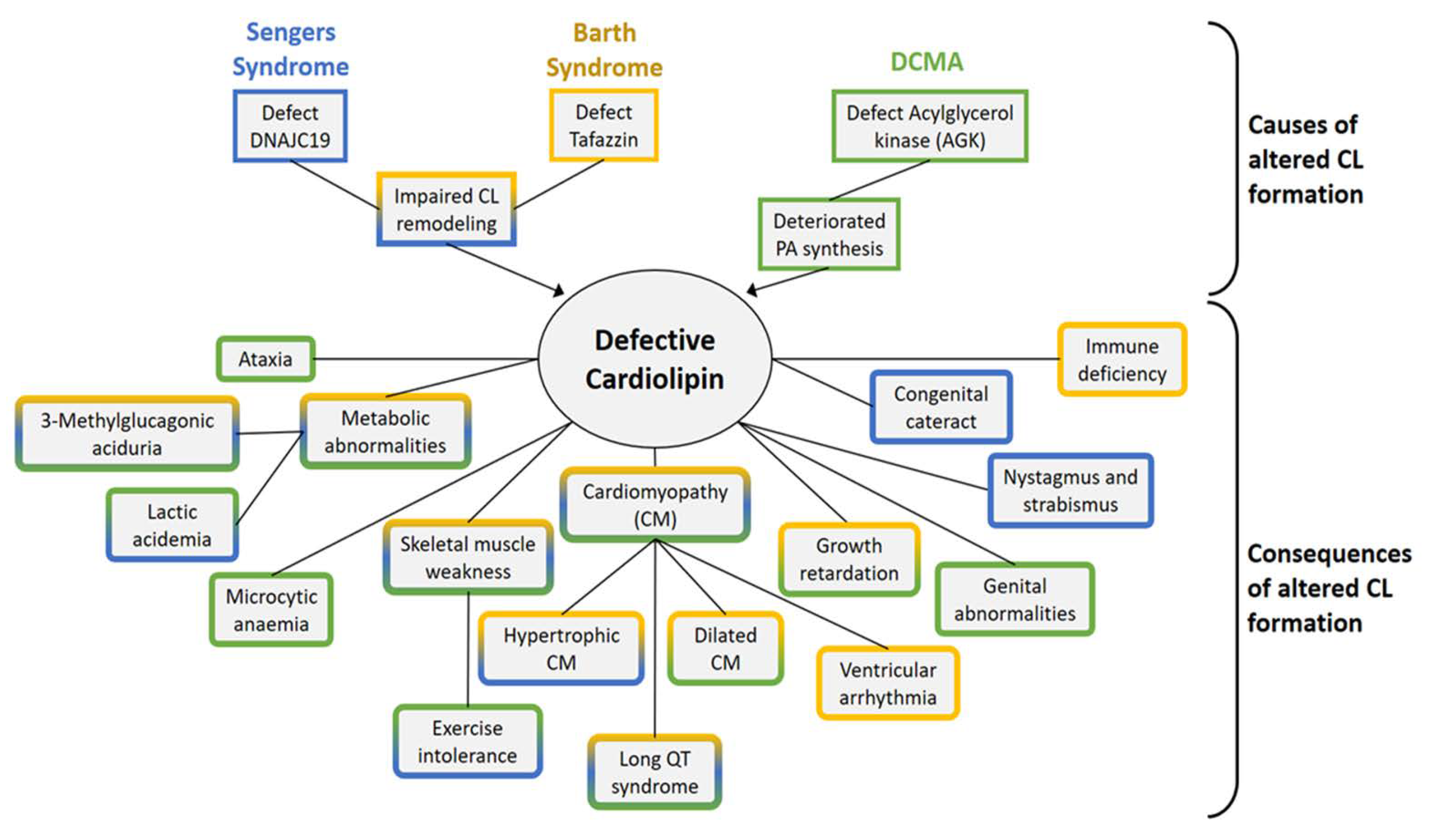
7. Barth Syndrome
8. Sengers Syndrome
9. Dilated Cardiomyopathy with Ataxia (DCMA)
10. Therapeutic Approaches
11. Conclusions
Funding
Conflicts of Interest
References
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Barth, E.; Stammler, G.; Speiser, B.; Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef]
- Schaper, J.; Meiser, E.; Stämmler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Maack, C. Barth syndrome cardiomyopathy. Cardiovasc. Res. 2017, 113, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Goldenthal, M.J. Mitochondrial centrality in heart failure. Heart Fail. Rev. 2008, 13, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K. Effects of elamipretide on skeletal muscle in dogs with experimentally induced heart failure. ESC Heart Fail. 2019, 6, 328–335. [Google Scholar] [CrossRef]
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; Van Der Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019, 38, e101056. [Google Scholar] [CrossRef]
- Zhang, R.; Krigman, J.; Luo, H.; Ozgen, S.; Yang, M.; Sun, N. Mitophagy in cardiovascular homeostasis. Mech. Ageing Dev. 2020, 188, 111245. [Google Scholar] [CrossRef]
- Schlame, M. Protein crowding in the inner mitochondrial membrane. Biochim. Biophys. Acta Bioenerg. 2020, 1862, 148305. [Google Scholar] [CrossRef]
- Gebert, N.; Joshi, A.S.; Kutik, S.; Becker, T.; McKenzie, M.; Guan, X.L.; Mooga, V.P.; Stroud, D.A.; Kulkarni, G.; Wenk, M.R.; et al. Mitochondrial Cardiolipin Involved in Outer-Membrane Protein Biogenesis: Implications for Barth Syndrome. Curr. Biol. 2009, 19, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Oemer, G.; Koch, J.; Wohlfarter, Y.; Alam, M.T.; Lackner, K.; Sailer, S.; Neumann, L.; Lindner, H.H.; Watschinger, K.; Haltmeier, M.; et al. Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Rep. 2020, 30, 4281–4291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, Z.; Murphy, A.N.; Wiley, S.E.; Perkins, G.A.; Worby, C.A.; Engel, J.L.; Heacock, P.; Nguyen, O.K.; Wang, J.H.; et al. Mitochondrial Phosphatase PTPMT1 Is Essential for Cardiolipin Biosynthesis. Cell Metab. 2011, 13, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Kubota-Sakashita, M.; Nagatsuka, Y.; Hirabayashi, Y.; Hanasaka, T.; Tohyama, K.; Kato, T. Cardiolipin is essential for early embryonic viability and mitochondrial integrity of neurons in mammals. FASEB J. 2019, 34, 1465–1480. [Google Scholar] [CrossRef]
- Dinca, A.A.; Chien, W.-M.; Chin, M.T. Identification of novel mitochondrial localization signals in human Tafazzin, the cause of the inherited cardiomyopathic disorder Barth syndrome. J. Mol. Cell. Cardiol. 2018, 114, 83–92. [Google Scholar] [CrossRef]
- Hijikata, A.; Yura, K.; Ohara, O.; Go, M. Structural and functional analyses of Barth syndrome-causing mutations and alternative splicing in the tafazzin acyltransferase domain. Meta Gene 2015, 4, 92–106. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, S.; Malhotra, A.; Edelman-Novemsky, I.; Ma, J.; Kruppa, A.; Cernicica, C.; Blais, S.; Neubert, T.A.; Ren, M.; et al. Characterization of Tafazzin Splice Variants from Humans and Fruit Flies. J. Biol. Chem. 2009, 284, 29230–29239. [Google Scholar] [CrossRef]
- Schlame, M.; Xu, Y.; Ren, M. The Basis for Acyl Specificity in the Tafazzin Reaction. J. Biol. Chem. 2017, 292, 5499–5506. [Google Scholar] [CrossRef]
- Xu, Y.; Anjaneyulu, M.; Donelian, A.; Yu, W.; Greenberg, M.L.; Ren, M.; Owusu-Ansah, E.; Schlame, M. Assembly of the complexes of oxidative phosphorylation triggers the remodeling of cardiolipin. Proc. Natl. Acad. Sci. USA 2019, 116, 11235–11240. [Google Scholar] [CrossRef]
- Taylor, W.A.; Hatch, G.M. Identification of the Human Mitochondrial Linoleoyl-coenzyme A Monolysocardiolipin Acyltransferase (MLCL AT-1). J. Biol. Chem. 2009, 284, 30360–30371. [Google Scholar] [CrossRef]
- Mejia, E.M.; Zegallai, H.; Bouchard, E.D.; Banerji, V.; Ravandi, A.; Hatch, G.M. Expression of human monolysocardiolipin acyltransferase-1 improves mitochondrial function in Barth syndrome lymphoblasts. J. Biol. Chem. 2018, 293, 7564–7577. [Google Scholar] [CrossRef] [PubMed]
- Miklas, J.W.; Clark, E.; Levy, S.; Detraux, D.; Leonard, A.; Beussman, K.; Showalter, M.R.; Smith, A.T.; Hofsteen, P.; Yang, X.; et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat. Commun. 2019, 10, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin Remodeling by ALCAT1 Links Oxidative Stress and Mitochondrial Dysfunction to Obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Baandrup, U.; Florio, R.A.; Roters, F.; Olsen, E.G. Electron microscopic investigation of endomyocardial biopsy samples in hypertrophy and cardiomyopathy. A semiquantitative study in 48 patients. Circulation 1981, 63, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Sharov, V.; Riddle, J.M.; Kono, T.; Lesch, M.; Goldstein, S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J. Mol. Cell. Cardiol. 1992, 24, 1333–1347. [Google Scholar] [CrossRef]
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 3–7. [Google Scholar] [CrossRef]
