
Estimation Algorithm for a Hybrid PDE–ODE Model Inspired by Immunocompetent Cancer-on-Chip Experiment



Abstract
:1. Introduction
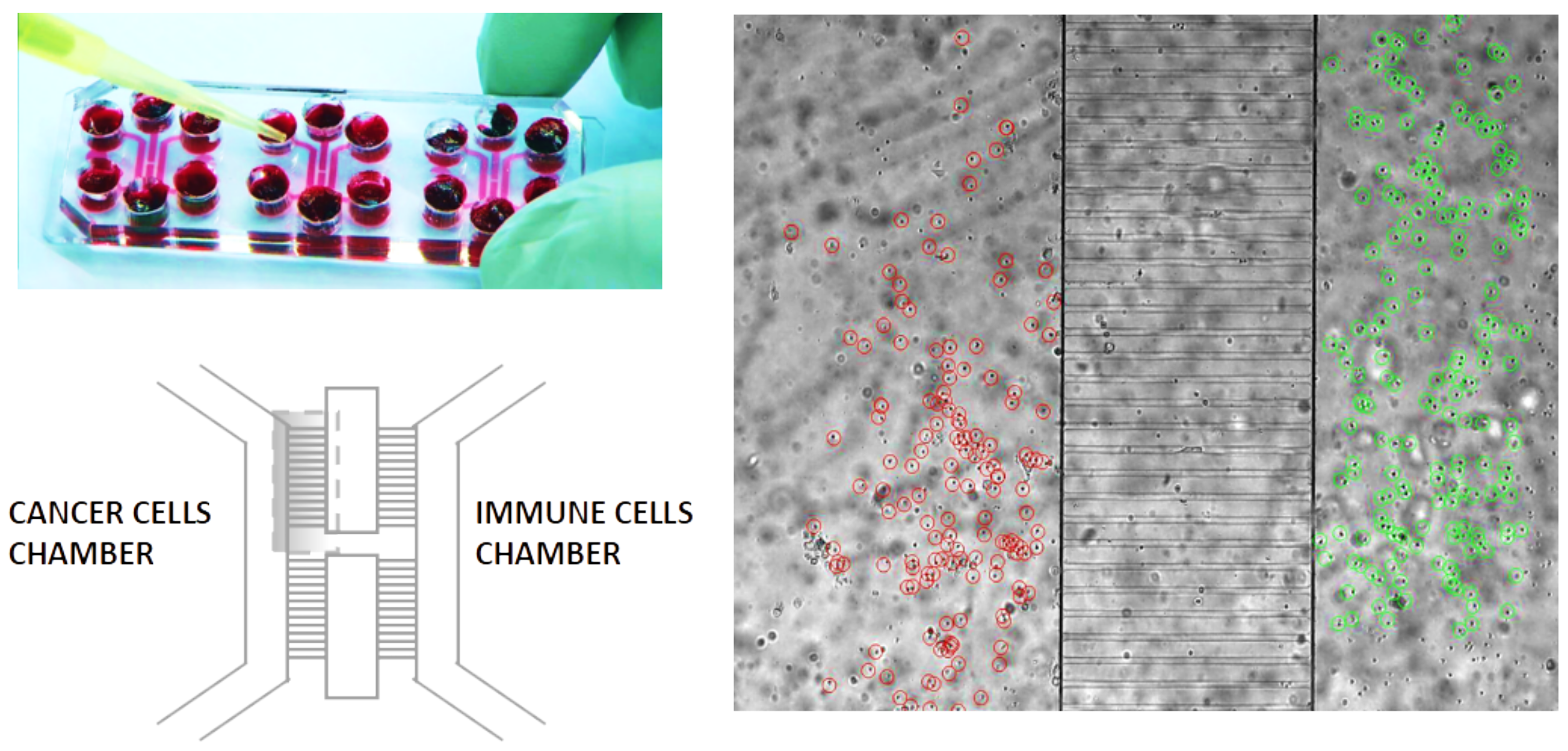
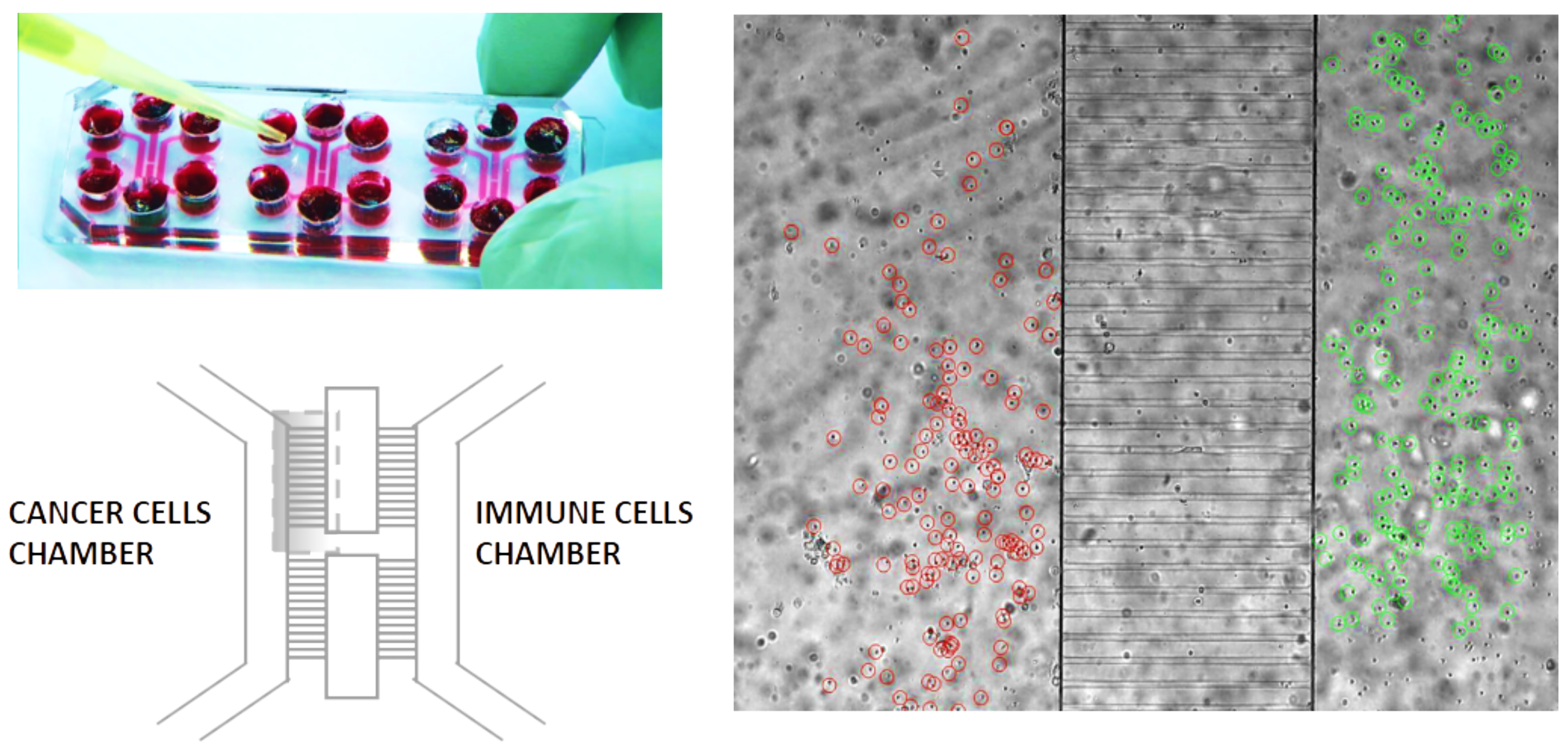
1.1. The Geometry of the Microfluidic Chip and the Related Computational Domain
- the reduction of the computational cost in view of the optimization procedure for the parameter estimation;
- the focus on short-range interactions.
1.2. Original Contribution of the Present Paper
- we model the presence of tumor cells, and we also take into account the repulsion forces to avoid overlapping. Eventually, a slight overlay may occur between tumor and immune cell in the case of close interactions. However, it does not seem to occur in the video footage;
- a chemical gradient concentrated around sources represented by cancer cells is considered and diffused in the environment;
- Robin boundary conditions for the inflow of chemoattractant in the area under consideration are applied and adjusted to drive cell migration in a diagonal direction, as observed experimentally;
- the alignment effect between cells is discarded since in this context is not present;
- a chemotactic sensitivity term—i.e., receptor saturation—is added in the drift term of the equation of particles motion, with the effect that chemotaxis of cells is reduced in areas of high chemoattractant concentrations;
- a stochastic component in the particle velocities is added to have more realistic cell trajectories in terms of randomness.
1.3. Main Contents and Plan of the Paper
- the development of a mathematical model describing the behavior of ICs in short-range interactions in the microfluidic chip environment;
- the development of ad-hoc parameter estimation techniques for time-varying velocity fields.
