Quantum Mechanical Modeling of the Vibrational Spectra of Minerals with a Focus on Clays

Abstract

1. Introduction
2. Theory and Terminology
2.1. Frequencies
2.2. Intensities
2.3. Bandwidths
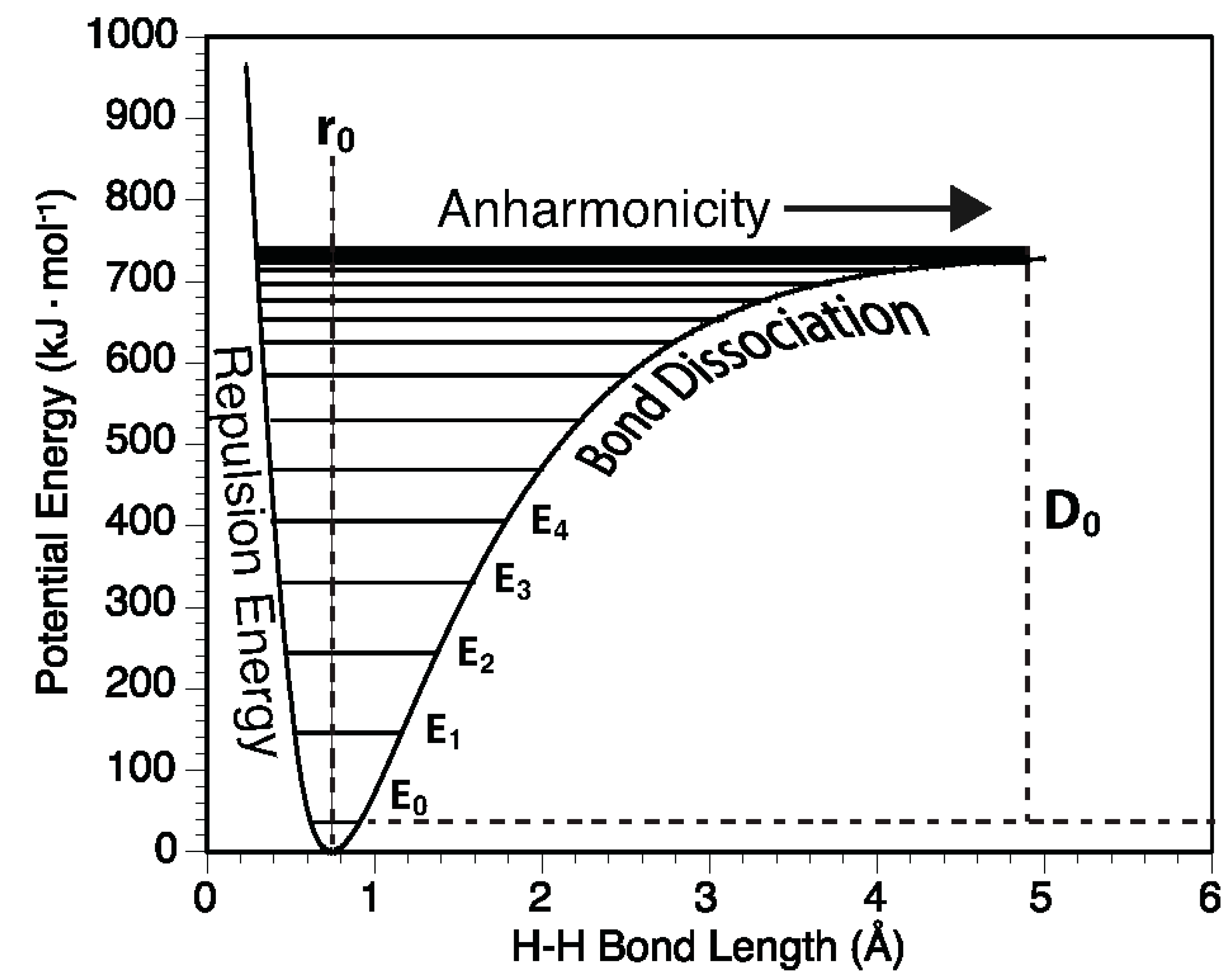
2.4. Anharmonicity
3. Practical Considerations
4. Examples
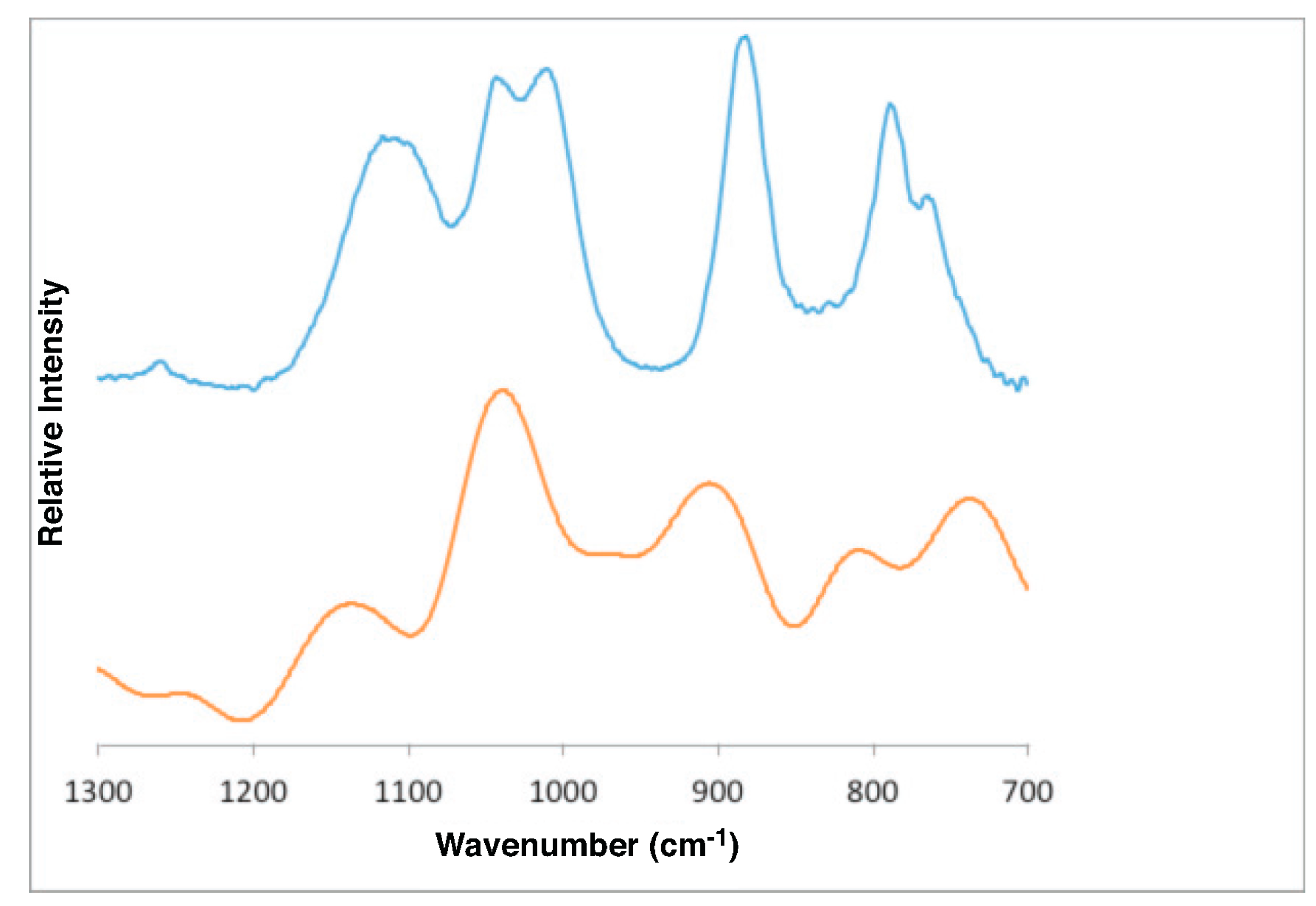
4.1. Gibbsite
- Gibbsite D [58]3623 3526 3456 3397 3367 3290 1054 1020 967 942 915
- DFT bulk gibbsite3673 3666 3549 3526 3508 3494 1119 1086 1058 1041 1010 990
- DFT (001) surface gibbsite3866 3835 3759 3736 3668 3522 3475 1110 1066 1027 994
4.2. Birnessites
4.3. Kaolinite–Acetic Acid
4.4. Silica Species in SiO2–H2O Liquids and Glass
4.5. Further Reading
5. Conclusions
- Make sure you are building a model that represents reality as closely as possible.