- Frohman, M.A. Role of mitochondrial lipids in guiding fission and fusion. J. Mol. Med. 2015, 93, 263–269. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Sharov, V.G.; Sabbah, H.N.; Shimoyama, H.; Goussev, A.V.; Lesch, M.; Goldstein, S. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am. J. Pathol. 1996, 148, 141–149. [Google Scholar]
- DeVay, R.M.; Dominguez-Ramirez, L.; Lackner, L.L.; Hoppins, S.; Stahlberg, H.; Nunnari, J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009, 186, 793–803. [Google Scholar] [CrossRef]
- Meglei, G.; McQuibban, G.A. The Dynamin-Related Protein Mgm1p Assembles into Oligomers and Hydrolyzes GTP To Function in Mitochondrial Membrane Fusion†. Biochemistry 2009, 48, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z.; Sims, H.F.; Cerqua, R.; Cade, W.T.; Han, X.; et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Vaz, F.; Houtkooper, R.H.; James, J.; Moore, V.; Tokunaga, C.; Kulik, W.; Wansapura, J.; Toth, M.J.; Strauss, A.; et al. Cardiac and Skeletal Muscle Defects in a Mouse Model of Human Barth Syndrome. J. Biol. Chem. 2010, 286, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Gonzalvez, F.; D’Aurelio, M.; Boutant, M.; Moustapha, A.; Puech, J.-P.; Landes, T.; Arnauné-Pelloquin, L.; Vial, G.; Taleux, N.; Slomianny, C.; et al. Barth syndrome: Cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1194–1206. [Google Scholar] [CrossRef]
- Acehan, D.; Xu, Y.; Stokes, D.L.; Schlame, M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Investig. 2007, 87, 40–48. [Google Scholar] [CrossRef]
- Dudek, J.; Cheng, I.F.; Chowdhury, A.; Wozny, K.; Balleininger, M.; Reinhold, R.; Grunau, S.; Callegari, S.; Toischer, K.; Wanders, R.J.; et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol Med 2016, 8, 139–154. [Google Scholar] [CrossRef]
- Dudek, J.; Cheng, I.-F.; Balleininger, M.; Vaz, F.M.; Streckfuss-Bömeke, K.; Hübscher, D.; Vukotic, M.; Wanders, R.J.A.; Rehling, P.; Guan, K. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 2013, 11, 806–819. [Google Scholar] [CrossRef]
- Chatzispyrou, I.A.; Guerrero-Castillo, S.; Held, N.M.; Ruiter, J.P.; Denis, S.W.; Ijlst, L.; Wanders, R.J.; Van Weeghel, M.; Ferdinandusse, S.; Vaz, F.M.; et al. Barth syndrome cells display widespread remodeling of mitochondrial complexes without affecting metabolic flux distribution. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3650–3658. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Xu, Y.; Ma, Q.; Lin, Z.; Schlame, M.; Bezzerides, V.J.; Strathdee, D.; Pu, W.T. AAV Gene Therapy Prevents and Reverses Heart Failure in a Murine Knockout Model of Barth Syndrome. Circ. Res. 2020, 126, 1024–1039. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.A.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008, 183, 681–696. [Google Scholar] [CrossRef]
- Rampelt, H.; Zerbes, R.M.; Van Der Laan, M.; Pfanner, N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.A.; Koob, S.; Heide, H.; Wittig, I.; Head, B.; Van Der Bliek, A.; Brandt, U.; Mittelbronn, M.; Reichert, A.S. APOOL Is a Cardiolipin-Binding Constituent of the Mitofilin/MINOS Protein Complex Determining Cristae Morphology in Mammalian Mitochondria. PLoS ONE 2013, 8, e63683. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Mourier, A.; Yamada, J.; McCaffery, J.M.; Nunnari, J.; Youle, R.J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. eLife 2015, 4, e07739. [Google Scholar] [CrossRef] [PubMed]
- Koob, S.; Barrera, M.; Anand, R.; Reichert, A.S. The non-glycosylated isoform of MIC26 is a constituent of the mammalian MICOS complex and promotes formation of crista junctions. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 1551–1563. [Google Scholar] [CrossRef]
- Van Strien, J.; Guerrero-Castillo, S.; Chatzispyrou, I.A.; Houtkooper, R.H.; Brandt, U.; Huynen, M.A. COmplexome Profiling ALignment (COPAL) reveals remodeling of mitochondrial protein complexes in Barth syndrome. Bioinformatics 2019, 35, 3083–3091. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of Mitochondrial Dynamics in the Failing Heart: Normalization Following Long-Term Therapy with Elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328. [Google Scholar] [CrossRef]
- Anand, R.; Kondadi, A.K.; Meisterknecht, J.; Golombek, M.; Nortmann, O.; Riedel, J.; Peifer-Weiß, L.; Brocke-Ahmadinejad, N.; Schlütermann, D.; Stork, B.; et al. MIC26 and MIC27 cooperate to regulate cardiolipin levels and the landscape of OXPHOS complexes. Life Sci. Alliance 2020, 3, e202000711. [Google Scholar] [CrossRef]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature 2016, 538, 406–410. [Google Scholar] [CrossRef]
- Sedlák, E.; Robinson, N.C. Phospholipase A2 Digestion of Cardiolipin Bound to Bovine CytochromecOxidase Alters Both Activity and Quaternary Structure†. Biochemistry 1999, 38, 14966–14972. [Google Scholar] [CrossRef]