2. Materials and Methods
2.1. Biological Framework
Setting of the Laboratory Experiments
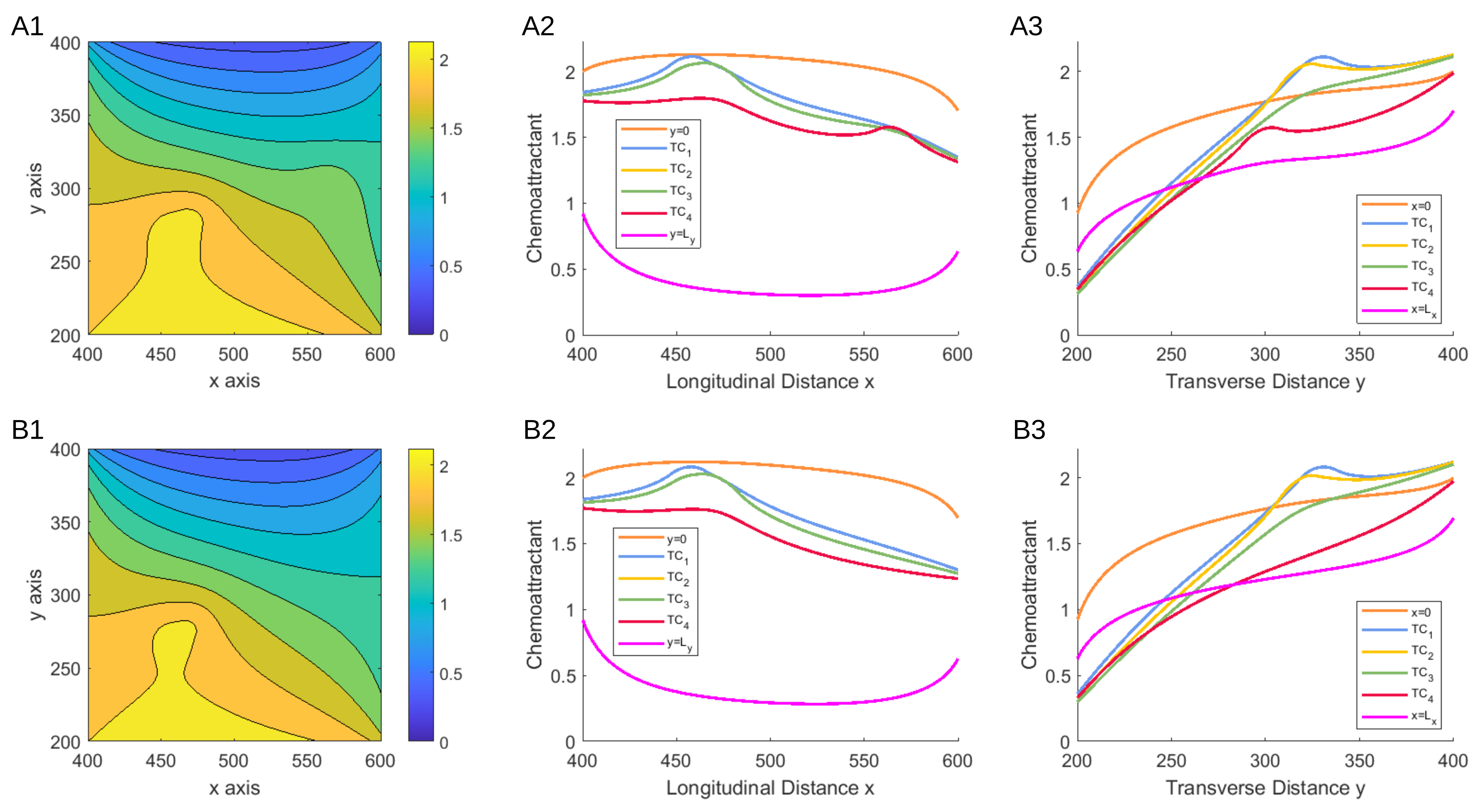
2.2. Mathematical Framework
The Model
2.3. Stochastic Model
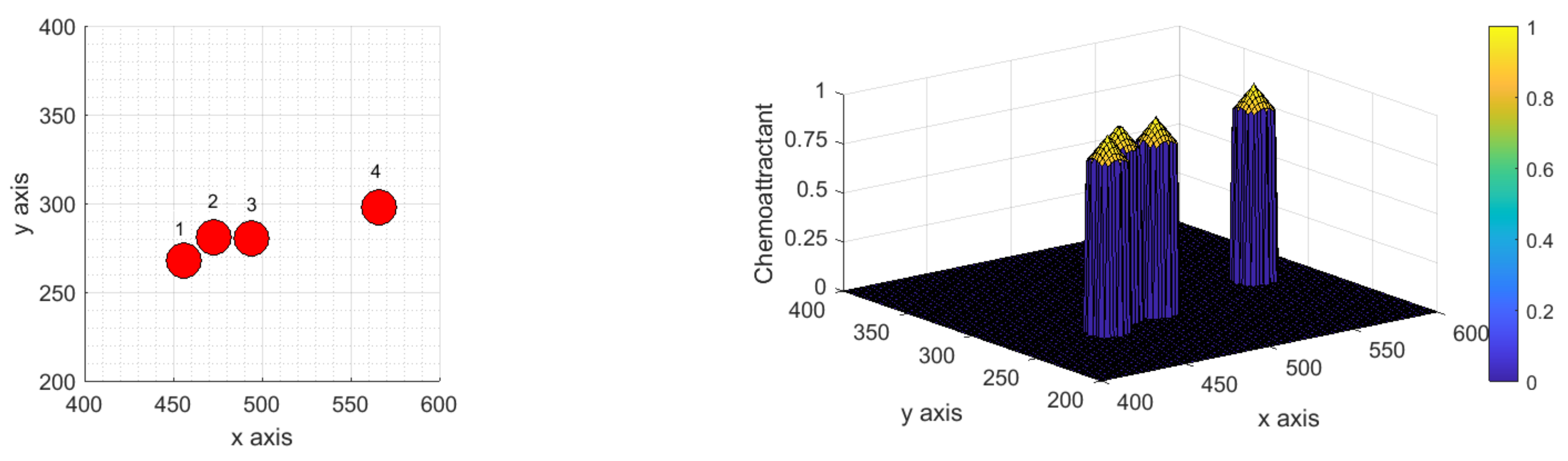
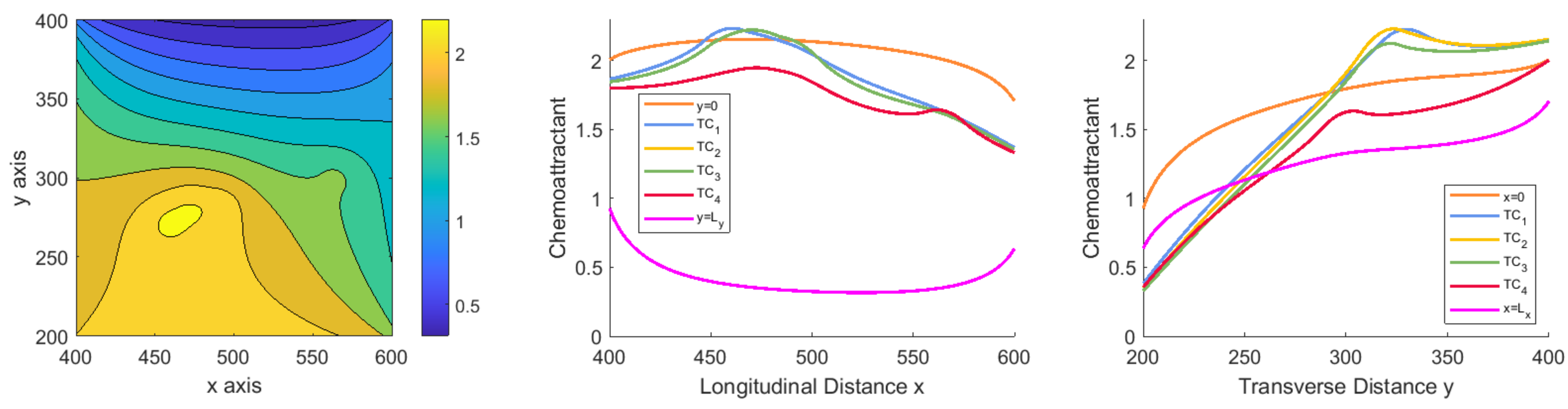
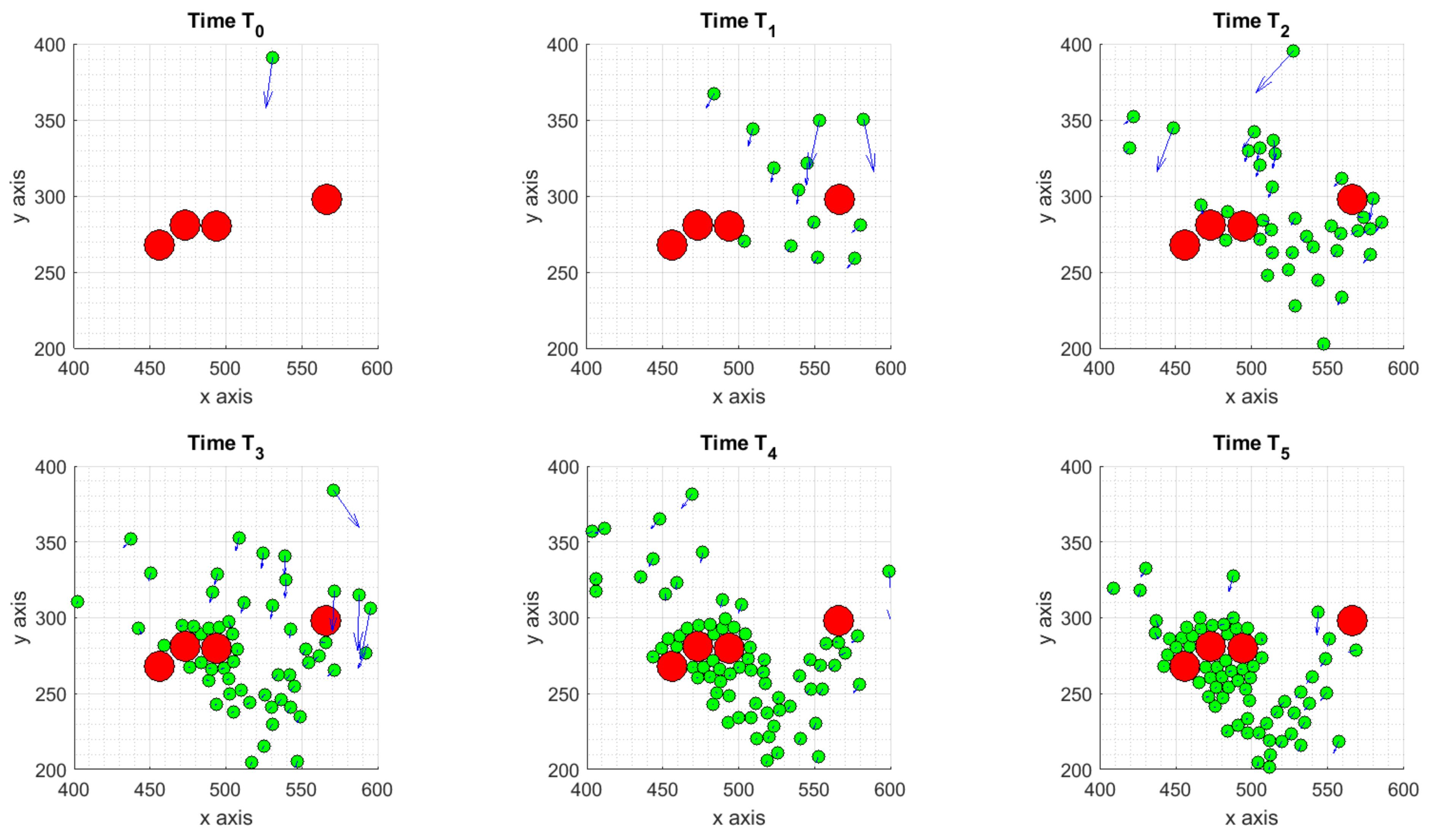
3. Study on Different Scenarios: Numerical Tests
3.1. Scenarios Representing Relevant Features of ICs Dynamics and Interactions
- 1.