- Search the literature for previous studies for guidance as to which method will be accurate for your purposes.
- Use periodic and cluster models to complement one another; each model type has strengths and weaknesses.
- Most classical force field methods will not be as accurate as quantum methods. Many are parameterized to have bond force constants, k, obtained from experimental vibrational frequencies.
- Benchmark computational methods against well-understood systems wherever possible to estimate error in calculations.
- If possible, compare your calculated results with other sources of experimental data (e.g., NMR, XANES, EXAFS) to increase the certainty of your calculated results.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simon, J.D.; McQuarrie, D.A. The Boltzmann Factor and Partition Functions. In Physical Chemistry: A Molecular Approach; University Science Book: Sausalito, CA, USA, 1997; pp. 693–722. [Google Scholar]
- Schauble, E. Applying stable isotope fractionation theory to new systems. Rev. Mineral. Geochem. 2004, 55, 65–111. [Google Scholar] [CrossRef]
- Eiler, J.M.; Bergquist, B.; Bourg, I.; Cartigny, P.; Farquhar, J.; Gagnon, A.; Guo, W.; Halevy, I.; Hofmann, A.; Larson, T.E.; et al. Frontiers of stable isotope geoscience. Chem. Geol. 2014, 372, 119–143. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Rosso, K.M. Geochemical kinetics via computational chemistry. In Molecular Modeling of Geochemical Reactions: An Introduction.; Kubicki, J.D., Ed.; Wiley: Hoboken, NJ, USA, 2016; pp. 375–414. [Google Scholar]
- Philipona, R.; Dürr, B.; Marty, C.; Ohmura, A.; Wild, M. Radiative forcing-measured at Earth’s surface-corroborate the increasing greenhouse effect. Geophys. Res. Lett. 2004, 31, L03202. [Google Scholar] [CrossRef]
- Collins, W.D.; Ramaswamy, V.; Schwarzkopf, M.D.; Sun, Y.; Portmann, R.W.; Fu, Q.; Casanova, S.E.B.; Dufresne, J.-L.; Fillmore, D.W.; Forster, P.M.D.; et al. Radiative forcing by well-mixed greenhouse gases: Estimates from climate models in the Intergovernmental Panel on Climate Change (IPCC) Fourth Assessment Report (AR4). J. Geophys. Res. 2006, 111, D14317. [Google Scholar] [CrossRef]
- Harries, J.E.; Brindley, H.E.; Sagoo, P.J.; Bantges, R.J. Increases in greenhouse forcing inferred from the outgoing longwave radiation spectra of the Earth in 1970 and 1997. Nature 2001, 410, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.; Jacobson, A.R.; Sweeney, C.; Andrews, A.E.; Conway, T.J.; Masarie, K.; Miller, J.B.; Bruhwiler, L.M.P.; Pétron, G.; Hirsch, A.I.; et al. An atmospheric perspective on North American carbon dioxide exchange: CarbonTracker. Proc. Natl. Acad. Sci. USA 2007, 104, 18925–18930. [Google Scholar] [CrossRef] [PubMed]
- IPCC. Climate Change 2007: Working Group I: The Physical Science Basis Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, 2007; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Morman, S.A.; Plumlee, G.S. The role of airborne mineral dusts in human disease. Aeolian Res. 2013, 9, 203–212. [Google Scholar] [CrossRef]
- Fröhlich-Nowoisky, J.; Kampf, C.J.; Weber, B.; Huffman, J.A.; Pöhlker, C.; Andreae, M.O.; Lang-Yona, N.; Burrows, S.M.; Gunthe, S.S.; Elbert, W.; et al. Bioaerosols in the Earth system: Climate, health, and ecosystem interactions. Atmos. Res. 2016, 182, 346–376. [Google Scholar] [CrossRef]
- Groot Zwaaftink, C.D.; Grythe, H.; Skov, H.; Stohl, A. Substantial contribution of northern high-latitude sources to mineral dust in the Arctic. J. Geophys. Res. 2016, 121, 13678–13697. [Google Scholar] [CrossRef]