- Sedlák, E.; Panda, M.; Dale, M.P.; Weintraub, S.T.; Robinson, N.C. Photolabeling of Cardiolipin Binding Subunits within Bovine Heart CytochromecOxidase†. Biochemistry 2006, 45, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Shinzawa-Itoh, K.; Seiyama, J.; Terada, H.; Nakatsubo, R.; Naoki, K.; Nakashima, Y.; Yoshikawa, S. Bovine Heart NADH−Ubiquinone Oxidoreductase Contains One Molecule of Ubiquinone with Ten Isoprene Units as One of the Cofactors. Biochemistry 2010, 49, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Sharpley, M.S.; Shannon, R.J.; Draghi, A.F.; Hirst, J. Interactions between Phospholipids and NADH:Ubiquinone Oxidoreductase (Complex I) from Bovine Mitochondria†. Biochemistry 2006, 45, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.C.; Zborowski, J.; Talbert, L.H. Cardiolipin-depleted bovine heart cytochrome c oxidase: Binding stoichiometry and affinity for cardiolipin derivatives. Biochemistry 1990, 29, 8962–8969. [Google Scholar] [CrossRef]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the dimeric ATP synthase from bovine mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef]
- Laird, D.M.; Eble, K.S.; Cunningham, C.C. Reconstitution of mitochondrial F0.F1-ATPase with phosphatidylcholine using the nonionic detergent, octylglucoside. J. Biol. Chem. 1986, 261, 14844–14850. [Google Scholar]
- Duncan, A.L.; Robinson, A.J.; Walker, J.E. Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl. Acad. Sci. USA 2016, 113, 8687–8692. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Penczek, P.A.; Fang, J.; Mallampalli, V.K.P.S.; Sparagna, G.C.; Dowhan, W. Arrangement of the Respiratory Chain Complexes in Saccharomyces cerevisiae Supercomplex III2IV2 Revealed by Single Particle Cryo-Electron Microscopy. J. Biol. Chem. 2012, 287, 23095–23103. [Google Scholar] [CrossRef]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Cardiolipin Is Essential for Organization of Complexes III and IV into a Supercomplex in Intact Yeast Mitochondria. J. Biol. Chem. 2005, 280, 29403–29408. [Google Scholar] [CrossRef]
- Jiang, F.; Ryan, M.T.; Schlame, M.; Zhao, M.; Gu, Z.; Klingenberg, M.; Pfanner, N.; Greenberg, M.L. Absence of Cardiolipin in thecrd1Null Mutant Results in Decreased Mitochondrial Membrane Potential and Reduced Mitochondrial Function. J. Biol. Chem. 2000, 275, 22387–22394. [Google Scholar] [CrossRef] [PubMed]
- Gómez, L.A.; Hagen, T.M. Age-related decline in mitochondrial bioenergetics: Does supercomplex destabilization determine lower oxidative capacity and higher superoxide production? Semin. Cell Dev. Biol. 2012, 23, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, D.B.; Sparagna, G.C.; Amoscato, A.A.; McMillin, J.B.; Dowhan, W. Decreased Cardiolipin Synthesis Corresponds with Cytochromec Release in Palmitate-induced Cardiomyocyte Apoptosis. J. Biol. Chem. 2001, 276, 38061–38067. [Google Scholar] [PubMed]
- Gadicherla, A.K.; Stowe, D.F.; Antholine, W.E.; Yang, M.; Camara, A.K. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 419–429. [Google Scholar] [CrossRef]
- Saini-Chohan, H.K.; Holmes, M.G.; Chicco, A.J.; Taylor, W.A.; Moore, R.L.; McCune, S.A.; Hickson-Bick, D.L.; Hatch, G.M.; Sparagna, G.C. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J. Lipid Res. 2008, 50, 1600–1608. [Google Scholar] [CrossRef]
- Nickel, A.G.; Von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.-C.; Kazakov, A.V.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.-H.; Campbell, M.D.; et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. eLife 2020, 9. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Conrad, M.; Jakupoglu, C.; Moreno, S.G.; Lippl, S.; Banjac, A.; Schneider, M.; Beck, H.; Hatzopoulos, A.K.; Just, U.; Sinowatz, F.; et al. Essential Role for Mitochondrial Thioredoxin Reductase in Hematopoiesis, Heart Development, and Heart Function. Mol. Cell. Biol. 2004, 24, 9414–9423. [Google Scholar] [CrossRef]
- Ho, Y.-S.; Magnenat, J.-L.; Gargano, M.; Cao, J. The Nature of Antioxidant Defense Mechanisms: A Lesson from Transgenic Studies. Environ. Health Perspect. 1998, 106, 1219. [Google Scholar] [CrossRef]
- Yen, H.C.; Oberley, T.D.; Vichitbandha, S.; Ho, Y.S.; Clair, D.K.S. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Investig. 1996, 98, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Klingenberg, M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by phosphorus-31 nuclear magnetic resonance. Biochemistry 1985, 24, 3821–3826. [Google Scholar] [CrossRef] [PubMed]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nat. Cell Biol. 2003, 426, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Senoo, N.; Kandasamy, S.; Ogunbona, O.B.; Baile, M.G.; Lu, Y.; Claypool, S.M. Cardiolipin, conformation, and respiratory complex-dependent oligomerization of the major mitochondrial ADP/ATP carrier in yeast. Sci. Adv. 2020, 6, eabb0780. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Bisaccia, F.; Capobianco, L.; Iacobazzi, V.; Indiveri, C.; Zara, V. Structural and functional properties of mitochondrial anion carriers. Biochim. Biophys. Acta Bioenerg. 1990, 1018, 147–150. [Google Scholar] [CrossRef]
