Abstract
In this work, we aimed to experimentally study the nucleation and growth of CaCO3 phases precipitated from supersaturated aqueous solutions in the presence of varying concentrations of sulphate oxyanion. The experiments were conducted under pH conditions close to neutral (7.6) and by considering a wide range of initial (SO42−)/(CO32−) ratios (0 to approx. 68) in the aqueous solution. We paid special attention to the evolution of the precipitates during ageing within a time framework of 14 days. The mineralogy, morphology, and composition of the precipitates were studied by X-ray diffraction, Fourier transform infrared spectroscopy, scanning electron microscopy, and EDX microanalysis. The concentration of sulphate ions in the reacted aqueous solution was studied by ICPs. The experimental results showed that the mineral composition of the precipitate recovered in each run varied with the (SO42−)/(CO32−) ratio in the parental solution, which influenced the mineral evolution of the precipitates during ageing. We observed that high concentrations of sulphate in the aqueous solution stabilized the vaterite precipitates and inhibited calcite formation. Furthermore, aragonite never precipitated directly from the solution, and it was only formed via a dissolution-precipitation process in solutions with a high (SO42−)/(CO32−) ratio after long reaction times. Finally, gypsum only precipitated after long ageing in those aqueous solutions with the highest concentration of sulphate. The reaction pathways during ageing, the morphology of the calcite crystals, and the composition of vaterite and calcite were discussed considering both kinetic and thermodynamic factors. These results showed a considerably more complex behavior of the system than that observed in experiments conducted under higher pHs and supersaturation levels and lower (SO42−)/(CO32−) ratios in the aqueous phase.
1. Introduction
The presence of calcium carbonate mineral phases is ubiquitous in surface and subsurface environments in the Earth. Calcium carbonate phases in such environments range from amorphous to crystalline and from anhydrous (calcite, aragonite, and vaterite (CaCO3)) to differently hydrated (monohydrocalcite (CaCO3·H2O) and ikaite (CaCO3·6H2O)) [1]. In these environments, the only stable phase of calcium carbonate is calcite, which appears as the main constituent of sedimentary carbonate rocks [1,2] and hard tissues in marine organisms [2,3,4]. However, all other phases can also form and often remain metastable over long periods due to kinetic factors and/or the influence of specific chemical species in a variety of bio-geological contexts [5,6,7]. Thus, amorphous CaCO3 (ACC) plays a main role in the early stages of the formation of carbonate hard tissues, where it forms at far-from-equilibrium conditions and, following Ostwald’s step rule, subsequently transforms into other CaCO3 phases through different reaction pathways [8,9,10]. In carbonate hard tissues, ACC can also remain stabilised in specific locations under the influence of organic molecules and/or inorganic foreign ions like magnesium [11] as a sort of CaCO3 storage which can eventually be disposed for hard tissue-damage reparation [12]. The appearance of the hydrated forms of calcium carbonate ikaite and monohydrocalcite is restricted to very specific environments like near-freezing conditions aqueous environments where the presence of magnesium or phosphate ions has been proposed to promote their formation [13,14]. Kinetic effects facilitate the formation of vaterite in natural environments where this phase represents an intermediate step in the reaction pathway that leads from ACC to calcite following Ostwald’s steps rule [15]. The seasonal formation and temporal stabilization of vaterite in evaporite-rich geological settings has also been linked to the influence of high concentrations of dissolved sulphate [16]. Finally, aragonite is the second most abundant CaCO3 phase in surface and subsurface environments after calcite [17]. Aragonite also is a main component of carbonate biominerals, often coexisting with calcite in the same hard tissue [4,18,19]. The main role played by high Mg2+/Ca2+ ratios in aqueous environments as the determining factor in the crystallization and stabilization of aragonite has been long known [5]. This role has been attributed to the inhibitory effect of dissolved magnesium on the growth of calcite [20,21,22,23]. Changes in the Mg2+/Ca2+ ratio in seawater have been proposed to explain the recurrent calcite-aragonite switches in marine CaCO3 biominerals along Earth’s history [24]. Consequently, the influence of different foreign ions in the crystallization of CaCO3 and, specifically, in its polymorph selection has long been a main topic of research in carbonate mineralogy. Understandably, most research efforts have been focused on shedding light on the role of magnesium and, to a lesser extent, that of other cations like Sr2+ or Co2+ [5,23,25,26,27]. However, much less attention has been paid to the influence of different anions on CaCO3 polymorph selection, even though there exists strong evidences that this influence, especially that of tetrahedral oxyanions, could also be most relevant [28]. Indeed, as mentioned above, phosphate oxyanions have been linked to the formation of ikaite [13]. A combined role of sulphate and Mg2+ in the switch from calcite to aragonite seas has been claimed on both geochemical and experimental evidences [29]. The notion that sulphate (SO42−), chromate (CrO42−), and selenate (SeO42−) promote the formation of vaterite and inhibit its transformation into calcite is supported by overwhelming experimental evidence [30,31,32,33,34,35,36,37,38]. Furthermore, this notion has been given theoretical support by computational studies that relate the effect of tetrahedral oxyanions on CaCO3 polymorph selection to thermodynamic effects arising from the different impacts that their incorporation-substituting carbonate groups have on the structure of the three CaCO3 polymorphs calcite, aragonite, and vaterite [28,31]. This incorporation produces a most significant disruption of the local structure of aragonite. Although less so, this disruption still is very marked in calcite, while the vaterite structure is the least disturbed by the isomorphic incorporation of tetrahedral oxyanions.
In an earlier work, the specific influence of the presence of sulphate oxyanions in the crystallization medium on the formation and evolution upon ageing of CaCO3 precipitates was studied at high pHs (10.9) by conducting batch experiments [31]. High pH conditions are restricted to limited geological settings such as hyper alkaline lakes in connection to tectonic rifts [39]. Even though other authors have studied the influence of sulphate ions on the precipitation of CaCO3 under lower pHs [37,38], these studies only explored a narrow range of SO42−/CO32− ratio conditions and a comprehensive study of close-to-neutrality conditions is still missing. In this study, we aim to improve our current understanding of the influence of sulphate oxyanions in CaCO3 formation and evolution by filling this gap. We present here the results of precipitation experiments conducted under circa neutral conditions (initial pH = 7.6), closer to those commonly found in surface and subsurface waters, and by considering a wide range of initial SO42−/CO32− ratios (0 to approx. 68) in an aqueous solution. Furthermore, we study the evolution of the precipitates upon ageing within a time framework of 14 days, regarding its mineralogy, morphology, and composition of the different phases that constitute the precipitate at each stage. Different reaction pathways during ageing are unraveled depending on the initial (SO42−)/(CO32−) ratio in the solution. These pathways are discussed considering both the kinetic and thermodynamic arguments. Our goal is to draw general conclusions on the influence of sulphate oxyanions and the (SO42−)/(CO32−) ratio in aqueous environments on the polymorphic selection of CaCO3.