- Prospero, J.M.; Collard, F.X.; Molinié, J.; Jeannot, A. Characterizing the annual cycle of African dust transport to the Caribbean Basin and South America and its impact on the environment and air quality. Glob. Biogeochem. Cycle. 2014, 28, 757–773. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Sykes, D. Molecular orbital calculations of vibrations in three-membered aluminosilicate rings. Phys. Chem. Miner. 1993, 19, 381–391. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Paul, K.W.; Kabalan, L.; Zhu, Q.; Mrozik, M.K.; Aryanpour, M.; Pierre-Louis, A.M.; Strongin, D.R. ATR-FTIR and density functional theory study of the structures, energetics, and vibrational spectra of phosphate adsorbed onto goethite. Langmuir 2012, 28, 14573–14587. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Neogi, S.; Kent, P.R.C.; Bandura, A.V.; Kubicki, J.D.; Wesolowski, D.J.; Cole, D.; Sofo, J.O. Hydrogen Bonds and Vibrations of Water on (110) Rutile. J. Phys. Chem. C 2009, 113, 13732–13740. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Frisch, M.; Gaw, J.; Schaefer, H.F.; Binkley, J.S. Analytic evaluation and basis set dependence of intensities of infrared spectra. J. Chem. Phys. 1986, 84, 2262–2278. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09 Revision E.01. Wallingford, CT. 2009. Available online: http://gaussian.com/ (accessed on 15 January 2019).
- Kubicki, J.; Sykes, D.; Rossman, G.R. Calculated Trends of OH Infrared Stretching Vibrations with Composition and Structure in Aluminosilicate Molecules. Phys. Chem. Miner. 1993, 20, 425–432. [Google Scholar] [CrossRef]
- Radom, L.; Schleyer, P.V.R.; Pople, J.A.; Hehre, W.J. Ab Initio Molecular Orbital Theory; John Wiley and Sons: San Francisco, CA, USA, 1986. [Google Scholar]
- Ogilvie, J.F. The Vibrational and Rotational Spectrometry of Diatomic Molecules; Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- Paterson, M.S. The Determination of Hydroxyl by Infrared Absorption in Quartz, Silicate Glasses and Similar Minerals. Bull. Minéral. 1982, 105, 20–29. [Google Scholar]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided. Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Padma Kumar, P. Ab initio molecular dynamics investigation of structural, dynamic and spectroscopic aspects of Se(VI) species in the aqueous environment. Phys. Chem. Chem. Phys. 2016, 18, 14561–14568. [Google Scholar] [CrossRef] [PubMed]
- Bloino, J.; Barone, V. A second-order perturbation theory route to vibrational averages and transition properties of molecules: General formulation and application to infrared and vibrational circular dichroism spectroscopies. J. Chem. Phys. 2012, 136. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, R.L.; Johnson, R.D.; Irikura, K.K.; Kacker, R.N. Anharmonic vibrational frequency calculations are not worthwhile for small basis sets. J. Chem. Theory Comput. 2013, 9, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.; Stolper, E.M.; Epstein, S. Measurement of water in rhyolitic glasses-calibration of an infrared spectroscopic technique. Am. Mineral. 1986, 71, 1527–1541. [Google Scholar]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Wong, M.W. Vibrational frequency prediction using density functional theory. Chem. Phys. Lett. 1996, 256, 391–399. [Google Scholar] [CrossRef]
- Schmidt, J.R.; Polik, W.F. WebMO Enterprise, Version 13.0; WebMO LLC: Holland, MI, USA, 2013. [Google Scholar]