- Nałęcz, K.A.; Bolli, R.; Wojtczak, L.; Azzi, A.; Nałcz, K.A. The monocarboxylate carrier from bovine heart mitochondria: Partial purification and its substrate-transporting properties in a reconstituted system. Biochim. Biophys. Acta Bioenerg. 1986, 851, 29–37. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Prezioso, G.; Palmieri, F. Kinetic characterization of the reconstituted carnitine carrier from rat liver mitochondria. Biochim. Biophys. Acta Biomembr. 1991, 1065, 231–238. [Google Scholar] [CrossRef]
- Nałecz, K.A.; Kamińska, J.; Nałecz, M.J.; Azzi, A. The activity of pyruvate carrier in a reconstituted system: Substrate specificity and inhibitor sensitivity. Arch. Biochem. Biophys. 1992, 297, 162–168. [Google Scholar] [CrossRef]
- Kaplan, R.S.; A Mayor, J.; Johnston, N.; Oliveira, D.L. Purification and characterization of the reconstitutively active tricarboxylate transporter from rat liver mitochondria. J. Biol. Chem. 1990, 265, 13379–13385. [Google Scholar]
- Vukotic, M.; Nolte, H.; König, T.; Saita, S.; Ananjew, M.; Krüger, M.; Tatsuta, T.; Langer, T. Acylglycerol Kinase Mutated in Sengers Syndrome Is a Subunit of the TIM22 Protein Translocase in Mitochondria. Mol. Cell 2017, 67, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Mårtensson, C.U.; Becker, T. Acylglycerol Kinase: Mitochondrial Protein Transport Meets Lipid Biosynthesis. Trends Cell Biol. 2017, 27, 700–702. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Cheneval, D.; Carafoli, E. The Mitochondrial Creatine Phosphokinase is Associated with Inner Membrane Cardiolipin. Results Probl. Cell Differ. 1986, 194, 151–156. [Google Scholar] [CrossRef]
- Müller, M.; Moser, R.; Cheneval, D.; Carafoli, E. Cardiolipin is the membrane receptor for mitochondrial creatine phosphokinase. J. Biol. Chem. 1985, 260, 3839–3843. [Google Scholar]
- Fritz-Wolf, K.; Schnyder, T.; Wallimann, T.; Kabsch, W. Structure of mitochondrial creatine kinase. Nat. Cell Biol. 1996, 381, 341–345. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Ramirez, S.; Brückner, A.; Kay, L.; Polge, C.; Epand, R.F.; Lee, R.M.; Lacombe, M.-L.; Epand, R.M. Mitochondrial kinases and their molecular interaction with cardiolipin. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2032–2047. [Google Scholar] [CrossRef]
- Li, Y.; Lou, W.; Raja, V.; Denis, S.; Yu, W.; Schmidtke, M.W.; Reynolds, C.A.; Schlame, M.; Houtkooper, R.H.; Greenberg, M.L. Cardiolipin-induced activation of pyruvate dehydrogenase links mitochondrial lipid biosynthesis to TCA cycle function. J. Biol. Chem. 2019, 294, 11568–11578. [Google Scholar] [CrossRef]
- Fatica, E.; Deleonibus, G.A.; House, A.; Kodger, J.V.; Pearce, R.; Shah, R.; Levi, L.; Sandlers, Y. Barth Syndrome: Exploring Cardiac Metabolism with Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Metabolites 2019, 9, 306. [Google Scholar] [CrossRef]
- Li, Y.; Lou, W.; Grevel, A.; Böttinger, L.; Liang, Z.; Ji, J.; Patil, V.A.; Liu, J.; Ye, C.; Hüttemann, M.; et al. Cardiolipin-deficient cells have decreased levels of the iron–sulfur biogenesis protein frataxin. J. Biol. Chem. 2020, 295, 11928–11937. [Google Scholar] [CrossRef]
- Patil, V.A.; Fox, J.L.; Gohil, V.M.; Winge, D.R.; Greenberg, M.L.; Suzuki, K.; Harada, N.; Yamane, S.; Nakamura, Y.; Sasaki, K.; et al. Loss of Cardiolipin Leads to Perturbation of Mitochondrial and Cellular Iron Homeostasis. J. Biol. Chem. 2012, 288, 1696–1705. [Google Scholar] [CrossRef]
- Raja, V.; Salsaa, M.; Joshi, A.S.; Li, Y.; Van Roermund, C.W.; Saadat, N.; Lazcano, P.; Schmidtke, M.; Hüttemann, M.; Gupta, S.V.; et al. Cardiolipin-deficient cells depend on anaplerotic pathways to ameliorate defective TCA cycle function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Le, C.H.; Benage, L.G.; Specht, K.S.; Puma, L.C.L.; Mulligan, C.M.; Heuberger, A.L.; Prenni, J.E.; Claypool, S.M.; Chatfield, K.C.; Sparagna, G.C.; et al. Tafazzin deficiency impairs CoA-dependent oxidative metabolism in cardiac mitochondria. J. Biol. Chem. 2020, 295. [Google Scholar] [CrossRef]
- Cole, L.K.; Mejia, E.M.; Sparagna, G.C.; Vandel, M.; Xiang, B.; Han, X.; Dedousis, N.; Kaufman, B.A.; Dolinsky, V.W.; Hatch, G.M. Cardiolipin deficiency elevates susceptibility to a lipotoxic hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2020, 144, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Sandlers, Y.; Mercier, K.; Pathmasiri, W.; Carlson, J.; McRitchie, S.; Sumner, S.; Vernon, H.J. Metabolomics Reveals New Mechanisms for Pathogenesis in Barth Syndrome and Introduces Novel Roles for Cardiolipin in Cellular Function. PLoS ONE 2016, 11, e0151802. [Google Scholar] [CrossRef] [PubMed]
- Korman, S.H.; Andresen, B.S.; Zeharia, A.; Gutman, A.; Boneh, A.; Pitt, J. 2-Ethylhydracrylic Aciduria in Short/Branched-Chain Acyl-CoA Dehydrogenase Deficiency: Application to Diagnosis and Implications for the R-Pathway of Isoleucine Oxidation. Clin. Chem. 2005, 51, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Altered Cardiac Myocyte Ca Regulation In Heart Failure. Physiology 2006, 21, 380–387. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Lokuta, A.J.; Maertz, N.A.; Meethal, S.V.; Potter, K.T.; Kamp, T.J.; Valdivia, H.H.; Haworth, R.A. Increased Nitration of Sarcoplasmic Reticulum Ca 2+ -ATPase in Human Heart Failure. Circulation 2005, 111, 988–995. [Google Scholar] [CrossRef]