2. Materials and Methods
Stock solutions of CaCl2 (1M, Fluka, Sigma-Aldrich, St. Louis, MO, USA), NaHCO3 (99.7%, Panreac, Barcelona, Spain), and Na2SO4 (>99%, Sigma-Aldrich, St. Louis, MO, USA) were prepared with deionized water (Millipore, Burlington, MA, USA; resistivity = 18.2 MΩ·cm). The CaCO3 precipitates were obtained by mixing 50 mL of a CaCl2 solution with 50 mL of a stirred NaHCO3 + Na2SO4 solution ((SO42−)/(CO32−) = 0–67.6) in Erlenmeyer flasks (Pyrex). The flasks were immediately closed and kept at 25 ± 0.5 °C while continuously stirred by means of a magnetic stirrer. PHREEQC software [40] was used to calculate the solution speciation and supersaturation with respect to the different solid phases that can form in the system using the phreeqc.dat database. This database includes a Ksp of 10−8.48 for calcite, of 10−8.34 for aragonite, of 10−4.58 for gypsum, and of 10−4.36 for anhydrite. The Ksp of 10−7.91 for vaterite [41] and of 10−6.0 for ACC [42] were manually added to this database.
Table 1 shows the initial concentration of the reactants, the initial calculated pH, the activities of the relevant species, and the saturation indexes (SI) with respect to calcite (Cal), aragonite (Arg), vaterite (Vtr), gypsum (Gp), and anhydrite (Anh). In all the experiments, the solutions were initially highly supersaturated with respect to the three CaCO3 polymorphs. In order to maintain the supersaturation at an approximately constant value with respect to the three CaCO3 polymorphs (SICal ≈ 2.35, SIArg ≈ 2.2, and SIVtr ≈ 1.8), the CaCl2 concentrations were adjusted. Only the solutions in experiments A15 to A25 were supersaturated with respect to gypsum (SIGp ≤ 0.25). In all experiments, the solutions were undersaturated with respect to anhydrite. It is important to note that the mentioned SI values are those that would be reached if mixing were instantaneous and if no crystallization occurred before the complete homogenization of the solution were reached after mixing. This is an unlikely situation, and it can be assumed that some CaCO3 precipitation will take place before a complete mixing of the reactant solutions [43,44,45]. This might lead to precipitates that are chemically inhomogeneous regarding their concentration of impurities.

Table 1.
A summary of the initial fluid chemistry.
Three independent runs were conducted for each experiment. The experimental runs were terminated after six different ageing times (5 min; 10 h; and 1, 3, 7, and 14 days). The precipitates were separated by filtering the aqueous phase through a 0.45-μm cellulose acetate membrane (Sartorius stedim; Albet), thoroughly rinsed with deionized water, subsequently rinsed with 10 mL of ethanol, and left to dry overnight at 30 °C in a drying chamber. The filtered aqueous solutions were stored in closed Pyrex flasks at 4 °C in a refrigerator until they were prepared for analysis.
Sample Characterization
The filtered solid phases recovered from each experiment were characterized by different techniques to determine their mineralogy, their chemical composition, and the morphology and size of their crystalline constituents. The precipitates were first analyzed by powder X-ray diffraction (XRD) to identify the mineral phases by means of a Panalytical X`Pert PRO MRD microprocessor-controlled X-ray diffractometer (version, Panalytical B.V., Almelo, The Netherlands). Most of the diffraction patterns were recorded between 20° and 60° and a few of them between 10° and 60° with a step size of 0.001° and using Cu Kα radiation. Diffractograms were processed with the X’Pert HighScore Plus (PANalytical B.V.) software. The diffraction patterns were compared to standard powder diffraction files from the ICDD-PDF2 data base (release 2007): 00–005–0586 for calcite, 01–080–2790 for aragonite, 04–017–8634 for vaterite, and 04–016–3025 for gypsum.
The precipitates were further analyzed by Fourier Transform Infrared (FTIR) spectroscopy using a BRUKER IFS 66v/S spectrophotometer (BRUKER, Billerica, MA, USA) equipped with a triglycine sulphate (TGS) detector. FTIR spectra were recorded on samples diluted in KBr by the co-addition of 64 scans, with a precision of 0.2 cm−1. The morphology of the mineral phases of the precipitate was imaged using a JEOL JSM 6400 Scanning Electron Microscope (SEM) (JEOL Ltd., Akishima, Tokyo, Japan). Semiquantitative chemical analyses of the precipitates were obtained using an Oxford Instrument Energy Dispersive X-ray (EDX) detector. KristalShaper software [46] was used to model the habit of calcite crystals.
In order to determine the content of S in the aqueous solution from each experiment, the solutions were analyzed immediately after filtering using inductively coupled plasma-optical emission spectrometry (ICP-OES) (SPECTRO Arcos, SPECTRO, Kleve, Germany).
3. Results
3.1. Mineralogical Evolution of the Precipitation during Ageing
The mineralogical evolution of the precipitates during ageing is summarized in Table 2 and illustrated in Figure 1, Figure 2, Figure 3 and Figure 4.
Table 2.
The mineral identity of the solid phases recovered in each experiment after different ageing times: Note that the phases in brackets indicate that they are very scarce but that they are unequivocally identified at least in one of the analytical techniques used.
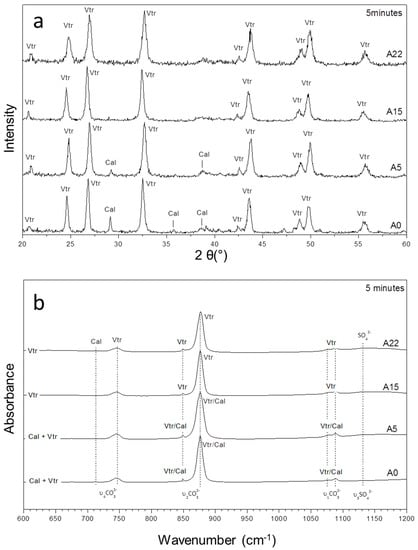
Figure 1.
The XRD patterns (a) and infrared spectra (b) of precipitates sampled after 5 min of reaction in the case of experiments A0, A5, A15, and A22 with initial (SO42−)/(CO32−) ratios of 0, 14.0, 41.8, and 60.0, respectively.