- Deterministic Motion,
- 2.
- Deterministic Motion including Cell Death;
- 3.
- Stochastic Motion.
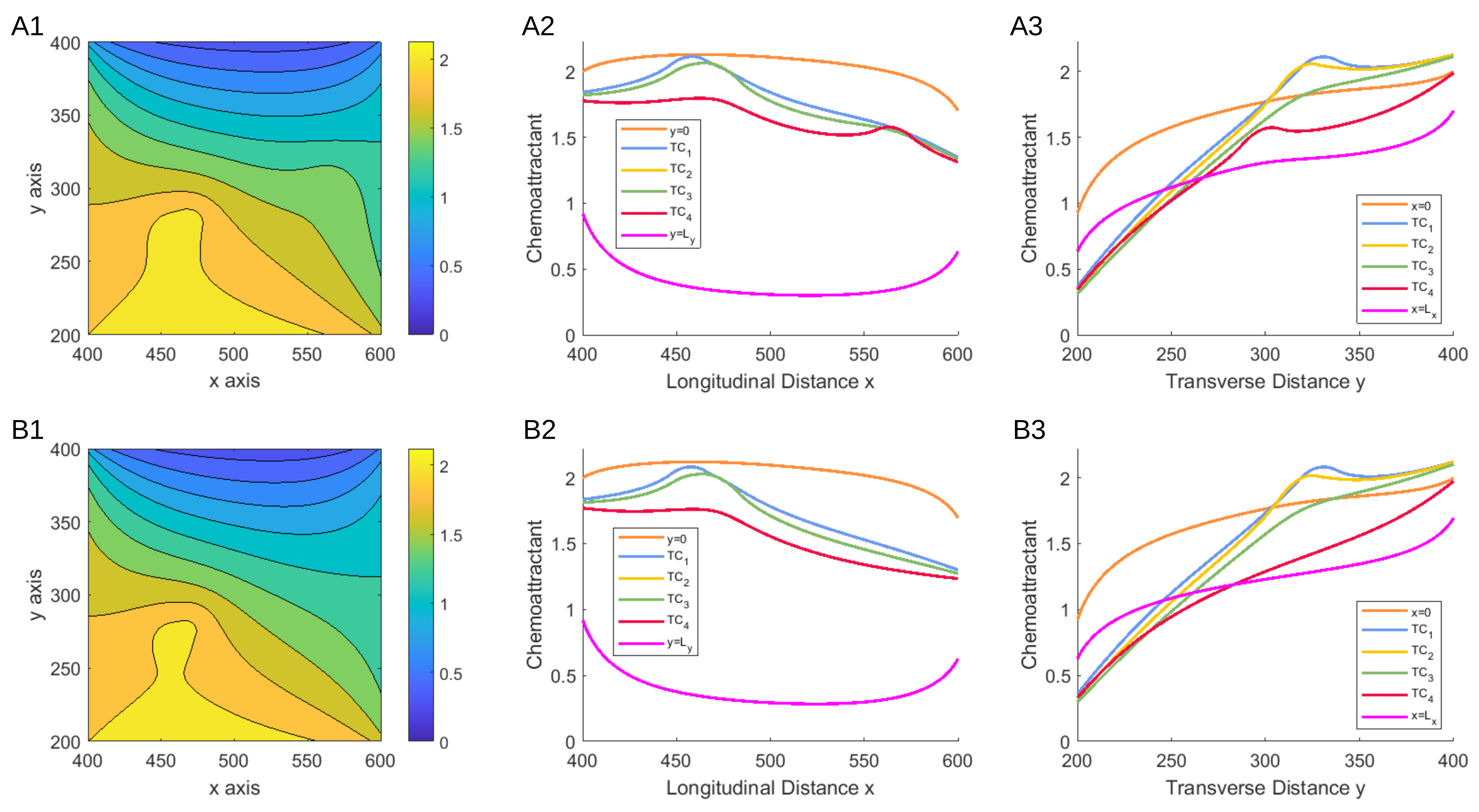
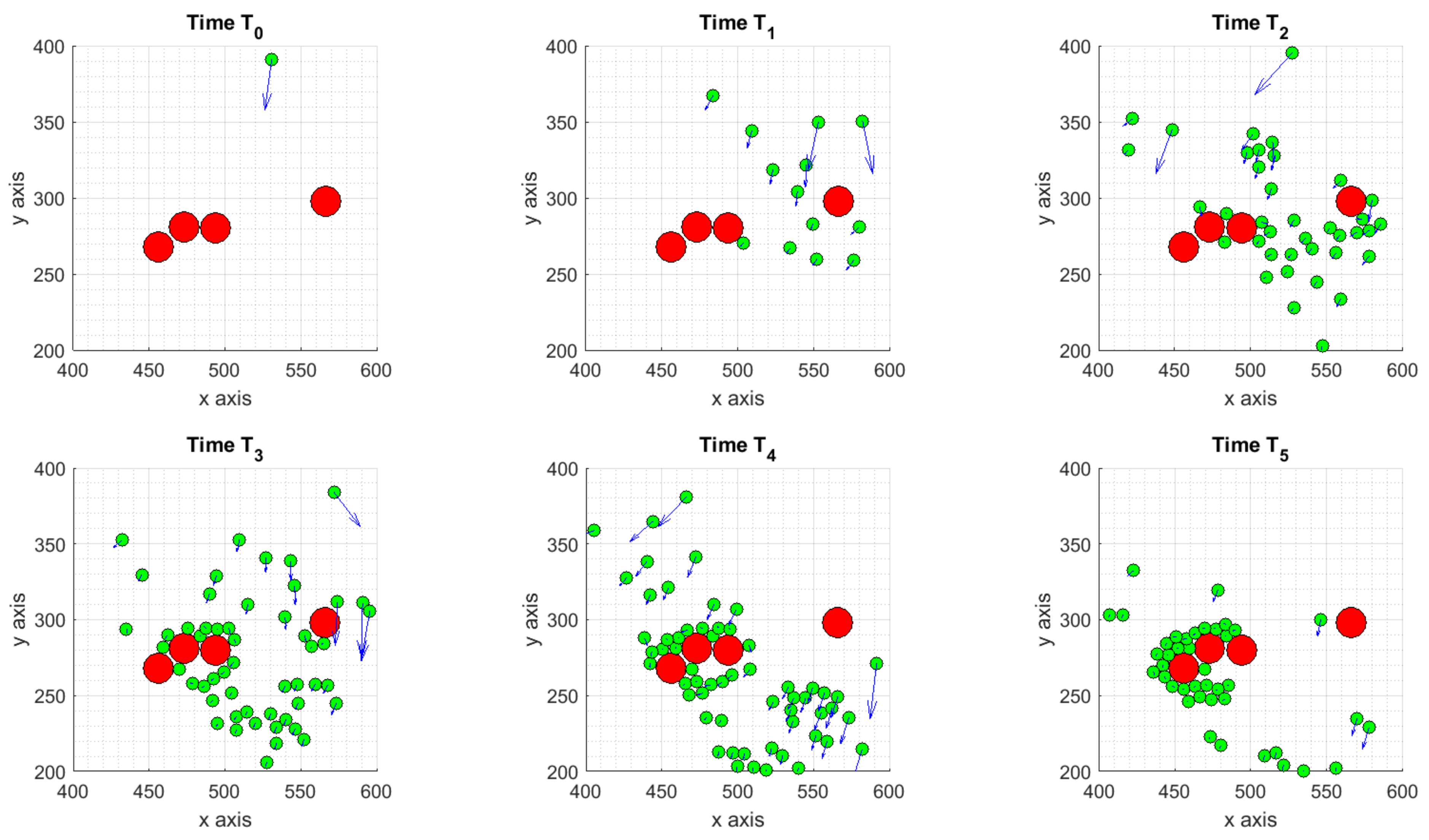
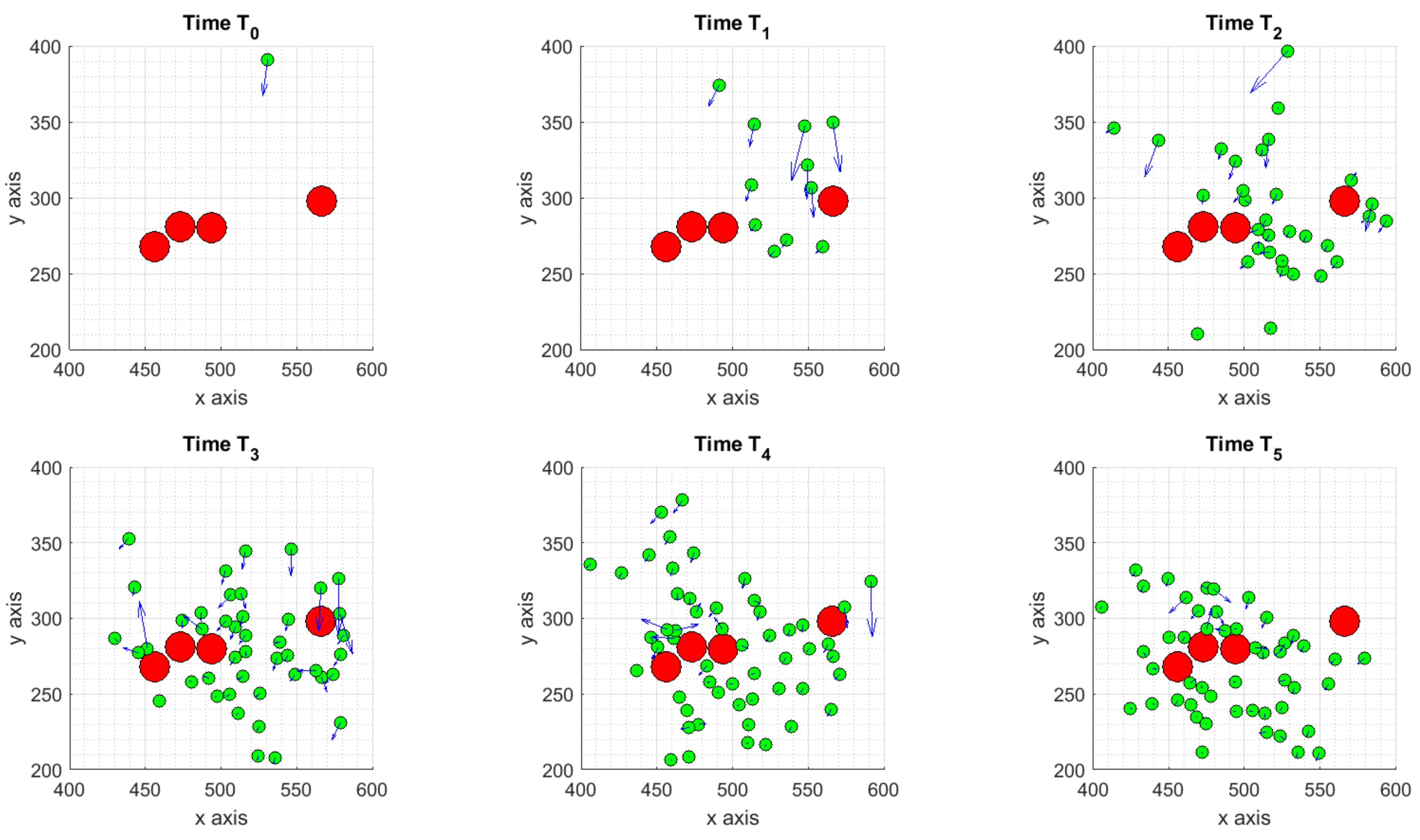
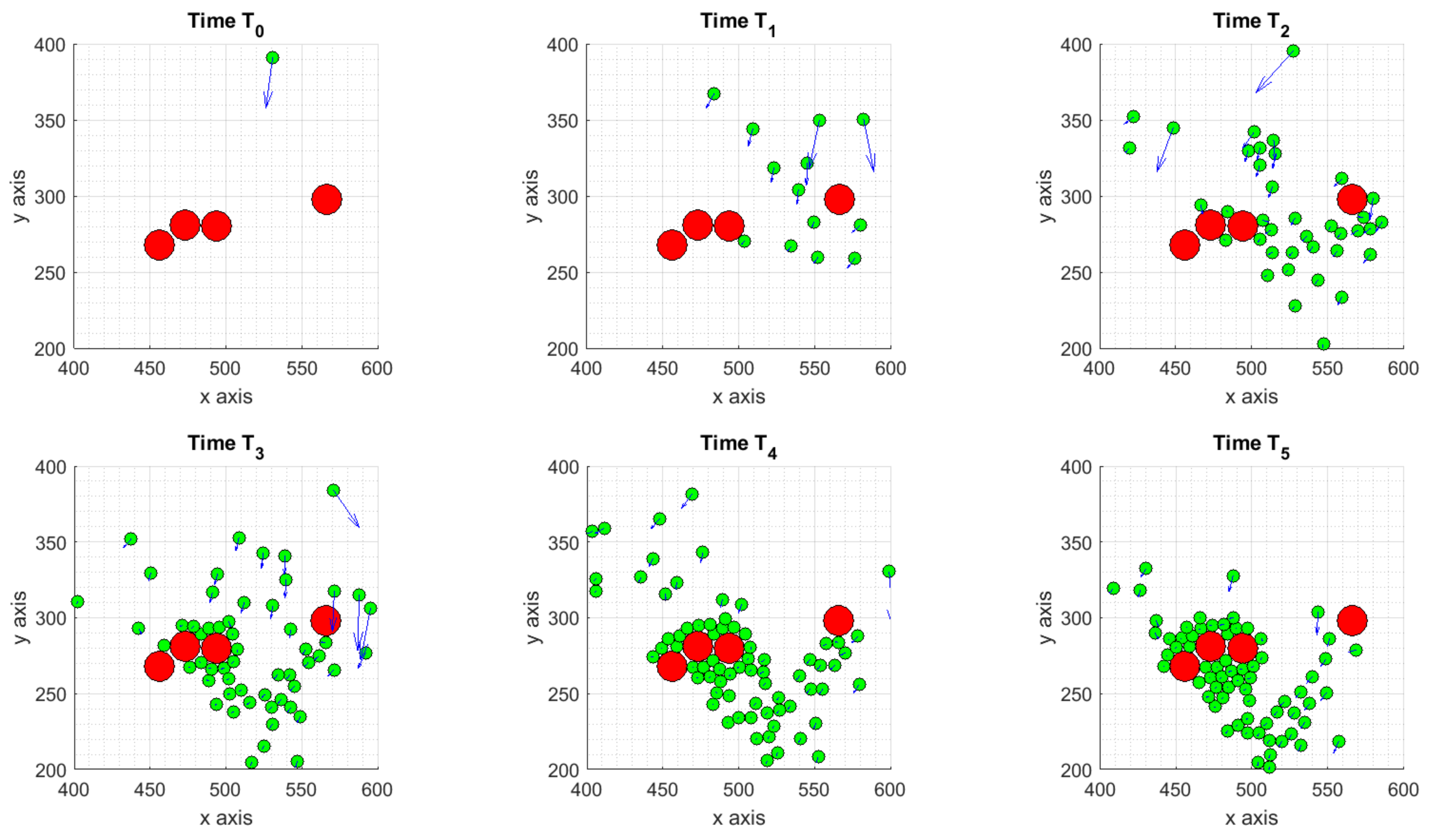
3.1.1. Scenario 1: Deterministic Motion
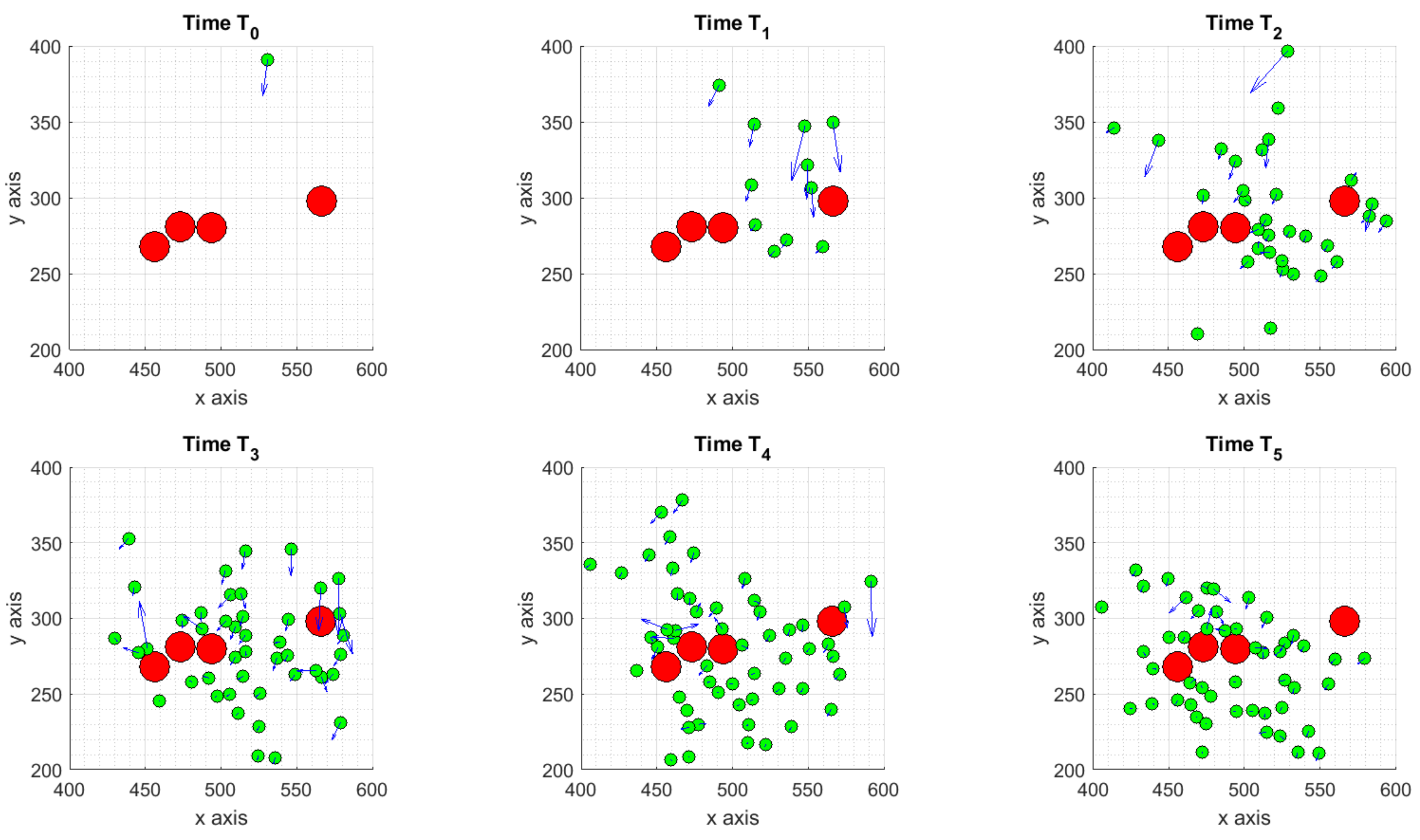
3.1.2. Scenario 2: Deterministic Motion including Cell Death
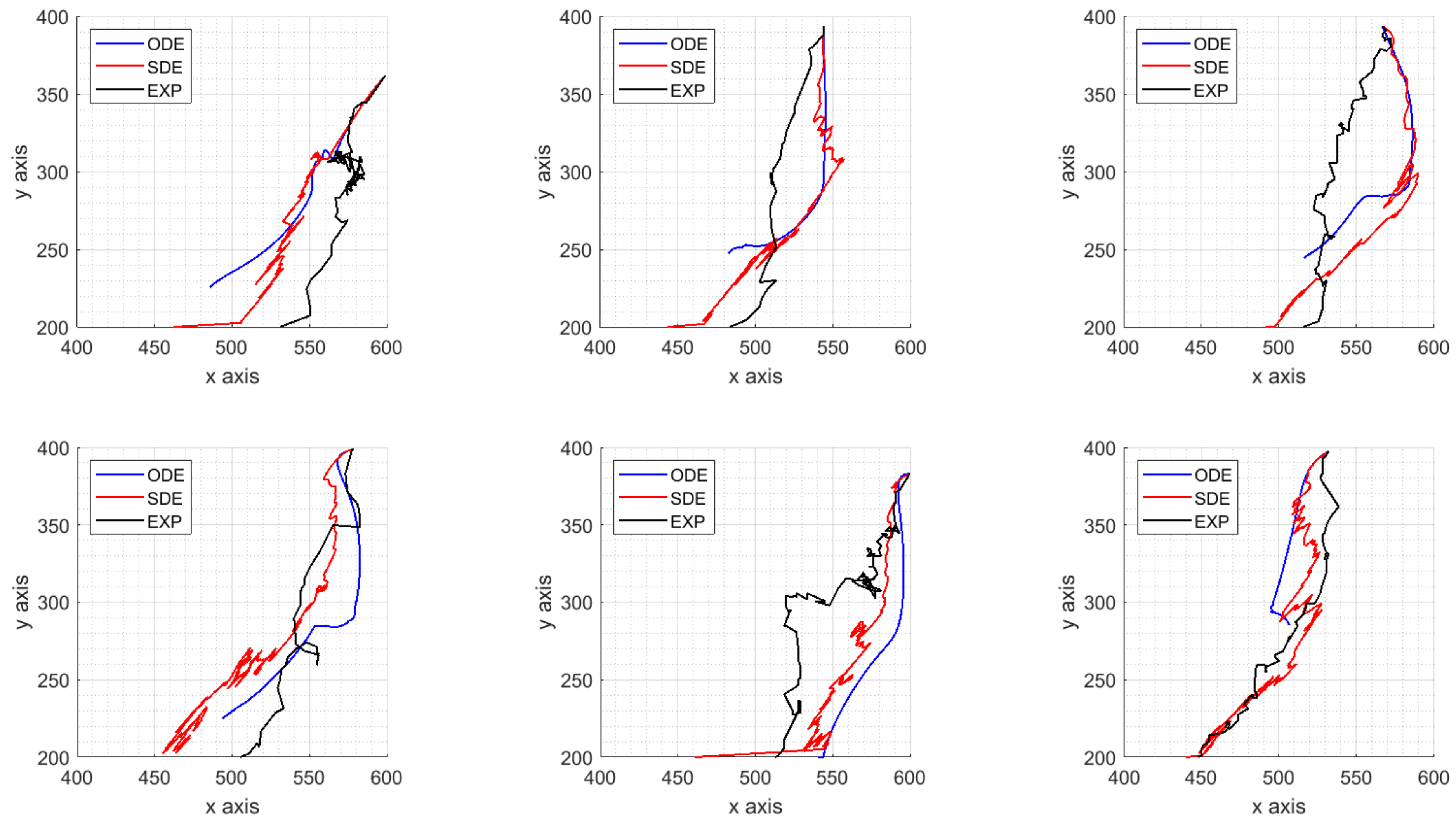
3.1.3. Scenario 3: Stochastic Motion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Units | Value | Ref. |
|---|---|---|---|---|
| Number of ICs in during 24 h | 97 | Experimental Data | ||
| Number of TCs in during 24 h | 4 | Experimental Data | ||
| Horizontal size of | Experimental Data | |||
| Vertical size of | Experimental Data | |||
| IC radius | [57] | |||
| TC radius | [58,59] | |||
| Detection radius of chemicals | Biological Assumption | |||