- Periasamy, M.; Janssen, P.M. Molecular Basis of Diastolic Dysfunction. Hear. Fail. Clin. 2008, 4, 13–21. [Google Scholar] [CrossRef]
- Dorn, G.W.; Maack, C. SR and mitochondria: Calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 2013, 55, 42–49. [Google Scholar] [CrossRef]
- Szabadkai, G.; Simoni, A.M.; Chami, M.; Wieckowski, M.R.; Youle, R.J.; Rizzuto, R. Drp-1-Dependent Division of the Mitochondrial Network Blocks Intraorganellar Ca2+ Waves and Protects against Ca2+-Mediated Apoptosis. Mol. Cell 2004, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Maack, C. Calcium release microdomains and mitochondria. Cardiovasc. Res. 2013, 98, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nat. Cell Biol. 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; A Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, R.; Wang, C.; Siliciano, A.F.; Long, S.B. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nat. Cell Biol. 2018, 559, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ball, W.B.; Madaris, T.R.; Srikantan, S.; Madesh, M.; Mootha, V.K.; Gohil, V.M. An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2020, 117, 16383–16390. [Google Scholar] [CrossRef]
- Christodoulou, J.; McInnes, R.R.; Jay, V.; Wilson, G.; Becker, L.E.; Lehotay, D.C.; Platt, B.-A.; Bridge, P.J.; Robinson, B.H.; Clarke, J.T.R. Barth syndrome: Clinical observations and genetic linkage studies. Am. J. Med. Genet. 1994, 50, 255–264. [Google Scholar] [CrossRef]
- Ades, L.C.; Gedeon, A.K.; Wilson, M.J.; Latham, M.; Partington, M.W.; Mulley, J.C.; Nelson, J.; Lui, K.; Sillence, D.O. Barth syndrome: Clinical features and confirmation of gene localisation to distal Xq28. Am. J. Med. Genet. 1993, 45, 327–334. [Google Scholar] [CrossRef]
- Roberts, A.E.; Nixon, C.; Steward, C.G.; Gauvreau, K.; Maisenbacher, M.; Fletcher, M.; Geva, J.; Byrne, B.J.; Spencer, C.T. The Barth Syndrome Registry: Distinguishing disease characteristics and growth data from a longitudinal study. Am. J. Med. Genet. Part A 2012, 158, 2726–2732. [Google Scholar] [CrossRef]
- Spencer, C.T.; Bryant, R.M.; Day, J.; Gonzalez, I.L.; Colan, S.D.; Thompson, W.R.; Berthy, J.; Redfearn, S.P.; Byrne, B.J. Cardiac and Clinical Phenotype in Barth Syndrome. Pediatrics 2006, 118, e337–e346. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.T.; Byrne, B.J.; Bryant, R.M.; Margossian, R.; Maisenbacher, M.; Breitenger, P.; Benni, P.B.; Redfearn, S.; Marcus, E.; Cade, W.T. Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2122–H2129. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Kluijtmans, L.A.J.; Rodenburg, R.J.; Sass, J.O.; Nouws, J.; Van Kaauwen, E.P.; Kleefstra, T.; Tranebjaerg, L.; De Vries, M.C.; Isohanni, P.; et al. 3-Methylglutaconic aciduria—Lessons from 50 genes and 977 patients. J. Inherit. Metab. Dis. 2013, 36, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.; Scholte, H.; Berden, J.; Moorsel, J.V.D.K.-V.; Luyt-Houwen, I.; Veer-Korthof, E.V.; Van Der Harten, J.; Sobotka-Plojhar, M. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Hornby, B.; McClellan, R.; Buckley, L.; Carson, K.; Gooding, T.; Vernon, H.J. Functional exercise capacity, strength, balance and motion reaction time in Barth syndrome. Orphanet J. Rare Dis. 2019, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bissler, J.J.; Tsoras, M.; Göring, H.H.H.; Hug, P.; Chuck, G.; Tombragel, E.; A McGraw, C.; Schlotman, J.; A Ralston, M.; Hug, G. Infantile Dilated X-Linked Cardiomyopathy, G4.5 Mutations, Altered Lipids, and Ultrastructural Malformations of Mitochondria in Heart, Liver, and Skeletal Muscle. Lab. Investig. 2002, 82, 335–344. [Google Scholar] [CrossRef]
- Xu, Y.; Phoon, C.K.; Berno, B.; D’Souza, K.; Hoedt, E.; Zhang, G.; A Neubert, E.H.G.Z.T.; Epand, K.D.R.M.; Ren, M.; Schlame, Y.X.M.R.M. Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat. Chem. Biol. 2016, 12, 641–647. [Google Scholar] [CrossRef]
- Kushmerick, M.J.; Moerland, T.S.; Wiseman, R.W. Mammalian skeletal muscle fibers distinguished by contents of phosphocreatine, ATP, and Pi. Proc. Natl. Acad. Sci. USA 1992, 89, 7521–7525. [Google Scholar] [CrossRef]
- Takahashi, H.; Kuno, S.-Y.; Katsuta, S.; Shimojo, H.; Masuda, K.; Yoshioka, H.; Anno, I.; Itai, Y. Relationships between Fiber Composition and NMR Measurements in Human Skeletal Muscle. NMR Biomed. 1996, 9, 8–12. [Google Scholar] [CrossRef]
- Cade, W.T.; Spencer, C.T.; Reeds, D.N.; Waggoner, A.D.; O’Connor, R.; Maisenbacher, M.; Crowley, J.R.; Byrne, B.J.; Peterson, L.R. Substrate metabolism during basal and hyperinsulinemic conditions in adolescents and young-adults with Barth syndrome. J. Inherit. Metab. Dis. 2013, 36, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure: A Translational Perspective. JACC Basic Transl. Sci. 2020, 5, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-W.; Galbraith, L.; Herndon, J.D.; Lu, Y.-L.; Pras-Raves, M.; Vervaart, M.; Van Kampen, A.; Luyf, A.; Koehler, C.M.; McCaffery, J.M.; et al. Defining functional classes of Barth syndrome mutation in humans. Hum. Mol. Genet. 2016, 25, 1754–1770. [Google Scholar] [CrossRef]