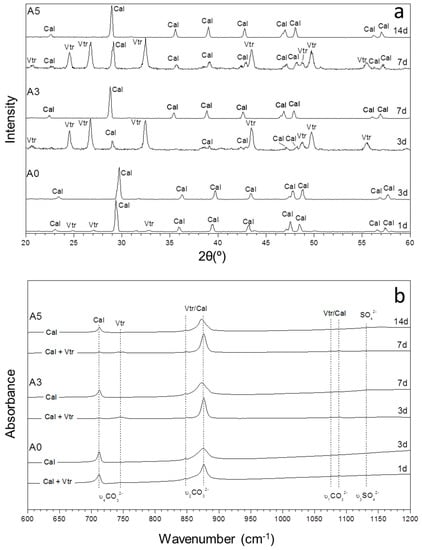
Figure 2.
The XRD patterns (a) and infrared spectra (b) of precipitates sampled from aqueous solutions with low (SO42−)/(CO32−) ratios (A0–A5) as a function of time.
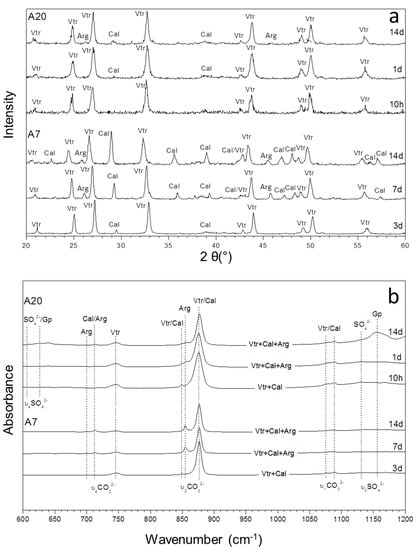
Figure 3.
The XRD patterns (a) and infrared spectra (b) of precipitates sampled from aqueous solutions with (SO42−)/(CO32−) ratios of 19.51 and 54.87 (A7 and A20) as a function of time.
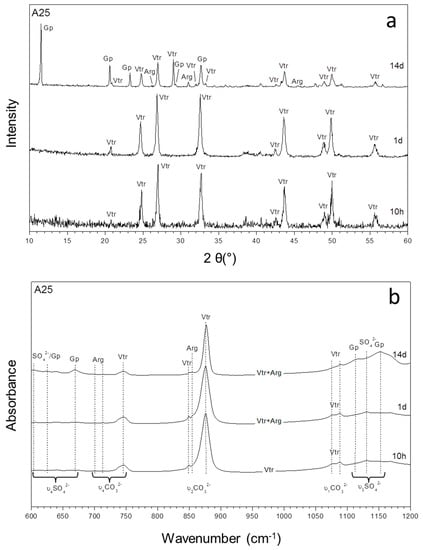
Figure 4.
The XRD patterns (a) and infrared spectra (b) of precipitates sampled from aqueous solutions with the highest (SO42−)/(CO32−) ratio (67.6) (A25) as a function of time.
The XRD and FTIR analyses of the precipitates evidence that, soon after their formation (ageing time = 5 min), they mainly consist of vaterite, with calcite present as a minor phase. The amount of calcite in the precipitate in a 5 min reaction progressively decreases as the initial (SO42−)/(CO32−) ratio in the aqueous solution increases. This is clearly evidenced by the increase in the intensity of the peaks assigned to vaterite and the decrease of those attributed to calcite in the diffraction patterns recorded in the samples corresponding to this ageing time (Figure 1a). Thus, the amount of calcite is significant in A0, where the initial (SO42−)/(CO32−) ratio in the aqueous solution is 0, while no calcite is detected in experiment A22 when the initial (SO42−)/(CO32−) ratio is 60.0. The FTIR spectra show absorbance band characteristics of vaterite and/or calcite, whereas the presence of aragonite is not detected (Figure 1b). A very weak band at approx. 1130 cm−1 (found in the spectrum of precipitates from experiments A5–A22 but absent in that corresponding to the solids formed from sulphate-free solution A0) can be assigned to sulphate ions incorporated in the structure of vaterite and/or calcite.
Furthermore, the mineralogical composition of the precipitates evolves with ageing time, following trends with characteristics varying depending on the initial (SO42−)/(CO32−) ratio in the solution. The composition of precipitates formed in experiments A0 to A5, where the initial (SO42−)/(CO32−) ratio in the aqueous solution varies between 0 and 14.03, shows a similar evolution trend. This trend is characterized by the progressive decrease of the amount of vaterite present in the precipitate as ageing progresses (Figure 2). This decrease takes place at a slower rate with increasing initial (SO42−)/(CO32−) ratios in the aqueous solution, as confirmed by both XRD (Figure 2a) and FTIR spectroscopy (Figure 2b). Thus, in the absence of SO42− in the solution (A0 in Table 1 and Table 2; (SO42−)/(CO32−) = 0), the precipitate exclusively consists of calcite after 3 days of ageing. In experiment A3 ((SO42−)/(CO32−) = 8.48), vaterite and calcite are detected after 3 days but calcite is the only CaCO3 phase present in the precipitate after 7 days of ageing. In contrast, vaterite is still present in the precipitate formed in A5 ((SO42−)/(CO32−) = 14.0) after 7 days of ageing (Figure 2; Table 2) but is undetected in the solid phase recovered after 14 days of reaction. This trend is observed in all these three experiments, i.e., vaterite can remain for longer ageing periods when the (SO42−)/(CO32−) ratio increases.
A different mineralogical evolution trend is observed in experiments A7 to A20, where the initial (SO42−)/(CO32−) ratio in the aqueous solution varies between 19.51 and 54.87 (Figure 3). In this case, the formation of aragonite is also observed during ageing. This CaCO3 polymorph appears as a constituent of the precipitate formed after ageing times that are shorter, as the initial (SO42−)/(CO32−) ratio in the aqueous solution is higher. Thus, aragonite is detected in the precipitate formed in A7 and A15 after 7 days of ageing, while this phase is found much earlier in the precipitate formed in A20 after only 1 day of ageing. This evolution trend is particularly evident in the FTIR spectra, which show weak bands at approx. 699 cm−1 and 712 cm−1 and at approx. 855 cm−1, consistent with the positions of the υ4 antisymmetric bending and υ2 out-of-plane vibrations of carbonate ion in aragonite, respectively. These bands are absent in the precipitates formed after short times of ageing (3 days and shorter for A7 and A15 and 10 h and shorter for A20). It is worthwhile to note that after 14 days of ageing, these precipitates consist of the three CaCO3 polymorphs, vaterite, aragonite, and calcite, together with gypsum as a minor phase. Calcite is still the most abundant phase in A7, whereas vaterite is the most abundant one in A15 and A20 (Figure 3a). Bands at 1130, 628, and 608 cm−1, particularly evident in the precipitate formed in A20 after 14 days of ageing, can be assigned to sulphate ions incorporated into the lattice of the CaCO3 polymorphs.