| Radius of action of repulsion between ICs | Biological Assumption | |||
| Radius of action of adhesion between ICs | Biological Assumption | |||
| Radius of action of Repulsion between ICs and TCs | Biological Assumption | |||
| D | Diffusion Coefficient | [60] | ||
| Growth Rate of f | [61] | |||
| Consumption Rate of f | [61] | |||
| Coefficient of Adhesion between ICs | Biological Assumption | |||
| Coefficient of Repulsion between ICs | Biological Assumption | |||
| Coefficient of Repulsion between ICs and TCs | Biological Assumption | |||
| Damping Coefficient | Biological Assumption | |||
| Cellular Drift Velocity | [60] | |||
| Receptor Dissociation Constant | [60] | |||
| a | Rate of exchange of the Chemoattractant with the external environment | 10 | Biological Assumption | |
| Coefficient of Chemotactic Effect. Scenarios 1 and 2 | Biological Assumption | |||
| Flux condition on . Scenarios 1 and 2 | Biological Assumption | |||
| Flux condition on | Biological Assumption | |||
| Flux condition on | Biological Assumption | |||
| Flux condition on . Scenarios 1 and 2 | Biological Assumption | |||
| Standard Deviation of x-trajectories | Experimental Data | |||
| Standard Deviation of y-trajectories | Experimental Data |
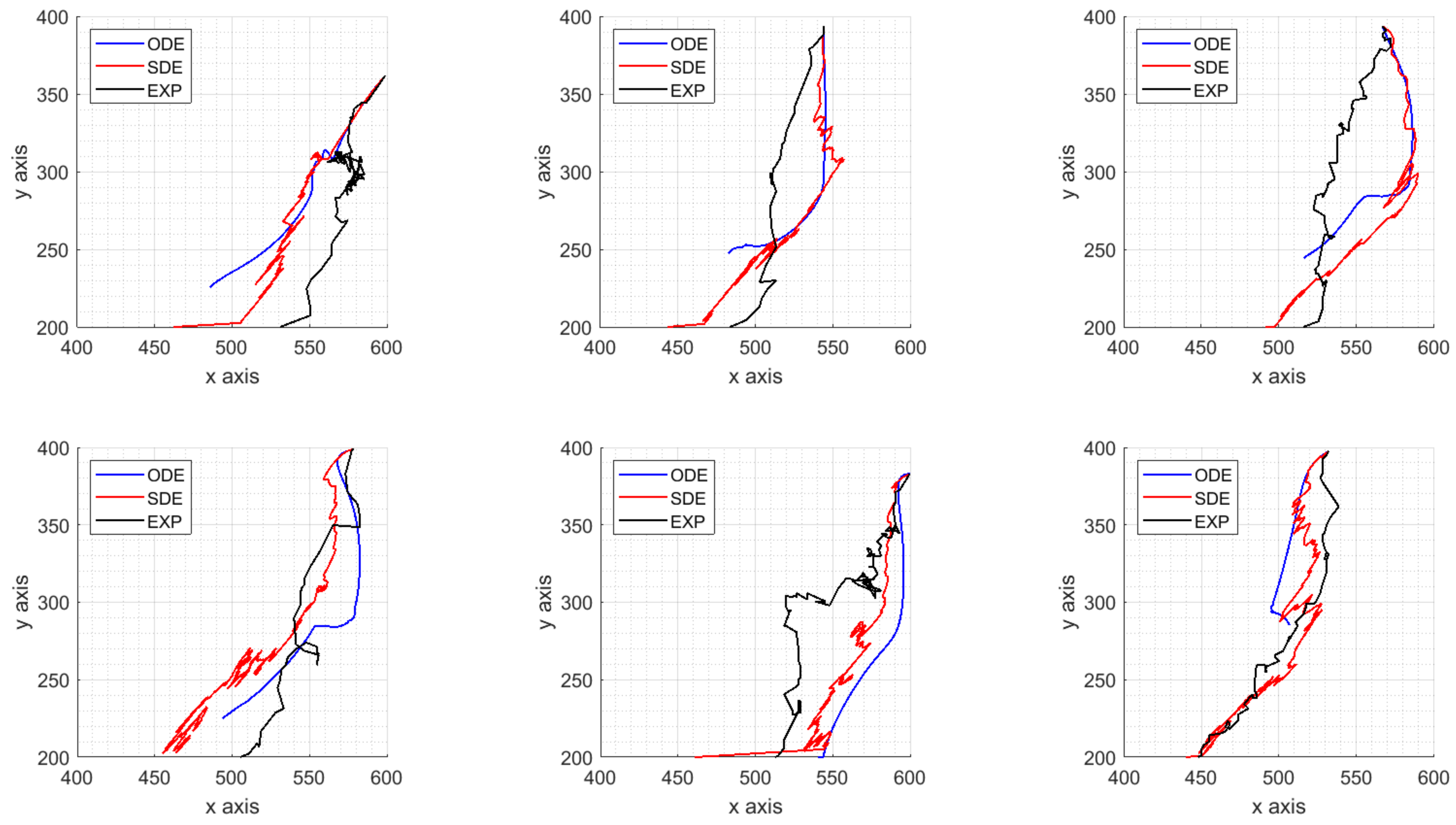
4. Parameters Estimation on Synthetic Data
- the dataset representing the target solutions of our procedure computed from synthetic ICs trajectories is obtained by the model with fixed parameters;
- a time-space approximation of the velocity fields is carried out with the spline technique presented in Section 4.1 and used as target solution of the calibration algorithm;
- perturbed model parameters are used as initial guess of a global search algorithm minimizing the norm of the difference between the target velocity fields and the estimated ones, as explained in Section 4.2.