- Whited, K.; Baile, M.G.; Currier, P.; Claypool, S.M. Seven functional classes of Barth syndrome mutation. Hum. Mol. Genet. 2012, 22, 483–492. [Google Scholar] [CrossRef]
- Schlame, M.; I Kelley, R.; Feigenbaum, A.; A Towbin, J.; Heerdt, P.M.; Schieble, T.; A Wanders, R.J.; DiMauro, S.; Blanck, T.J.J. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003, 42, 1994–1999. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Rodenburg, R.J.; Thiels, C.; Van Lenthe, H.; Stet, F.; Poll-The, B.T.; Stone, J.E.; Steward, C.G.; Wanders, R.J.; Smeitink, J.A.M.; et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography–mass spectrometry as a diagnostic test for Barth syndrome. Anal. Biochem. 2009, 387, 230–237. [Google Scholar] [CrossRef]
- Xu, Y.; Sutachan, J.J.; Plesken, H.; I Kelley, R.; Schlame, Y.X.M.R.M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab. Investig. 2005, 85, 823–830. [Google Scholar] [CrossRef]
- Cade, W.T.; Laforest, R.; Bohnert, K.L.; Reeds, D.N.; Bittel, A.J.; Fuentes, L.D.L.; Bashir, A.; Woodard, P.K.; Pacak, C.A.; Byrne, B.J.; et al. Myocardial glucose and fatty acid metabolism is altered and associated with lower cardiac function in young adults with Barth syndrome. J. Nucl. Cardiol. 2019, 1–11. [Google Scholar] [CrossRef]
- Cade, W.T.; Bohnert, K.L.; Peterson, L.R.; Patterson, B.W.; Bittel, A.J.; Okunade, A.L.; Fuentes, L.D.L.; Steger-May, K.; Bashir, A.; Schweitzer, G.G.; et al. Blunted fat oxidation upon submaximal exercise is partially compensated by enhanced glucose metabolism in children, adolescents, and young adults with Barth syndrome. J. Inherit. Metab. Dis. 2019, 42, 480–493. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the Mitochondrial Protein Acylglycerol Kinase Causes Sengers Syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, A.; Haack, T.B.; Atiq, M.; Mottaghi, H.; Haghighi-Kakhki, H.; A Bashir, R.; Ahting, U.; Feichtinger, R.G.; A Mayr, J.; Rötig, A.; et al. Sengers syndrome: Six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J. Rare Dis. 2014, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Stroud, D.A.; Baker, M.J.; De Souza, D.P.; Frazier, A.E.; Liem, M.; Tull, D.; Mathivanan, S.; McConville, M.J.; Thorburn, D.R.; et al. Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 2017, 67, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Jordens, E.Z.; Palmieri, L.; Huizing, M.; Heuvel, L.V.D.; Sengers, R.C.A.; Dörner, A.; Ruitenbeek, W.; Trijbels, F.J.; Valsson, J.; Sigfusson, G.; et al. Adenine nucleotide translocator 1 deficiency associated with Sengers syndrome. Ann. Neurol. 2002, 52, 95–99. [Google Scholar] [CrossRef]
- Davey, K.M.; Parboosingh, J.S.; McLeod, D.R.; Chan, A.; Casey, R.; Ferreira, P.; Snyder, F.F.; Bridge, P.J.; Bernier, F.P. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J. Med. Genet. 2005, 43, 385–393. [Google Scholar] [CrossRef]
- Shen, Z.; Ye, C.; McCain, K.; Greenberg, M.L. The Role of Cardiolipin in Cardiovascular Health. Biomed. Res. Int. 2015, 2015, 1–12. [Google Scholar] [CrossRef]
- Sparkes, R.; Patton, D.; Bernier, F.P. Cardiac features of a novel autosomal recessive dilated cardiomyopathic syndrome due to defective importation of mitochondrial protein. Cardiol. Young 2007, 17, 215–217. [Google Scholar] [CrossRef]
- Janz, A.; Chen, R.; Regensburger, M.; Ueda, Y.; Rost, S.; Klopocki, E.; Günther, K.; Edenhofer, F.; Duff, H.J.; Ergün, S.; et al. Generation of two patient-derived iPSC lines from siblings (LIBUCi001-A and LIBUCi002-A) and a genetically modified iPSC line (JMUi001-A-1) to mimic dilated cardiomyopathy with ataxia (DCMA) caused by a homozygous DNAJC19 mutation. Stem Cell Res. 2020, 46, 101856. [Google Scholar] [CrossRef]
- Rohani, L.; Machiraju, P.; Sabouny, R.; Meng, G.; Liu, S.; Zhao, T.; Iqbal, F.; Wang, X.; Ravandi, A.; Wu, J.C.; et al. Reversible Mitochondrial Fragmentation in iPSC-Derived Cardiomyocytes From Children With DCMA, a Mitochondrial Cardiomyopathy. Can. J. Cardiol. 2020, 36, 554–563. [Google Scholar] [CrossRef]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a Mitochondrial Cochaperone Associated with Cardiomyopathy, Forms a Complex with Prohibitins to Regulate Cardiolipin Remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef]
- Jefferies, J.L. Barth syndrome. Am. J. Med Genet. Part C Semin. Med. Genet. 2013, 163, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Morales, D.L. Mechanical Circulatory Support in Children: Bridge to Transplant Versus Recovery. Curr. Heart Fail. Rep. 2012, 9, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.D.; Jaquiss, R.D.; Rosenthal, D.N.; Humpl, T.; Canter, C.E.; Blackstone, E.H.; Naftel, D.C.; Ichord, R.N.; Bomgaars, L.; Tweddell, J.S.; et al. Prospective Trial of a Pediatric Ventricular Assist Device. N. Engl. J. Med. 2012, 367, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Dedieu, N.; Giardini, A.; Steward, C.G.; Fenton, M.; Karimova, A.; Hsia, T.Y.; Burch, M. Successful mechanical circulatory support for 251 days in a child with intermittent severe neutropenia due to Barth syndrome. Pediatr. Transpl. 2012, 17, E46–E49. [Google Scholar] [CrossRef] [PubMed]