- Lee, C.M.; Kubicki, J.D.; Fan, B.; Zhong, L.; Jarvis, M.C.; Kim, S.H. Hydrogen-Bonding Network and OH Stretch Vibration of Cellulose: Comparison of Computational Modeling with Polarized IR and SFG Spectra. J. Phys. Chem. B 2015, 119, 15138–15149. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Mohamed, N.M.A.; Watts, H.D.; Kubicki, J.D.; Kim, S.H. Sum-frequency-generation vibration spectroscopy and density functional theory calculations with dispersion corrections (DFT-D2) for cellulose Iα and Iβ. J. Phys. Chem. B 2013, 117, 6681–6692. [Google Scholar] [CrossRef] [PubMed]
- Gogonea, V. Self-Consistent Reaction Field Methods: Cavities. In Encyclopedia of Computational Chemistry; von Rague Schleyer, P., Allinger, N.L., Clark, T., Gastiger, J., Kollman, P.A., Schaefer, H.F.I., Schreiner, P.R., Eds.; Wiley: New York, NY, USA, 1998; pp. 2560–2574. [Google Scholar]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Delley, B. The conductor-like screening model for polymers and surfaces. Mol. Simul. 2006, 32, 117–123. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V.; Bürger, T.; Lohrenz, J.C.W. Refinement and Parametrization of COSMO-RS. J. Phys. Chem. A 1998, 102, 5074–5085. [Google Scholar] [CrossRef]
- Covert, P.A.; Hore, D.K. Geochemical Insight from Nonlinear Optical Studies of Mineral–Water Interfaces. Annu. Rev. Phys. Chem. 2016, 67, 233–257. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Ruiz, M.; Ferrage, E.; Blanchard, M.; Fernandez-Castanon, J.; Delville, A.; Johnson, M.R.; Michot, L.J. Combination of Inelastic Neutron Scattering Experiments and Ab-Initio Quantum Calculations for the Study of the Hydration Properties of Oriented Saponites. J. Phys. Chem. C 2017, 121, 5029–5040. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B. Condens. Matter 1993, 48, 13115–13118. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J.; Hafner, J. Theory of the crystal structures of selenium and tellurium: The effect of generalized-gradient corrections to the local-density approximation. Phys. Rev. B 1994, 50, 13181–13185. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; López Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The calculation of the vibrational frequencies of crystalline compounds and its implementation in the CRYSTAL code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; Garcia, A.; Junquera, J.; Ordejon, P.; Sanchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2001, 14, 2745–2779. [Google Scholar] [CrossRef]
- Saalfeld, H.; Wedde, M. Refinement of the crystal structure of gibbsite, Al(OH)3. Z. Krist. Cryst. Mater. 1974, 139, 120–135. [Google Scholar] [CrossRef]
- Frost, R.A.Y.L.; Kloprogge, J.T.; Russell, S.C.; Szetu, J.L. Vibrational Spectroscopy and Dehydroxylation of Aluminum (Oxo) hydroxides: Gibbsite. Appl. Spectrosc. 1999, 53, 423–434. [Google Scholar] [CrossRef]
- Balan, E.; Lazzeri, M.; Morin, G.; Mauri, F. First-principles study of the OH-stretching modes of gibbsite. Am. Mineral. 2006, 91, 115–119. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Apitz, S.E. Molecular cluster models of aluminum oxide and aluminum hydroxide surfaces. Am. Mineral. 1998, 83, 1054–1066. [Google Scholar] [CrossRef]
- Ling, F.T.; Post, J.E.; Heaney, P.J.; Kubicki, J.D.; Santelli, C.M. Fourier-transform infrared spectroscopy (FTIR) analysis of triclinic and hexagonal birnessites. Spectroc. Acta Pt. A Molec. Biomolec. Spectr. 2017, 178, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Alstadt, V.J.; Kubicki, J.D.; Freedman, M.A. Competitive Adsorption of Acetic Acid and Water on Kaolinite. J. Phys. Chem. A 2016, 120, 8339–8346. [Google Scholar] [CrossRef] [PubMed]