Another mineralogical evolution trend is observed in experiments A22 and A25, where the initial (SO42−)/(CO32−) ratios in the aqueous solution are 60.00 and 67.61, respectively (Figure 4). In these experiments, vaterite is always the first and only phase that precipitates whereas calcite, never forms part of the precipitate regardless of the ageing time. It also is worthwhile to note that the formation of aragonite is unequivocally observed after 1 day of ageing in both experiments A25 and A22. After 7 days, gypsum precipitates and coexists with vaterite and aragonite, also in both experiments.
3.2. Characterization of the Precipitate and the Aqueous Solution
The SEM micrographs in Figure 5 show that vaterite appears as cauliflower-like aggregates (Figure 5a,b) or radial aggregates that consist of lens-shaped crystallites (Figure 5c,d). As is apparent, no significant evolution of the vaterite morphology is observed as a function of the initial (SO42−)/(CO32−) ratio in the aqueous solution or the ageing time. In contrast, calcite appears as single crystals with morphologies and sizes highly dependent on both reaction time and initial (SO42−)/(CO32−) ratio in the solution. Thus, in experiment A0, where there is no sulphate in the solution, calcite crystals are bounded by flat {10–14} rhombohedron faces regardless of the reaction time and show sizes in the 10−25 μm range (Figure 5a,c,e). In experiments A3 to A7, where the initial (SO42−)/(CO32−) ratio varies between 8.5 and 19.5, the calcite crystals that formed early after the mixing of the aqueous solution also show the typical rhombohedron-like shape with flat {10–14} faces (Figure 5f). However, their morphology undergoes an evolution as ageing progresses. This evolution is characterized by a progressively increasing elongation along [001] (Figure 5f−h) as well as by the emergence of rough pseudofacets that lead to the development of a new form, the more acute {02–21} rhombohedrum (see inset in Figure 5h). This evolution seems to be more marked in precipitates A7 and A5 than A3, which points to a positive relationship between the higher initial (SO42−)/(CO32−) ratio in the aqueous solution and more elongated and rougher-faced calcite crystals. Thus, after 14 days of ageing, calcite crystals formed in experiment A3 show a length/width ratio around 2.4. This ratio is around 3.3 and 3.5 in calcite crystals formed in experiments A5 and A7, respectively (Figure 5i,j). As mentioned above, calcite crystals are hardly noticeable in experiments A15 to A20 and are absent in experiment A22 and A25 regardless of the reaction time.

Figure 5.
SEM images of the precipitates recovered from solutions with different (SO42−)/(CO32−) ratios after various elapsed times ranging from 5 min to 14 days. Vaterite appears as cauliflower-like aggregates (a,b) or lens-shaped crystallites (c,d). When there is no sulphate in the solution, the calcite crystals are bounded by flat {10–14} rhombohedron faces (a,c,e). In solutions with a moderate concentration of sulphate, the calcite crystal habit undergoes an evolution characterized by a progressive increase in elongation along [001] (f–j) and by the emergence of the {02–21} form (see inset in h). Aragonite appears as crystals with the so-called “morning-star” morphologies (k). Gypsum crystals exhibit tabular habits with the {010} form as the main face (l).
Aragonite is observed in experiments A7–A25 ((SO42−)/(CO32−) = 19.51–67.61). In all cases, aragonite appears as crystals bounded by curved prismatic faces. These crystals grow on the surface of vaterite aggregates, forming so-called “morning-star” aggregates (Figure 5k). Finally, gypsum crystals, which are observed in experiments A22 and A25, typically exhibit tabular habits with the {010} form as the main face with an accentuated elongation along [001] (Figure 5l).
EDX analyses conducted on vaterite and calcite crystals evidence that they contain S in all the cases, except for the vaterite and calcite crystals formed in the absence of SO42− (experiment A0) in the aqueous solution. For both vaterite and calcite, a direct correlation between the [S] (% at) and the initial (SO42−)/(CO32−) ratio in the aqueous solution is detected (Figure 6a). Thus, after 5 min of reaction, a [S] (% at) of 0.91 was measured in vaterite aggregates formed in experiment A3, while the S/Ca ratios were as high as 1.54 and 3.23 in vaterite formed in experiments A7 and A25, respectively. A similar trend was observed in calcite crystals, where the S/Ca ratio after 5 min of reaction were 2.81 in experiment A3, 4.48 in experiment A5, and 5.76 in experiment A20. It is interesting to note that for both calcite and vaterite for a given (SO42−)/(CO32−) initial ratio, the highest S content is always detected in the precipitates with shorter ageing (5 min of reaction) and progressively decreases as ageing progresses. This evolution is depicted in Figure 6b for calcite formed in experiment A5. It is also striking that, regardless of the initial (SO42−)/(CO32−) ratio and the ageing time, calcite crystals always show higher S contents than vaterite aggregates. The small size of aragonite crystals formed in experiments A7 to A25 and gypsum crystals formed in experiments A22 and A25 prevented EDX analyses from being conducted on them.
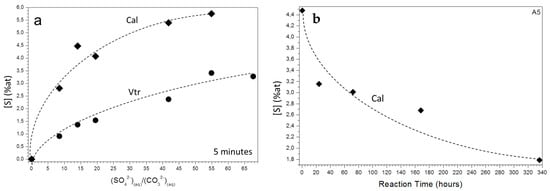
Figure 6.
(a) The concentration of S (% at) in calcite and vaterite sampled after 5 min of reaction as a function of the initial (SO42−)/(CO32−) ratio and (b) the variation of [S] (% at) with ageing in calcite crystals sampled from aqueous solutions with a (SO42−)/(CO32−) ratio of 14.03 (A5).
Finally, ICP-OES analyses of the S concentration in the aqueous solution show that at 5 min of reaction, the concentration of S in the aqueous solution is progressively lower than the initial aqueous solution as the initial (SO42−)/(CO32−) ratio in the aqueous solution increases (Figure 7a). Furthermore, the concentration of S in the aqueous solution varies with ageing time, following similar trends independently of the initial (SO42−)/(CO32−) ratio in the solution (Figure 7b). Thus, the concentration of S in the aqueous solution is characterized by a rapid drop followed by a progressive increase as ageing progresses.