- Coman, D.J.; Yaplito-Lee, J.; Boneh, A. New indications and controversies in arginine therapy. Clin. Nutr. 2008, 27, 489–496. [Google Scholar] [CrossRef]
- Rigaud, C.; Lebre, A.-S.; Touraine, R.; Beaupain, B.; Ottolenghi, C.; Chabli, A.; Ansquer, H.; Ozsahin, H.; Di Filippo, S.; De Lonlay, P.; et al. Natural history of Barth syndrome: A national cohort study of 22 patients. Orphanet J. Rare Dis. 2013, 8, 70. [Google Scholar] [CrossRef]
- Suzuki-Hatano, S.; Saha, M.; Rizzo, S.A.; Witko, R.L.; Gosiker, B.J.; Ramanathan, M.; Soustek, M.S.; Jones, M.D.; Kang, P.B.; Byrne, B.J.; et al. AAV-Mediated TAZ Gene Replacement Restores Mitochondrial and Cardioskeletal Function in Barth Syndrome. Hum. Gene Ther. 2019, 30, 139–154. [Google Scholar] [CrossRef]
- Malhotra, A.; Edelman-Novemsky, I.; Xu, Y.; Plesken, H.; Ma, J.; Schlame, M.; Ren, M. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 2337–2341. [Google Scholar] [CrossRef]
- Schaloske, R.H.; Dennis, E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2006, 1761, 1246–1259. [Google Scholar] [CrossRef]
- Huang, Y.; Powers, C.; Moore, V.; Schafer, C.; Ren, M.; Phoon, C.K.L.; James, J.F.; Glukhov, A.V.; Javadov, S.; Vaz, F.M.; et al. The PPAR pan-agonist bezafibrate ameliorates cardiomyopathy in a mouse model of Barth syndrome. Orphanet J. Rare Dis. 2017, 12, 1–9. [Google Scholar] [CrossRef]
- Berger, J.P.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.H.; Jebessa, Z.H.; Kreusser, M.M.; Horsch, A.; He, T.; Kronlage, M.; Dewenter, M.; Sramek, V.; Oehl, U.; Krebs-Haupenthal, J.; et al. Faculty Opinions recommendation of A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat. Med. 2018, 24, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.; Moore, V.; Dasgupta, N.; Javadov, S.; James, J.F.; Glukhov, A.I.; Strauss, A.W.; Khuchua, Z. The Effects of PPAR Stimulation on Cardiac Metabolic Pathways in Barth Syndrome Mice. Front. Pharmacol. 2018, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Harris, N.; Ren, J.; Han, X. Mitochondria-Targeted Antioxidant Prevents Cardiac Dysfunction Induced by Tafazzin Gene Knockdown in Cardiac Myocytes. Oxid. Med. Cell. Longev. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Aich, A.; Jain, G.; Wozny, K.; Lüchtenborg, C.; Hartmann, M.; Bernhard, O.; Balleiniger, M.; Alfar, E.A.; Zieseniss, A.; et al. Defective Mitochondrial Cardiolipin Remodeling Dampens HIF-1α Expression in Hypoxia. Cell Rep. 2018, 25, 561–570.e6. [Google Scholar] [CrossRef]
- Grosberg, A.; Alford, P.W.; McCain, M.L.; Parker, K.K. Ensembles of engineered cardiac tissues for physiological and pharmacological study: Heart on a chip. Lab Chip 2011, 11, 4165–4173. [Google Scholar] [CrossRef]
- Agarwal, A.; Goss, J.A.; Cho, A.; McCain, M.L.; Parker, K.K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip 2013, 13, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Grosberg, A.; Nesmith, A.P.; Goss, J.A.; Brigham, M.D.; McCain, M.L.; Parker, K.K. Muscle on a chip: In vitro contractility assays for smooth and striated muscle. J. Pharmacol. Toxicol. Methods 2012, 65, 126–135. [Google Scholar] [CrossRef]
- Johnson, J.M.; Ferrara, P.J.; Verkerke, A.R.P.; Coleman, C.B.; Wentzler, E.J.; Neufer, P.D.; Kew, K.A.; Brás, L.E.D.C.; Funai, K. Targeted overexpression of catalase to mitochondria does not prevent cardioskeletal myopathy in Barth syndrome. J. Mol. Cell. Cardiol. 2018, 121, 94–102. [Google Scholar] [CrossRef]
- Rajagopal, B.S.; Silkstone, G.G.; Nicholls, P.; Wilson, M.T.; Worrall, J.A.R. An investigation into a cardiolipin acyl chain insertion site in cytochrome c. Biochim. Biophys. Acta Gen. Subj. 2012, 1817, 780–791. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Molkentin, J.D. Identifying the components of the elusive mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10396–10397. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochromec/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic Therapy With Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs With Advanced Heart Failure. Circ. Heart Fail. 2016, 9, e002206. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.-Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-Targeted Peptide Accelerates ATP Recovery and Reduces Ischemic Kidney Injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox Signal. 2009, 11, 2095–2104. [Google Scholar] [CrossRef]
- Machiraju, P.; Wang, X.; Sabouny, R.; Huang, J.; Zhao, T.; Iqbal, F.; King, M.; Prasher, D.; Lodha, A.; Jimenez-Tellez, N.; et al. SS-31 Peptide Reverses the Mitochondrial Fragmentation Present in Fibroblasts From Patients With DCMA, a Mitochondrial Cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 167. [Google Scholar] [CrossRef]