- Molina-Montes, E.; Timón, V.; Hernández-laguna, A.; Sainz-díaz, C.I. Dehydroxylation mechanisms in Al3+/Fe3+ dioctahedral phyllosilicates by quantum mechanical methods with cluster models. Geochim. Cosmochim. Acta 2008, 72, 3929–3938. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, G.; Steele-Macinnis, M.; Schmidt, C.; Jahn, S. Vibrational mode frequencies of silica species in SiO₂-H₂O liquids and glasses from ab initio molecular dynamics. J. Chem. Phys. 2012, 136, 154501. [Google Scholar] [CrossRef] [PubMed]
- Marx, D.; Hutter, J. Modern Methods and Algorithms of Quantum Chemistry. Forsch. Jülich NIC Ser. 2000, 1, 301–449. [Google Scholar]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, G.; Steele-MacInnis, M.; Kowalski, P.M.; Schmidt, C.; Jahn, S. Vibrational properties of silica species in MgO-SiO₂ glasses obtained from ab initio molecular dynamics. Chem. Geol. 2013, 346, 22–33. [Google Scholar] [CrossRef]
- Spiekermann, G.; Steele-MacInnis, M.; Kowalski, P.M.; Schmidt, C.; Jahn, S. Vibrational mode frequencies of H4SiO4, D4SiO4, H6Si2O7, and H6Si3O9 in aqueous environment, obtained from ab initio molecular dynamics. J. Chem. Phys. 2012, 137, 164506. [Google Scholar] [CrossRef] [PubMed]
- Scholtzová, E.; Madejová, J.; Jankovič, L.; Tunega, D. Structural and spectroscopic characterization of montmorillonite intercalated with N-butylammonium cations (N = 1–4)-Modeling and experimental study. Clay. Clay Miner. 2016, 64, 401–412. [Google Scholar] [CrossRef]
- Tunega, D.; Benco, L.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. Ab initio molecular dynamics study of adsorption sites on the (001) surfaces of 1:1 dioctahedral clay minerals. J. Phys. Chem. B 2002, 106, 11515–11525. [Google Scholar] [CrossRef]
- Scholtzová, E.; Tunega, D.; Madejová, J.; Pálková, H.; Komadel, P. Theoretical and experimental study of montmorillonite intercalated with tetramethylammonium cation. Vib. Spectrosc. 2013, 66, 123–131. [Google Scholar] [CrossRef]
- Balan, E.; Lazzeri, M.; Saitta, A.M.; Allard, T.; Fuchs, Y.; Mauri, F. First-principles study of OH-stretching modes in kaolinite, dickite, and nacrite. Am. Mineral. 2005, 90, 50–60. [Google Scholar] [CrossRef]
- Balan, E.; Saitta, A.M.; Mauri, F.; Calas, G. First-principles modeling of the infrared spectrum of kaolinite. Am. Mineral. 2001, 86, 1321–1330. [Google Scholar] [CrossRef]
- Balan, E.; Saitta, A.M.; Mauri, F.; Lemaire, C.; Guyot, F. First-principles calculation of the infrared spectrum of lizardite. Am. Mineral. 2002, 87, 1286–1290. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | ν(Harm) (cm−1) | ν(Anharm) (cm−1) |
|---|---|---|
| Fundamental Bands | ||
| 1(1) | 3913.897 | 3726.279 |
| 2(1) | 3800.402 | 3628.674 |
| 3(1) | 1664.814 | 1615.055 |
| Overtones | ||
| 1(2) | 7827.794 | 7355.593 |
| 2(2) | 7600.805 | 7172.676 |
| 3(2) | 3329.628 | 3193.999 |
| Combination Bands | ||
| 2(1) 1(1) | 7714.299 | 7190.895 |
| 3(1) 1(1) | 5578.711 | 5324.089 |
| 3(1) 2(1) | 5465.217 | 5233.679 |
| ZPE(Harm) = 56.10 kJ/mol ZPE(Anharm) = 55.24 kJ/mol | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubicki, J.D.; Watts, H.D. Quantum Mechanical Modeling of the Vibrational Spectra of Minerals with a Focus on Clays. Minerals 2019, 9, 141. https://doi.org/10.3390/min9030141
Kubicki JD, Watts HD. Quantum Mechanical Modeling of the Vibrational Spectra of Minerals with a Focus on Clays. Minerals. 2019; 9(3):141. https://doi.org/10.3390/min9030141
Chicago/Turabian StyleKubicki, James D., and Heath D. Watts. 2019. "Quantum Mechanical Modeling of the Vibrational Spectra of Minerals with a Focus on Clays" Minerals 9, no. 3: 141. https://doi.org/10.3390/min9030141
APA StyleKubicki, J. D., & Watts, H. D. (2019). Quantum Mechanical Modeling of the Vibrational Spectra of Minerals with a Focus on Clays. Minerals, 9(3), 141. https://doi.org/10.3390/min9030141