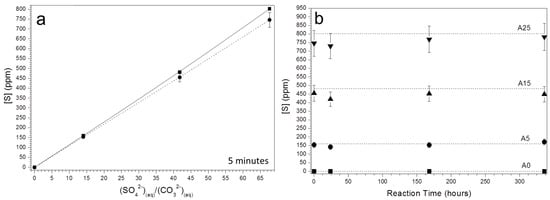
Figure 7.
(a) The concentration of S (from inductively coupled plasma-optical emission spectrometry (ICP-OES) analyses) in aqueous solutions sampled after 5 min of reaction as a function of the initial (SO42−)/(CO32−) ratios (dashed lines): The initial concentrations of S in the solutions are connected by a solid line. (b) The evolution of the concentration of S (from ICP-OES) of aqueous solutions with initial (SO42−)/(CO32−) ratios of 0 (A0), 14.3 (A5), 41.8 (A15), and 67.7 (A25), respectively.
4. Discussion
The experimental results described in the previous section (XRD and FTIR analyses) show that the mineral composition of the precipitate recovered in each run varied as a function of two main factors: the high supersaturation of the parental aqueous solution, which controls the phases that are formed at the initial stages of crystallization, and the (SO42−)/(CO32−) ratio in the parental solution, which influences the mineral evolution of the precipitates during ageing. After the mixing of the reactants, the aqueous solutions are highly supersaturated with respect to all the crystalline CaCO3 polymorphs (Table 1), and spontaneous precipitation takes place. According with the Ostwald step rule [15], the formation of the most soluble and disorder phase is kinetically favored [47]. However, under the conditions used in these experiments, the precipitation of the amorphous calcium carbonate, the most soluble CaCO3 phase, can be discarded since the aqueous solution is always undersaturated with respect to this phase (SIACC ≤ −0.1). Therefore, kinetics factors promote the formation of vaterite as the first phase that precipitates immediately after the mixing of the parental solutions. After vaterite nucleation, the system can further reduce its free energy by the subsequent transformation of vaterite into calcite through a solvent mediated transformation [48]. In addition, a reduction of supersaturation resulting from the growth of vaterite promotes the nucleation of calcite. Therefore, as the system approaches equilibrium, the most stable CaCO3 phase, calcite, should become the prevailing phase at the expense of vaterite. The mineralogical evolution observed in experiments with the lower initial (SO42−)/(CO32−) ratios (A0–A5) is in good agreement with this expected evolution. Furthermore, it is in good consonance with the precipitate mineralogical evolution upon ageing previously observed in similar mixing experiments conducted in more highly supersaturated systems (SICal approx. 3.7) under higher pH (approx. 10.9) [31]. Our current results are further evidence that the presence of a high amount of sulphate ions in the aqueous media slows down or even inhibits the direct precipitation of calcite and transformation of vaterite into calcite, thereby contributing to the stabilization of vaterite with respect to calcite. The prolonged existence of vaterite as a major constituent of the precipitate is observed in all the experiments. The stabilization of vaterite is more evident the higher the (SO42−)/(CO32−) ratio is in the aqueous solutions. Indeed, in experiments A22 and A25, where this ratio is highest, vaterite does not transform into calcite even after 14 days of ageing. These results are again consistent with those previously reported by Fernández-Díaz et al [31], who notice that vaterite can remain as a major constituent phase for long ageing periods when (SO42−)/(CO32−) ratios are higher than one. The stabilization of vaterite and the retardation of calcite crystallization in the presence of sulphate ions can be explained by a combination of both thermodynamics and kinetics factors. The computational modelling of the substitution of a small amount of carbonate groups by sulphate groups in the structures of vaterite and calcite [28,31] showed that the vaterite structure is much less disrupted by the isomorphic incorporation of AO42− groups, including sulphate, chromate, and selenate, than the calcite structure. In the case of sulphate, the incorporation of this oxyanion results in an increment of calcite lattice energy, while it has the opposite effect on the lattice energy of vaterite. This different impact of sulphate isomorphic incorporation on the energetics of calcite and vaterite structures is especially relevant for the S content range up to 3% molar fraction, where it translates into an effective reduction of the energy difference between the two calcium carbonate polymorphs. The larger capability of vaterite to accommodate sulphate oxyanions in its structure compared to calcite seems to be in contradiction with the experimental evidence provided by the EDX analyses of calcite and vaterite grains in the precipitates obtained in these experiments. These analyses steadily show a higher S content in calcite crystals than in vaterite aggregates formed in the same experiment. This is regardless of the initial (SO42−)/(CO32−) ratio and the ageing time (see Figure 6a). Similar results were reported by Fernández-Díaz et al. [31] for precipitates formed under higher supersaturations and pHs. These authors explain the apparent contradiction between the experimental results and the energy-based expectations considering that the higher S contents measured in calcite most likely were the consequence of this phase forming later than vaterite. In such a scenario, calcite crystals would have grown from a CO32−-depleted medium after vaterite nucleation and, consequently, in a solution with a (SO42−)/(CO32−) ratio significantly higher than the initial one.
Although the results of computer simulations of sulphate incorporation do not predict stability crossovers between vaterite and calcite, the approximation of the lattice energy of both polymorphs can explain the increasingly larger duration of vaterite as a precipitate component with a growing initial sulphate concentration in the solution.
While the incorporation of impurities from the aqueous solution can change the lattice energy of the bulk crystal, surface-related phenomena like impurity adsorption can modify the surface chemistry of the different polymorphs, thereby changing their surface energies [49]. The impact of this phenomenon is particularly relevant in the case of small particles, where the surface/volume ratio is very high, and the effect of lattice energy in the stability of the different polymorphs is comparatively much less important than that of surface energy. A small increase of the surface energy of calcite nanoclusters and nanoparticles due to sulphate adsorption could dramatically increase the barrier for its nucleation, since such a barrier is proportional to the square of the surface energy. Were this to happen, the main outcome would also be a progressively slower transformation of vaterite into calcite as the (SO42−)/(CO32−) ratio in the parent aqueous solutions increases. Kinetics arguments involving the adsorption of impurities have often been invoked to explain the stabilization of vaterite with respect to calcite and the retardation of calcite nucleation and growth. Phenomena observed during the precipitation of CaCO3 from a supersaturated aqueous solution bearing phosphate ions are an example [50]. In this system, the stabilization of vaterite was rather attributed to the blocking of active sites for dissolution on the surface of this phase due to the adsorption of phosphate ions. A similar phosphate adsorption onto surface sites was claimed to explain calcite growth inhibition. Indeed, a similar reasoning can be applied to explain the observed effect of sulphate ions. Another alternative thermodynamic and mechanistic argument has been invoked to explain the inhibition of calcite growth by the effect of sulphate ions. In situ atomic force microscopy AFM experiments of calcite growth performed using a fluid cell and flowing supersatutared sulphate-bearing aqueous solutions showed that a low concentration of sulphate in the aqueous solution (5 mM) is enough to decrease the rate of step spread on the calcite surface [51]. These nanoscale AFM observations supported the interpretation that the trapping of sulphate ions by nanometric growing layers resulted in the generation of interfacial strain energy. A strain relaxation perpendicular to the growing layer introduces local variations in bond lengths, giving rise to local departures from calcite surface ideal nanotopography. The direct consequence of this phenomenon is a decrease of the step advancement rate according to the so-called “template effect” model [20,25,52], which is more marked the larger the amount of sulphate ions trapped within calcite monolayers.