- Allen, M.E.; Pennington, E.R.; Perry, J.B.; Dadoo, S.; Makrecka-Kuka, M.; Dambrova, M.; Moukdar, F.; Patel, H.D.; Han, X.; Kidd, G.K.; et al. The cardiolipin-binding peptide elamipretide mitigates fragmentation of cristae networks following cardiac ischemia reperfusion in rats. Commun. Biol. 2020, 3, 1–12. [Google Scholar] [CrossRef]
- Xu, Y.; Condell, M.; Plesken, H.; Edelman-Novemsky, I.; Ma, J.; Ren, M.; Schlame, M. A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 11584–11588. [Google Scholar] [CrossRef]
- Acehan, D.; Khuchua, Z.; Houtkooper, R.H.; Malhotra, A.; Kaufman, J.; Vaz, F.M.; Ren, M.; Rockman, H.A.; Stokes, D.L.; Schlame, M. Distinct effects of tafazzin deletion in differentiated and undifferentiated mitochondria. Mitochondrion 2009, 9, 86–95. [Google Scholar] [CrossRef]
- Sakamoto, T.; Inoue, T.; Otomo, Y.; Yokomori, N.; Ohno, M.; Arai, H.; Nakagawa, Y. Deficiency of Cardiolipin Synthase Causes Abnormal Mitochondrial Function and Morphology in Germ Cells ofCaenorhabditis elegans. J. Biol. Chem. 2011, 287, 4590–4601. [Google Scholar] [CrossRef] [PubMed]
- Serricchio, M.; Bütikofer, P. An essential bacterial-type cardiolipin synthase mediates cardiolipin formation in a eukaryote. Proc. Natl. Acad. Sci. USA 2012, 109, E954–E961. [Google Scholar] [CrossRef] [PubMed]
- Soustek, M.S.; Falk, D.J.; Mah, C.S.; Toth, M.J.; Schlame, M.; Lewin, A.S.; Byrne, B.J. Characterization of a Transgenic Short Hairpin RNA-Induced Murine Model of Tafazzin Deficiency. Hum. Gene Ther. 2011, 22, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Landriscina, C.; Megli, F.M.; Quagliariello, E. Turnover of fatty acids in rat liver cardiolipin: Comparison with other mitochondrial phospholipids. Lipids 1976, 11, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Wahjudi, P.N.; Yee, J.K.; Martinez, S.R.; Zhang, J.; Teitell, M.; Nikolaenko, L.; Swerdloff, R.; Wang, C.; Lee, W.N.P. Turnover of nonessential fatty acids in cardiolipin from the rat heart. J. Lipid Res. 2011, 52, 2226–2233. [Google Scholar] [CrossRef]
- Boynton, T.O.; Shimkets, L.J. MyxococcusCsgA, DrosophilaSniffer, and human HSD10 are cardiolipin phospholipases. Genes Dev. 2015, 29, 1903–1914. [Google Scholar] [CrossRef]
- Shen, Z.; Li, Y.; Gasparski, A.N.; Abeliovich, H.; Greenberg, M.L. Cardiolipin Regulates Mitophagy through the Protein Kinase C Pathway. J. Biol. Chem. 2017, 292, 2916–2923. [Google Scholar] [CrossRef]
- Konno, Y.; Ohno, S.; Akita, Y.; Kawasaki, H.; Suzuki, K. Enzymatic Properties of a Novel Phorbol Ester Receptor/Protein Kinase, nPKC1. J. Biochem. 1989, 106, 673–678. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Anmial | Human |
|---|---|---|
| Mitochondrial morphology | A tafazzin knockdown mouse model of Barth syndrome describes alteration of mitochondrial phospholipid compositions via lipidomics [32]. Morphology alterations as mitochondrial enlargement, concentric layers of cristae or large vacuoles were observed in tafazzin-deficient mice [33]. | BTHS patient-derived lymphoblasts (BTHS lymphoblasts) reveal enlarged mitochondria with a lower surface area of cristae and altered morphology [34]. Another study with BTHS lymphoblasts detected giant, partly onion-shaped mitochondria [35]. |
| Oxidative phosphorylation | A BTHS mouse model with an inducible systemic knockdown of tafazzin gene shows a reduced respiration on succinate as well as on pyruvate and malate. Furthermore, respirasome remodeling was detected [36]. | BTHS patient-derived induced pluripotent stem cells (BTHS-iPSC) reveal structural remodeling of respiratory chain complexes resulting in decreased mitochondrial respiration [37]. In a second study, mitochondria of BTHS lymphoblasts show a reduced respiratory activity on succinate and ascorbate [34]. |
| Krebs cycle | BTHS mouse model reveals a striking reduction in succinate dehydrogenase activity in cardiac mitochondria [36]. | BTHS skin fibroblasts reveal a significant destabilization of 2-oxoglutarate dehydrogenase and branched-chain ketoacid dehydrogenase [38]. |
| Apoptosis | Murine germline TAZ knockout mice model reveals significant increased cardiomyocyte apoptosis and fibrosis occurrence [39]. | BTHS lymphoblasts show a requirement of cardiolipin for apoptosis and an apoptotic defect [40]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasmus, C.; Dudek, J. Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies. Life 2020, 10, 277. https://doi.org/10.3390/life10110277
Wasmus C, Dudek J. Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies. Life. 2020; 10(11):277. https://doi.org/10.3390/life10110277
Chicago/Turabian StyleWasmus, Christina, and Jan Dudek. 2020. "Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies" Life 10, no. 11: 277. https://doi.org/10.3390/life10110277
APA StyleWasmus, C., & Dudek, J. (2020). Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies. Life, 10(11), 277. https://doi.org/10.3390/life10110277