4.1. Multidimensional Interpolation
4.2. The Calibration Algorithm
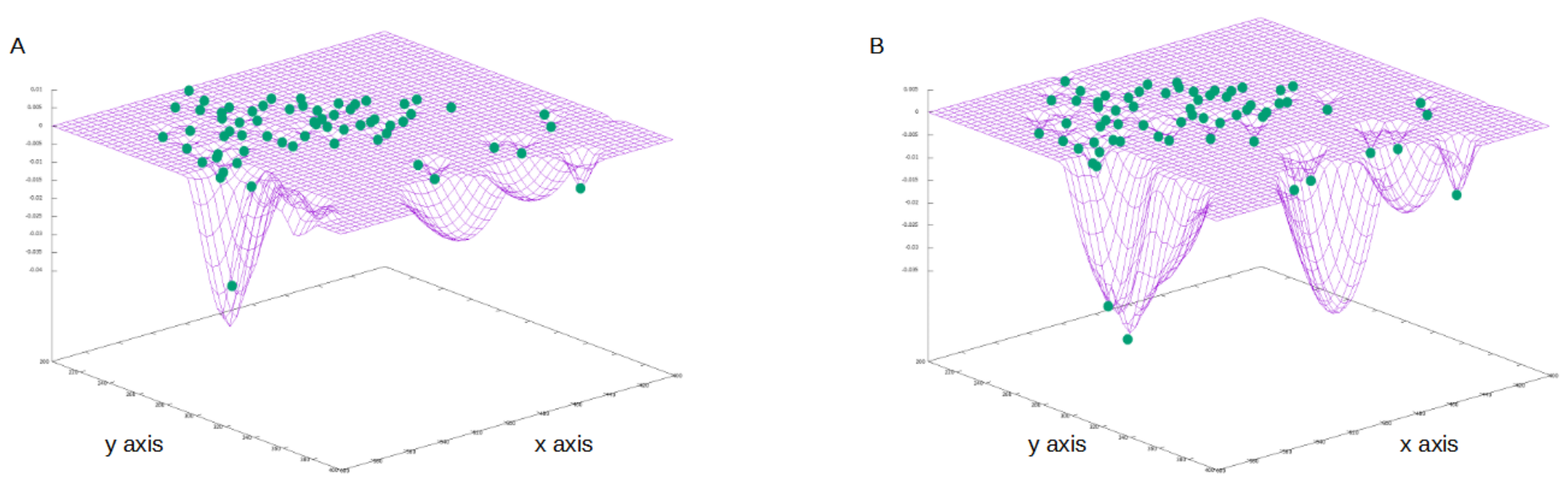
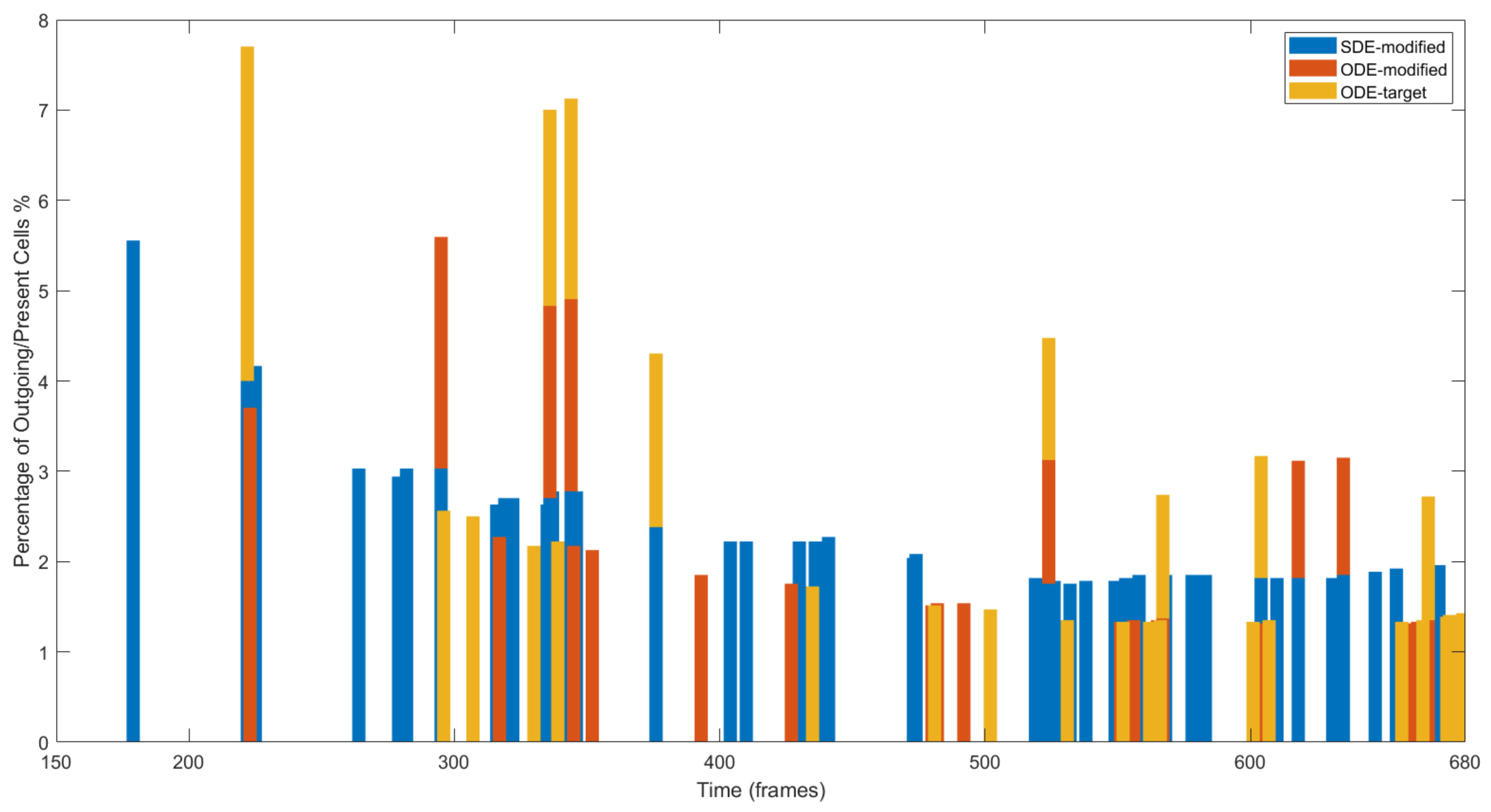
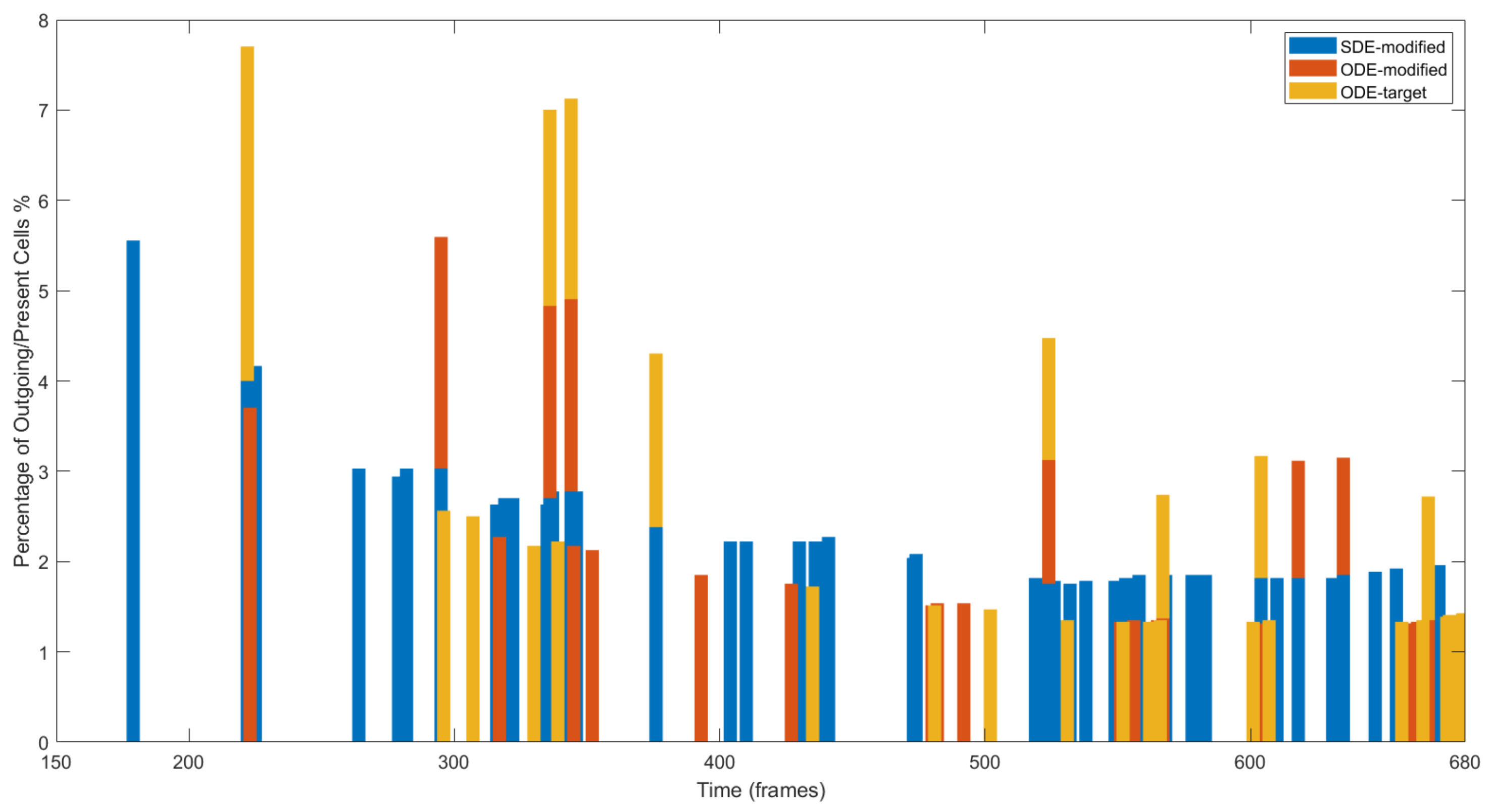
4.3. Results on Parameters Estimation
5. Conclusions and Future Work
- the deterministic scenario, with some ICs attracted by the tumor and others moving towards to the boundaries of the considered domain;
- the cell death scenario, as a subcase of the previous one, obtained assuming two TCs have died after their interaction with ICs, causing changes in the internal concentration of chemicals thus affecting ICs dynamics;
- the stochastic scenario, obtained adding to the equation of the motion a Brownian walk to mimic the randomness on ICs trajectories.