An interesting result observed in experiments A7 to A25, where the initial (SO42−)/(CO32−) ratio in the aqueous solution was >19.51, was the formation of aragonite. It took place through the solvent-mediated transformation of vaterite and could also be explained invoking both the thermodynamic and kinetics arguments. For equivalent compositions of sulphate in the polymorphs, the computer modelling of sulphate isomorphic incorporation predicts a significantly larger increment of aragonite lattice energy compared to that of both calcite and vaterite. This result indicates that the substitution of carbonate groups by sulphate in aragonite structure is very unfavorable [28,31]. As a result, even when this polymorph forms from highly supersaturated solutions with very high (SO42−)/(CO32−) ratios, it grows relatively sulphate-free compared to calcite. The direct consequence is a much more marked impact of the presence of sulphate in the growth medium on calcite stability. Indeed, the solubility of a sulphate-bearing calcite can overcome that of sulphate-free aragonite, as pointed out by Busenberg and Plummer [53], who reported this to be the case for calcites with sulphate contents above 3 mole%. Such a solubility crossover could explain the observation that, upon ageing of the vaterite precipitate, aragonite forms simultaneously to or even after the formation of the theoretically stable polymorph calcite when the concentration of sulphate in the aqueous solution is very high. This is the case in experiments A7 to A20. Unfortunately, the small size of the aragonite crystals formed in these experiments has prevented EDX analyses to obtain evidence that could give full support to our interpretation, although the high concentration of sulphate measured in calcite crystals point in this direction (see Figure 6). On the other hand, the fact that sulphate incorporation into aragonite structures is very unfavorable, it can be concluded that the inhibitory effect of sulphate ions during the growth of aragonite will arguably be less effective than in the case of calcite and so will be a hypothetical “template effect” mechanism.
Besides the precipitation of CaCO3 polymorphs, gypsum was unequivocally detected after 7 days of interaction in the experiments with the highest (SO42−)/(CO32−) ratios (A20–A25). The precipitation of this phase is not surprising since the initial solutions are supersaturated with respect to this phase (SIGp approx. 0.17–0.25). However, the early precipitation of CaCO3 phases after the mixing of the parent solutions depletes the solution in Ca2+ (and to a much lesser extent in SO42−), leading to a very important reduction of aqueous phase SIGp. Thus, systems initially close to equilibrium with respect to gypsum, as is the case of the solution in experiment A15, will become undersaturated soon after the precipitation of CaCO3 starts, making the formation of gypsum impossible. The systems initially more highly supersaturated with respect to gypsum will either approach equilibrium or become significantly less supersaturated, allowing gypsum precipitation to be prevented over large periods.
The presence of sulphate in the aqueous solutions and ageing not only appears to be a critical factor controlling the mineralogy of the recovered precipitates but also affects the morphology of calcite single crystals. As mentioned in Section 3.2, after a short ageing time, calcite single crystals exhibit the typical rhombohedral idiomorphic shape (see Figure 5a,c,e), which progressively become blockier and elongated along the c-axis as ageing progresses. This elongation is clearly more pronounced as the (SO42−)/(CO32−) ratio in the aqueous solution increases. Since there is a clear correlation between the concentration of sulphate in the aqueous solution and the amount of this anion that is incorporated into the calcite (and vaterite) structure, directly evidenced by EDX (see Figure 6a) and supported by an ICP analysis (Figure 7a), we can conclude that the modification of the calcite crystal habit is a direct result of the incorporation/sorption of this anion. The effect of several ions on calcite habit has been profusely discussed in the literature. In particular, the elongation of calcite crystals has been interpreted as the result of a significant lowering of the surface energy of certain faces other than {10014} due to a preferential sorption of foreign ions on these faces [54]. It has also been attributed to a modification of calcite surface nanotopography due to step-specific impurity interactions [55]. These interactions contribute to the stabilization of new crystal faces that become prevalent during growth. In the case under consideration here, the habit of calcite crystals grown in the presence of sulphate, we observe the stabilization of the {0221} form. The resulting habits strikingly resembles those of calcite crystals grown in the presence of small cations such as Mg2+, Co2+, or Mn2+ [56].
Finally, the progressive decrease of S content in both the calcite and vaterite precipitates with reaction time (Figure 6b), which is in good agreement with a parallel progressive increase of the sulphate concentration in the aqueous solution (Figure 7b), points to the precipitate undergoing recrystallization processes through a dissolution-precipitation mechanism. The driving force for this recrystallization process could be the reduction of the extra energy associated with an excess of sulphate incorporated in calcite and vaterite structures under the influence of the high supersaturations dominating the early staged of the precipitation process. The progress of this recrystallization process leads to a chemical purification of the precipitates that further contributes to the stabilization of calcite as ageing progresses.
Author Contributions
I.C.M. performed the laboratory experiments and the DRX analysis and prepared the samples for FTIR, SEM images, EDX, and ICP analysis. J.M.A. and L.F.-D. contributed to the conception of the study. All authors analyzed the data and were involved in manuscript preparation. All authors read and approved the manuscript.
Funding
This study was supported by the Ministry of Science, Innovation and Universities (CIENCIA) (Spain) under project CGL2016-77138-C2-1-P. I. Cuesta Mayorga acknowledges an FPI (BES-2014-070279) fellowship from the Spanish CIENCIA.