- the development of a simulation algorithm mimicking short-range dynamics of ICs in the neighborhood of TCs, as observed in Cancer-on-Chip experiment;
- the introduction of an ad-hoc methodology for the calibration of model parameters based on the time-space approximation of synthetic velocity fields computed from immune cell trajectories.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Discretization of the PDE
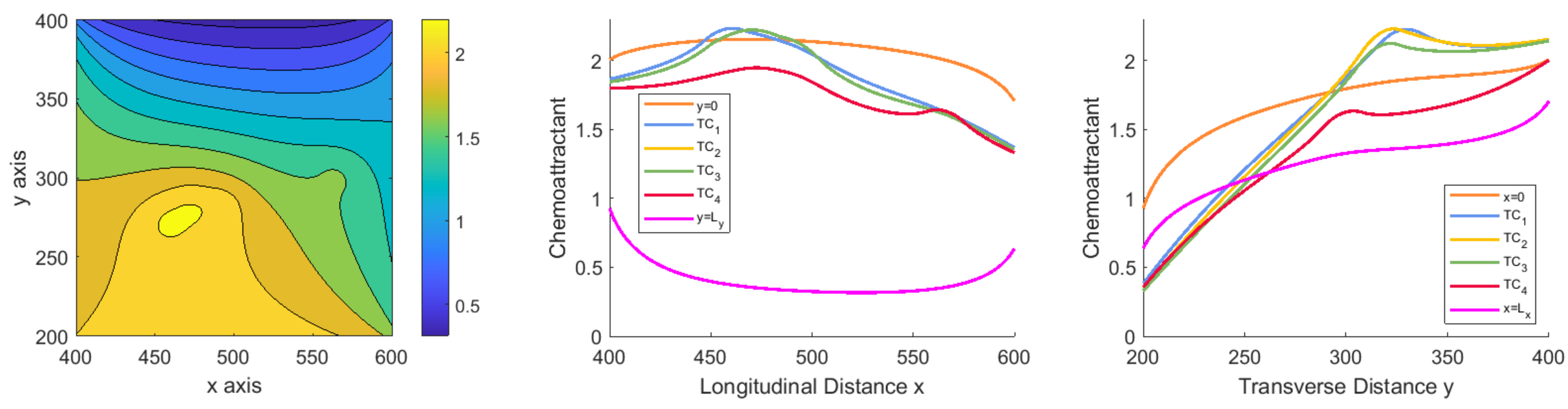
Appendix A.2. Boundary Conditions
Appendix A.3. Discretization of the ODE
Appendix A.4. Discretization of the SDE
References
- Businaro, L.; de Ninno, A.; Schiavoni, G.; Lucarini, V.; Ciasca, G.; Gerardino, A.; Belardelli, F.; Gabriele, L.; Mattei, F. Cross talk between cancer and immune cells: Exploring complex dynamics in a microfluidic environment. Lab Chip 2013, 13, 229–239. [Google Scholar] [CrossRef]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting Organ-Level Lung Functions on a Chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graney, P.L.; Tavakol, D.N.; Chramiec, A.; Ronaldson-Bouchard, K.; Vunjak-Novakovic, G. Engineered models of tumor metastasis with immune cell contributions. iScience 2021, 24, 102179. [Google Scholar] [CrossRef]
- Mattei, F.; Andreone, S.; Mencattini, A.; De Ninno, A.; Businaro, L.; Martinelli, E.; Schiavoni, G. Oncoimmunology Meets Organs-on-Chip. Front. Mol. Biosci. 2021, 8, 627454. [Google Scholar] [CrossRef]
- Maulana, T.I.; Kromidas, E.; Wallstabe, L.; Cipriano, M.; Alb, M.; Zaupa, C.; Hudecek, M.; Fogal, B.; Loskill, P. Immunocompetent cancer-on-chip models to assess immuno-oncology therapy. Adv. Drug Deliv. Rev. 2021, 173, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Parlato, S.; Grisanti, G.; Sinibaldi, G.; Peruzzi, G.; Casciola, C.M.; Gabriele, L. Tumor-on-a-chip platforms to study cancer–immune system crosstalk in the era of immunotherapy. Lab Chip 2020, 21, 234–253. [Google Scholar] [CrossRef]
- Montanez-Sauri, S.I.; Sung, K.E.; Berthier, E.; Beebe, D.J. Enabling screening in 3D microenvironments: Probing matrix and stromal effects on the morphology and proliferation of T47D breast carcinoma cells. Integr. Biol. 2013, 5, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Hassell, B.; Goyal, G.; Lee, E.; Sontheimer-Phelps, A.; Levy, O.; Chen, C.; Ingber, D.E. Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In Vitro. Cell Rep. 2017, 21, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.; Trappmann, B.; Stapleton, S.C.; Toro, E.; Chen, C. Microfluidics embedded within extracellular matrix to define vascular architectures and pattern diffusive gradients. Lab Chip 2013, 13, 3246–3252. [Google Scholar] [CrossRef] [Green Version]
- Bischel, L.L.; Young, E.W.; Mader, B.R.; Beebe, D.J. Tubeless microfluidic angiogenesis assay with three-dimensional endothelial-lined microvessels. Biomaterials 2012, 34, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.B.; Whisler, J.A.; Jeon, J.; Kamm, R.D. Mechanisms of tumor cell extravasation in an in vitro microvascular network platform. Integr. Biol. 2013, 5, 1262–1271. [Google Scholar] [CrossRef] [Green Version]
- Moya, M.L.; Hsu, Y.-H.; Lee, A.; Hughes, C.C.; George, S.C. In Vitro Perfused Human Capillary Networks. Tissue Eng. Part C Methods 2013, 19, 730–737. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.-H.T.; Stapleton, S.C.; Yang, M.T.; Cha, S.S.; Choi, C.K.; Galie, P.; Chen, C.S. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 6712–6717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Phan, D.T.T.; Sobrino, A.; George, S.C.; Hughes, C.C.W.; Lee, A.P. Engineering anastomosis between living capillary networks and endothelial cell-lined microfluidic channels. Lab Chip 2015, 16, 282–290. [Google Scholar] [CrossRef]
- Jeong, S.-Y.; Lee, J.-H.; Shin, Y.; Chung, S.; Kuh, H.-J. Co-Culture of Tumor Spheroids and Fibroblasts in a Collagen Matrix-Incorporated Microfluidic Chip Mimics Reciprocal Activation in Solid Tumor Microenvironment. PLoS ONE 2016, 11, e0159013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Kim, S.-K.; Khawar, I.A.; Jeong, S.-Y.; Chung, S.; Kuh, H.-J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2002, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Zervantonakis, I.; Hughes, S.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA 2012, 109, 13515–13520. [Google Scholar] [CrossRef] [Green Version]
- Braun, E.; Bretti, G.; Natalini, R. Mass-Preserving Approximation of a Chemotaxis Multi-Domain Transmission Model for Microfluidic Chips. Mathematics 2021, 9, 688. [Google Scholar] [CrossRef]
- Natalini, R.; Paul, T. On the Mean Field limit for Cucker-Smale models. Discret. Contin. Dyn.-Syst. B 2021. [Google Scholar] [CrossRef]
- Agliari, E.; Biselli, E.; De Ninno, A.; Schiavoni, G.; Gabriele, L.; Gerardino, A.; Mattei, F.; Barra, A.; Businaro, L. Cancer-driven dynamics of immune cells in a microfluidic environment. Sci. Rep. 2014, 4, 6639. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.X.; Fu, K.Y.; Sumalee, A.; Ngoduy, D.; Lam, W.H. A cross-entropy method and probabilistic sensitivity analysis framework for calibrating microscopic traffic models. Transp. Res. Part C Emerg. Technol. 2016, 63, 147–169. [Google Scholar] [CrossRef]
- Kirschner, D.; Panetta, J.C. Modeling immunotherapy of the tumor-immune interaction. J. Math. Biol. 1998, 37, 235–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.W.L.; Seager, R.J.; Litvak, F.; Spill, F.; Sieow, J.L.; Leong, P.H.; Kumar, D.; Tan, A.S.M.; Wong, S.C.; Adriani, G.; et al. Integrated in silico and 3D in vitro model of macrophage migration in response to physical and chemical factors in the tumor microenvironment. Integr. Biol. 2020, 12, 90–108. [Google Scholar] [CrossRef]
- Yang, T.D.; Park, J.-S.; Choi, Y.; Choi, W.; Ko, T.-W.; Lee, K.J. Zigzag Turning Preference of Freely Crawling Cells. PLoS ONE 2011, 6, e20255. [Google Scholar] [CrossRef] [Green Version]
- Checcoli, A.; Pol, J.G.; Naldi, A.; Noel, V.; Barillot, E.; Kroemer, G.; Thieffry, D.; Calzone, L.; Stoll, G. Dynamical Boolean Modeling of Immunogenic Cell Death. Front. Physiol. 2020, 11, 1320. [Google Scholar] [CrossRef]
- Braun, E.C. Organs-On-Chips: Mathematical Modelling and Parameter Estimation. Ph.D. Thesis, Universitá degli Studi di Roma Tre, Roma, Italy, 2021. [Google Scholar]
- Di Costanzo, E.; Natalini, R.; Preziosi, L. A hybrid mathematical model for self-organizing cell migration in the zebrafish lateral line. J. Math. Biol. 2014, 71, 171–214. [Google Scholar] [CrossRef] [Green Version]
- Peri, D. Easy-to-implement multidimensional spline interpolation with application to ship design optimisation. Ship Technol. Res. 2017, 65, 32–46. [Google Scholar] [CrossRef]
- Stevens, A.; Othmer, H.G. Aggregation, Blowup, and Collapse: The ABC’s of Taxis in Reinforced Random Walks. SIAM J. Appl. Math. 1997, 57, 1044–1081. [Google Scholar] [CrossRef]
- Pomeau, Y.; Piasecki, J. The Langevin equation. C. R. Phys. 2017, 18, 570–582. [Google Scholar] [CrossRef]
- Bai, J.; Tu, T.-Y.; Kim, C.; Thiery, J.P.; Kamm, R.D. Identification of drugs as single agents or in combination to prevent carcinoma dissemination in a microfluidic 3D environment. Oncotarget 2015, 6, 36603–36614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Gao, Y.; Hao, Y.; Li, E.; Wang, Y.; Zhang, J.; Wang, W.; Gao, Z.; Wang, Q. Application of a microfluidic chip-based 3D co-culture to test drug sensitivity for individualized treatment of lung cancer. Biomaterials 2013, 34, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- De Ninno, A.; Bertani, F.R.; Gerardino, A.; Schiavoni, G.; Musella, M.; Galassi, C.; Mattei, F.; Sistigu, A.; Businaro, L. Microfluidic Co-Culture Models for Dissecting the Immune Response in in vitro Tumor Microenvironments. J. Vis. Exp. 2021, 170, e61895. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Greenberg, J.M.; Alt, W. Stability results for a diffusion equation with functional drift approximating a chemotaxis model. Trans. Am. Math. Soc. 1987, 300, 235. [Google Scholar] [CrossRef]