Acknowledgments
The authors wish to thank the National Centre of Electronic Microscopy (CNME), the X-ray Diffraction Central Service, the Geological Techniques Research Centre of the Complutense University of Madrid (UCM), and the CSIC for kindly providing technical support to their research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anthony, J.W.; Bideaux, R.A.; Bladh, K.W.; Nichols, M.C. Borates, Carbonates, Sulfates. In Handbook of Mineralogy; Mineralogical Society of America: Chantilly, VA, USA, 2007; Volume V. [Google Scholar]
- Tucker, M.E. Sedimentary Petrology: An Introduction to the Origin of Sedimentary Rocks, 3rd ed.; Blackwell Science Ltd.: Oxford, UK, 2001. [Google Scholar]
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: New York, NY, USA, 1989; p. 324. [Google Scholar]
- Mann, S. Biomineralization. Principles and Concepts in Bioinorganic Materials Chemistry; Oxford University Press, Inc.: New York, NY, USA, 2001; p. 198. [Google Scholar]
- Lippmann, F. Sedimentary Carbonate Minerals; Springer: Berlin/Heidelberg, Germany, 1973; p. 228. [Google Scholar]
- Morse, J.W.; Arvidson, R.S.; Lüttge, A. Calcium carbonate formation and dissolution. Chem. Rev. 2007, 107, 342–381. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.W.; Mackenzie, F.T. Geochemistry of Sedimentary Carbonates; Elsevier: Amsterdam, The Netherlands, 1990. [Google Scholar]
- De Yoreo, J.J.; Gilbert, P.U.P.A.; Sommerdijk, N.A.J.M.; Penn, R.L.; Whitelam, S.; Joester, D.; Zhang, H.; Rimer, J.D.; Navrotsky, A.; Banfield, J.F.; et al. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 2015, 349, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Habraken, W.; Gur, D.; Fratzl, P.; Weiner, S.; Addadi, L. Calcite Crystal Growth by a Solid-State Transformation of Stabilized Amorphous Calcium Carbonate Nanospheres in a Hydrogel. Angew. Chem. 2013, 125, 4967–4970. [Google Scholar] [CrossRef]
- Meldrum, F.C.; Cölfen, H. Controlling Mineral Morphologies and Structures in Biological and Synthetic Systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Blanco, J.D.; Shaw, S.; Bots, P.; Roncal-Herrero, T.; Benning, L.G. The role of pH and Mg on the stability and crystallization of amorphous calcium carbonate. J. Alloys Compd. 2012, 536, S477–S479. [Google Scholar] [CrossRef]
- Griesshaber, E.; Kelm, K.; Sehrbrock, A.; Mader, W.; Mutterlose, J.; Brand, U.; Schmahl, W.W. Amorphous calcium carbonate in the shell material of the brachiopod Megerlia truncata. Eur. J. Mineral. 2009, 21, 715–723. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Wolthers, M.; Wolf-Gladrow, D.A.; Nehrke, G. Effect of pH and Phosphate on Calcium Carbonate Polymorphs Precipitated at near-Freezing Temperature. Cryst. Growth Des. 2015, 15, 1596–1601. [Google Scholar] [CrossRef]
- Tollefsen, E.; Stockmann, G.; Skelton, A.; Mörth, C.-M.; Dupraz, C.; Sturkell, E. Chemical controls on ikaite formation. Mineral. Mag. 2018, 82, 1119–1129. [Google Scholar] [CrossRef]
- Söhnel, O.; Garside, J. Precipitation: Basic Principles and Industrial Applications; Butterworth-Heinemann: Oxford, UK, 1992. [Google Scholar]
- Grasby, S.E. Naturally precipitating vaterite (μ-CaCO3) spheres: Unusual carbonates formed in an extreme environment. Geochim. Cosmochim. Acta 2003, 67, 1659–1666. [Google Scholar] [CrossRef]
- Radha, A.V.; Navrotsky, A. Thermodynamics of Carbonates. Rev. Mineral. Geochem. 2013, 77, 73–121. [Google Scholar] [CrossRef]
- Weiner, S.; Dove, P.M. An overview of biomineralization processes and the problem of the vital effect. Rev. Mineral. Geochem. 2003, 54, 1–29. [Google Scholar] [CrossRef]
- Radha, A.V.; Forbes, T.Z.; Killian, C.E.; Gilbert, P.U.P.A.; Navrotsky, A. Transformation and crystallization energetics of synthetic and biogenic amorphous calcium carbonate. Proc. Natl. Acad. Sci. 2010, 107, 16438–16443. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Fernández-Díaz, L.; Putnis, A. The role of magnesium in the growth of calcite: An AFM study. Chem. Geol. 2010, 271, 52–58. [Google Scholar] [CrossRef]
- Falini, G.; Gazzano, M.; Ripamonti, A. Crystallization of calcium carbonate in presence of magnesium and polyelectrolytes. J. Cryst. Growth 1994, 137, 577–584. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Putnis, A.; Prieto, M.; Putnis, C.V. The role of magnesium in the crystallization of calcite and aragonite in a porous medium. J. Sediment. Res. 1996, 66, 482–491. [Google Scholar] [CrossRef]
- Reddy, M.M.; Nancollas, G.H. The crystallization of calcium carbonate. IV. The effect of magnesium, strontium and sulfate ions. J. Cryst. Growth 1976, 35, 33–38. [Google Scholar] [CrossRef]
- Stanley, S.M.; Hardie, L.A. Secular oscillations in the carbonate mineralogy of reef-building and sediment-producing organisms driven by tectonically forced shifts in seawater chemistry. Palaeogeogr. Palaeoclimatol. Palaeoecol. 1998, 144, 3–19. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Pina, C.M.; Fernández-Díaz, L.; Putnis, A. Nanoscale growth of solids crystallising from multicomponent aqueous solutions. Surf. Sci. 2003, 545, L767–L773. [Google Scholar] [CrossRef]
- González-López, J.; Fernández-González, Á.; Jiménez, A. Precipitation behaviour in the system Ca2+-Co2+-CO32−-H2O at ambient conditions—Amorphous phases and CaCO3 polymorphs. Chem. Geol. 2018, 482, 91–100. [Google Scholar] [CrossRef]
- Reddy, M.M.; Wang, K. Crystallization of calcium carbonate in the presence of metal ions: I. Inhibition by magnesium ion at pH 8.8 and 25 °C. J. Cryst. Growth 1980, 50, 470–480. [Google Scholar] [CrossRef]
- Arroyo-de Dompablo, M.E.; Fernández-González, M.A.; Fernández-Díaz, L. Computational investigation of the influence of tetrahedral oxoanions (sulphate, selenate and chromate) on the stability of calcium carbonate polymorphs. RSC Adv. 2015, 5, 59845–59852. [Google Scholar] [CrossRef]
- Bots, P.; Benning, L.G.; Rickaby, R.E.M.; Shaw, S. The role of SO4 in the switch from calcite to aragonite seas. Geology 2011, 39, 331–334. [Google Scholar] [CrossRef]
- Cruz, J.A.; Sánchez-Pastor, N.; Gigler, A.M.; Fernández-Díaz, L. Vaterite Stability in the Presence of Chromate. Spectrosc. Lett. 2011, 44, 495–499. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Fernández-González, Á.; Prieto, M. The role of sulfate groups in controlling CaCO3 polymorphism. Geochim. Cosmochim. Acta 2010, 74, 6064–6076. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Pina, C.M.; Astilleros, J.M.; Sánchez-Pastor, N. The carbonatation of gypsum: Pathways and pseudomorph formation. Am. Mineral. 2009, 94, 1223–1234. [Google Scholar] [CrossRef]
- Fernández-González, Á.; Fernández-Díaz, L. Growth of calcium carbonate in the presence of Se(VI) in silica hydrogel. Am. Mineral. 2013, 98, 1824–1833. [Google Scholar] [CrossRef]
- Hua, B.; Deng, B.; Thornton, E.C.; Yang, J.; Amonette, J.E. Incorporation of Chromate into Calcium Carbonate Structure During Coprecipitation. Water Air Soil Pollut. 2007, 179, 381–390. [Google Scholar] [CrossRef]
- Jroundi, F.; Gonzalez-Muñoz, M.T.; Garcia-Bueno, A.; Rodriguez-Navarro, C. Consolidation of archaeological gypsum plaster by bacterial biomineralization of calcium carbonate. Acta Biomater. 2014, 10, 3844–3854. [Google Scholar] [CrossRef]
- Sánchez-Pastor, N.; Gigler, A.M.; Cruz, J.A.; Park, S.-H.; Jordan, G.; Fernández-Díaz, L. Growth of Calcium Carbonate in the Presence of Cr(VI). Cryst. Growth Des. 2011, 11, 3081–3089. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, F.; Cao, Z.; Jing, W.; Chen, Y. Crystallization of CaCO3 in the presence of sulfate and additives: Experimental and molecular dynamics simulation studies. J. Colloid Interface Sci. 2012, 377, 430–437. [Google Scholar] [CrossRef]
- Wagterveld, R.M.; Yu, M.; Miedema, H.; Witkamp, G.J. Polymorphic change from vaterite to aragonite under influence of sulfate: The “morning star” habit. J. Cryst. Growth 2014, 387, 29–35. [Google Scholar] [CrossRef]
- Chavagnac, V.; Ceuleneer, G.; Monnin, C.; Lansac, B.; Hoareau, G.; Boulart, C. Mineralogical assemblages forming at hyperalkaline warm springs hosted on ultramafic rocks: A case study of Oman and Ligurian ophiolites. Geochem. Geophys. Geosyst. 2013, 14, 2474–2495. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2): A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Water-Resources Investigations Report 99-4259; USGS: Denver, CO, USA, 1999.
- Plummer, L.N.; Busenberg, E. The solubilities of calcite, aragonite and vaterite in CO2-H2O solutions between 0 and 90 °C, and an evaluation of the aqueous model for the system CaCO3-CO2-H2O. Geochim. Cosmochim. Acta 1982, 46, 1011–1040. [Google Scholar] [CrossRef]
- Ogino, T.; Suzuki, T.; Sawada, K. The formation and transformation mechanism of calcium carbonate in water. Geochim. Cosmochim. Acta 1987, 51, 2757–2768. [Google Scholar] [CrossRef]
- Kile, D.E.; Eberl, D.D.; Hoch, A.R.; Reddy, M.M. An assessment of calcite crystal growth mechanisms based on crystal size distributions. Geochim. Cosmochim. Acta 2000, 64, 2937–2950. [Google Scholar] [CrossRef]
- Kowacz, M.; Putnis, C.V.; Putnis, A. The Control of Solution Composition on Ligand-Promoted Dissolution: DTPA−Barite Interactions. Cryst. Growth Des. 2009, 9, 5266–5272. [Google Scholar] [CrossRef]
- Söhnel, O.; Mullin, J.W. Influence of mixing on batch precipitation. Cryst. Res. Technol. 1987, 22, 1235–1240. [Google Scholar] [CrossRef]
- KrystalShaper. Available online: http://www.jcrystal.com/ (accessed on 10 February 2019).
- Sawada, K. The Mechanisms of Crystallization and Transformation of Calcium Carbonates. Pure Appl. Chem. 1997, 69, 921. [Google Scholar] [CrossRef]
- Cardew, P.T.; Davey, R.J. The Kinetics of Solvent-Mediated Phase Transformations. Proc. R. Soc. Lond. A Math. 1985, 398, 415–428. [Google Scholar] [CrossRef]
- Navrotsky, A. Energetic clues to pathways to biomineralization: Precursors, clusters, and nanoparticles. Proc. Natl. Acad. Sci. USA 2004, 101, 12096–12101. [Google Scholar] [CrossRef]
- Katsifaras, A.; Spanos, N. Effect of inorganic phosphate ions on the spontaneous precipitation of vaterite and on the transformation of vaterite to calcite. J. Cryst. Growth 1999, 204, 183–190. [Google Scholar] [CrossRef]
- Vavouraki, A.I.; Putnis, C.V.; Putnis, A.; Koutsoukos, P.G. An Atomic Force Microscopy study of the growth of calcite in the presence of sodium sulfate. Chem. Geol. 2008, 253, 243–251. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Pina, C.M.; Fernández-Díaz, L.; Putnis, A. Molecular-scale surface processes during the growth of calcite in the presence of manganese. Geochim. Cosmochim. Acta 2002, 66, 3177–3189. [Google Scholar] [CrossRef]
- Busenberg, E.; Plummer, N. Kinetic and thermodynamic factors controlling the distribution of SO32− and Na+ in calcites and selected aragonites. Geochim. Cosmochim. Acta 1985, 49, 713–725. [Google Scholar] [CrossRef]
- Titiloye, J.O.; Parker, S.C.; Mann, S. Atomistic Simulation of Calcite Surfaces and the Influence of Growth Additives on Their Morphology. J. Cryst. Growth 1993, 131, 533–545. [Google Scholar] [CrossRef]
- Davis, K.J.; Wasylenki, L.E.; Dove, P.M.; De Yoreo, J.J. Morphological consequences of differential Mg2+ incorporation at structurally distinct steps on calcite. Am. Mineral. 2004, 89, 714–720. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Astilleros, J.M.; Pina, C.M. The morphology of calcite crystals grown in a porous medium doped with divalent cations. Chem. Geol. 2006, 225, 314–321. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).