- Keller, E.F.; Segel, L.A. Initiation of slime mold aggregation viewed as an instability. J. Theor. Biol. 1970, 26, 399–415. [Google Scholar] [CrossRef]
- Péré, M.; Chaves, M.; Roux, J. Core Models of Receptor Reactions to Evaluate Basic Pathway Designs Enabling Heterogeneous Commitments to Apoptosis. In Proceedings of the International Conference on Computational Methods in Systems Biology, Konstanz, Germany, 23–25 September 2020; pp. 298–320. [Google Scholar] [CrossRef]
- Edalgo, Y.T.N.; Zornes, A.L.; Versypt, A.N.F. A hybrid discrete–continuous model of metastatic cancer cell migration through a remodeling extracellular matrix. AIChE J. 2019, 65, e16671. [Google Scholar] [CrossRef]
- Othmer, H.G.; Kim, Y. Hybrid models of cell and tissue dynamics in tumor growth. Math. Biosci. Eng. 2015, 12, 1141–1156. [Google Scholar] [CrossRef]
- Perfahl, H.; Hughes, B.; Alarcon, T.; Maini, P.K.; Lloyd, M.C.; Reuss, M.; Byrne, H.M. 3D hybrid modelling of vascular network formation. J. Theor. Biol. 2017, 414, 254–268. [Google Scholar] [CrossRef]
- Rousset, M.; Samaey, G. Simulating individual-based models of bacterial chemotaxis with asymptotic variance reduction. Math. Model. Methods Appl. Sci. 2013, 23, 2155–2191. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, F.R.; Chaplain, M.A.J.; Lorenzi, T. A hybrid discrete-continuum approach to model Turing pattern formation. Math. Biosci. Eng. 2017, 17, 7442–7479. [Google Scholar] [CrossRef]
- Othmer, H.G. Cell-Based, Continuum and Hybrid Models of Tissue Dynamics. In Mathematical Models and Methods for Living Systems Book Series: Lecture Notes in Mathematics; Springer International Publishing Switzerland: Cham, Switzerland, 2016; pp. 1–72. [Google Scholar] [CrossRef]
- Mertz, A.F.; Che, Y.; Banerjee, S.; Goldstein, J.M.; Rosowski, K.A.; Revilla, S.F.; Niessen, C.M.; Marchetti, M.C.; Dufresne, E.R.; Horsley, V. Cadherin-based intercellular adhesions organize epithelial cell-matrix traction forces. Proc. Natl. Acad. Sci. USA 2012, 110, 842–847. [Google Scholar] [CrossRef] [Green Version]
- D’Orsogna, M.R.; Chuang, Y.L.; Bertozzi, A.L.; Chayes, L.S. Self-Propelled Particles with Soft-Core Interactions: Patterns, Stability, and Collapse. Phys. Rev. Lett. 2006, 96, 104302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayly, P.V.; Taber, L.A.; Carlsson, A.E. Damped and persistent oscillations in a simple model of cell crawling. J. R. Soc. Interface 2011, 9, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Fournier, M.F.; Sauser, R.; Ambrosi, D.; Meister, J.-J.; Verkhovsky, A. Force transmission in migrating cells. J. Cell Biol. 2010, 188, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, B.; Fournier, M.F.; Jacobson, K.; Verkhovsky, A.; Mogilner, A. Actin-Myosin Viscoelastic Flow in the Keratocyte Lamellipod. Biophys. J. 2009, 97, 1853–1863. [Google Scholar] [CrossRef] [Green Version]
- Lapidus, I.; Schiller, R. Model for the chemotactic response of a bacterial population. Biophys. J. 1976, 16, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Cristiani, E.; Piccoli, B.; Tosin, A. Multiscale Modeling of Granular Flows with Application to Crowd Dynamics. Multiscale Model. Simul. 2011, 9, 155–182. [Google Scholar] [CrossRef] [Green Version]
- Scianna, M.; Tosin, A.; Preziosi, L. From discrete to continuous models of cell colonies: A measure-theoretic approach. arXiv 2011, arXiv:1108.1212. [Google Scholar]
- Weninger, W.; Biro, M.; Jain, R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat. Rev. Immunol. 2014, 14, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Wei, S.H.; Parker, I.; Cahalan, M.D. Two-Photon Imaging of Lymphocyte Motility and Antigen Response in Intact Lymph Node. Science 2002, 296, 1869–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiśniewski, J.R.; Hein, M.; Cox, J.; Mann, M. A “Proteomic Ruler” for Protein Copy Number and Concentration Estimation without Spike-in Standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [Green Version]
- Boulter, E.; Grall, D.; Cagnol, S.; Van Obberghen-Schilling, E. Regulation of cell-matrix adhesion dynamics and Rac-1 by integrin linked kinase. FASEB J. 2006, 20, 1489–1491. [Google Scholar] [CrossRef]
- Puck, T.T.; Marcus, P.I.; Cieciura, S.J. Clonal growth of mammalian cells in vitro. J. Exp. Med. 1956, 103, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.D. Mathematical Biology. II: Spatial Models and Biomedical Applications, 3rd ed.; Springer: Berlin, Germany, 2002. [Google Scholar]
- Curk, T.; Marenduzzo, D.; Dobnikar, J. Chemotactic Sensing towards Ambient and Secreted Attractant Drives Collective Behaviour of E. coli. PLoS ONE 2013, 8, e74878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engl, H.W.; Hanke, M.; Neubauer, A. Regularization of Inverse Problems; Springer: Dordrecht, The Netherlands, 2000; Volume 375. [Google Scholar]
- Kennedy, J.; Eberhart, R. Particle swarm optimization. In Proceedings of the ICNN’95–International Conference on Neural Networks, Perth, WA, Australia, 27 November–1 December 1995; Volume 4, pp. 1942–1948. [Google Scholar] [CrossRef]
- Harris, T.; Banigan, E.; Christian, D.A.; Konradt, C.; Wojno, E.T.; Norose, K.; Wilson, E.H.; John, B.; Weninger, W.; Luster, A.D.; et al. Generalized Lévy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 2012, 486, 545–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Qi, S.; Jin, H.; Qi, Z.; Zhang, Z.; Fu, L.; Luo, Q. Zigzag Generalized Lévy Walk: The In Vivo Search Strategy of Immunocytes. Theranostics 2015, 5, 1275–1290. [Google Scholar] [CrossRef] [Green Version]
- Strikwerda, J.C. Finite Difference Schemes and Partial Differential Equations, 2nd ed.; Society for Industrial & Applied Mathematics: Philadelphia, PA, USA, 2004. [Google Scholar] [CrossRef] [Green Version]
- Hundsdorfer, W.; Verwer, J.G. Numerical solution of time-dependent advection–diffusion–reaction equations. In Computational Mathematics; Springer: Berlin, Germany, 2003. [Google Scholar]
- Higham, D.J. An Algorithmic Introduction to Numerical Simulation of Stochastic Differential Equations. SIAM Rev. 2001, 43, 525–546. [Google Scholar] [CrossRef]



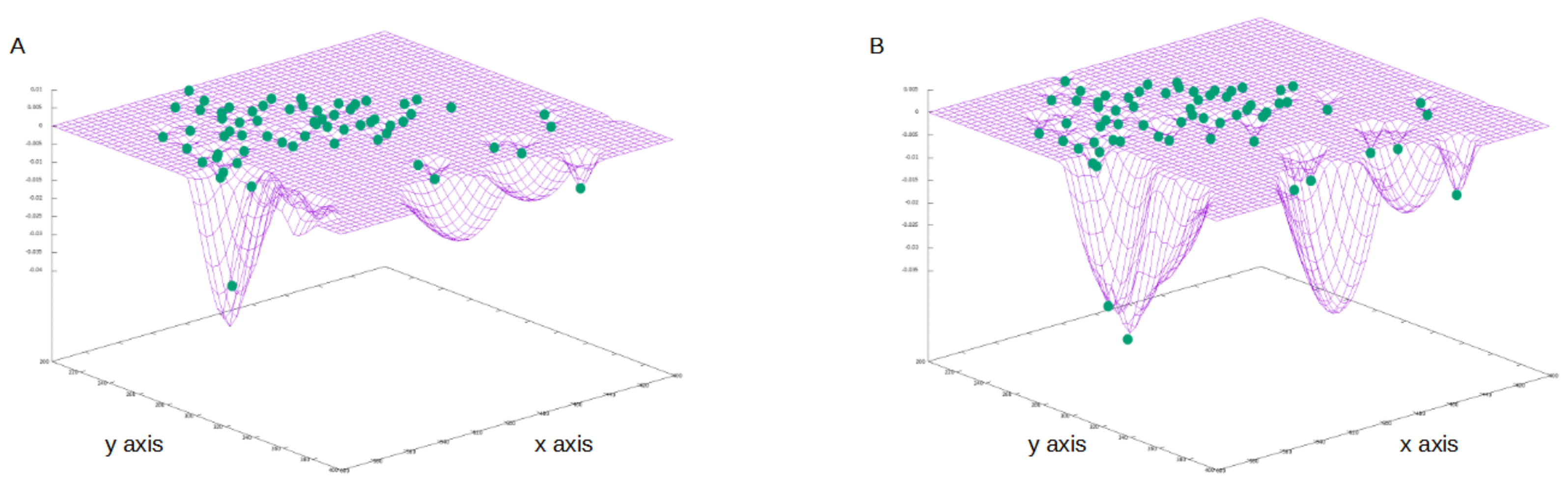
| Functional | RE % | RE % | RE % | RE % | Fval | ||
|---|---|---|---|---|---|---|---|
| - | - | - | |||||
| - | - | ||||||
| - | |||||||
| Functional | RE % | RE % | RE % | RE % | Fval | |||
|---|---|---|---|---|---|---|---|---|
| - | - | - | ||||||
| - | - | |||||||
| - | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bretti, G.; De Ninno, A.; Natalini, R.; Peri, D.; Roselli, N. Estimation Algorithm for a Hybrid PDE–ODE Model Inspired by Immunocompetent Cancer-on-Chip Experiment. Axioms 2021, 10, 243. https://doi.org/10.3390/axioms10040243
Bretti G, De Ninno A, Natalini R, Peri D, Roselli N. Estimation Algorithm for a Hybrid PDE–ODE Model Inspired by Immunocompetent Cancer-on-Chip Experiment. Axioms. 2021; 10(4):243. https://doi.org/10.3390/axioms10040243
Chicago/Turabian StyleBretti, Gabriella, Adele De Ninno, Roberto Natalini, Daniele Peri, and Nicole Roselli. 2021. "Estimation Algorithm for a Hybrid PDE–ODE Model Inspired by Immunocompetent Cancer-on-Chip Experiment" Axioms 10, no. 4: 243. https://doi.org/10.3390/axioms10040243
APA StyleBretti, G., De Ninno, A., Natalini, R., Peri, D., & Roselli, N. (2021). Estimation Algorithm for a Hybrid PDE–ODE Model Inspired by Immunocompetent Cancer-on-Chip Experiment. Axioms, 10(4), 243. https://doi.org/10.3390/axioms10040243