Hydration Mechanism and Hardening Property of α-Hemihydrate Phosphogypsum

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of HH Powders
2.3. Preparation of Hydration Products
2.4. Characterization Methods
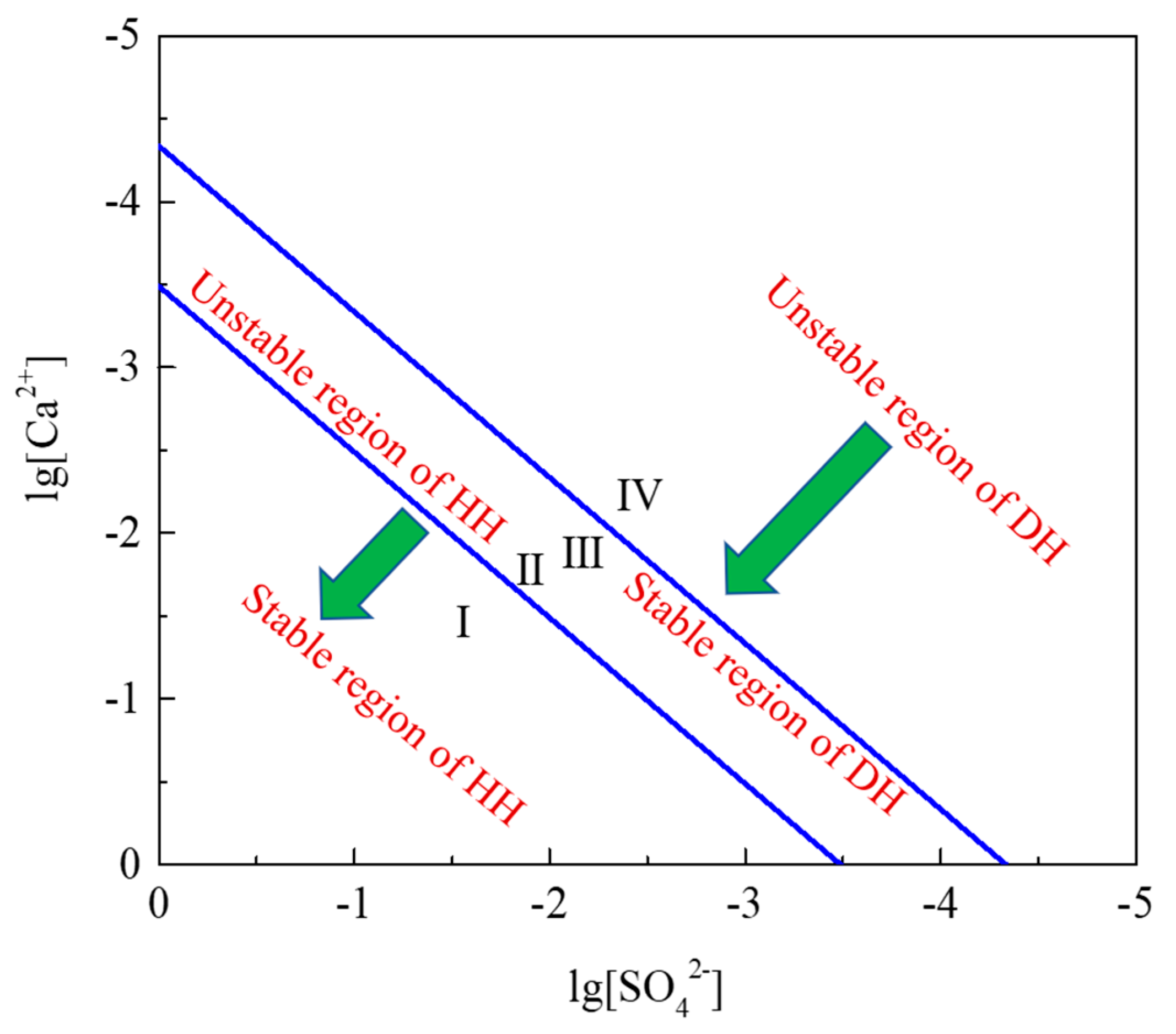
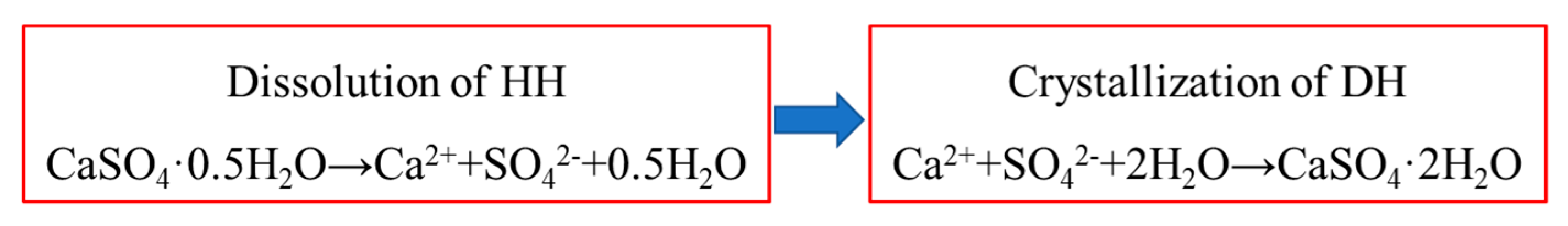
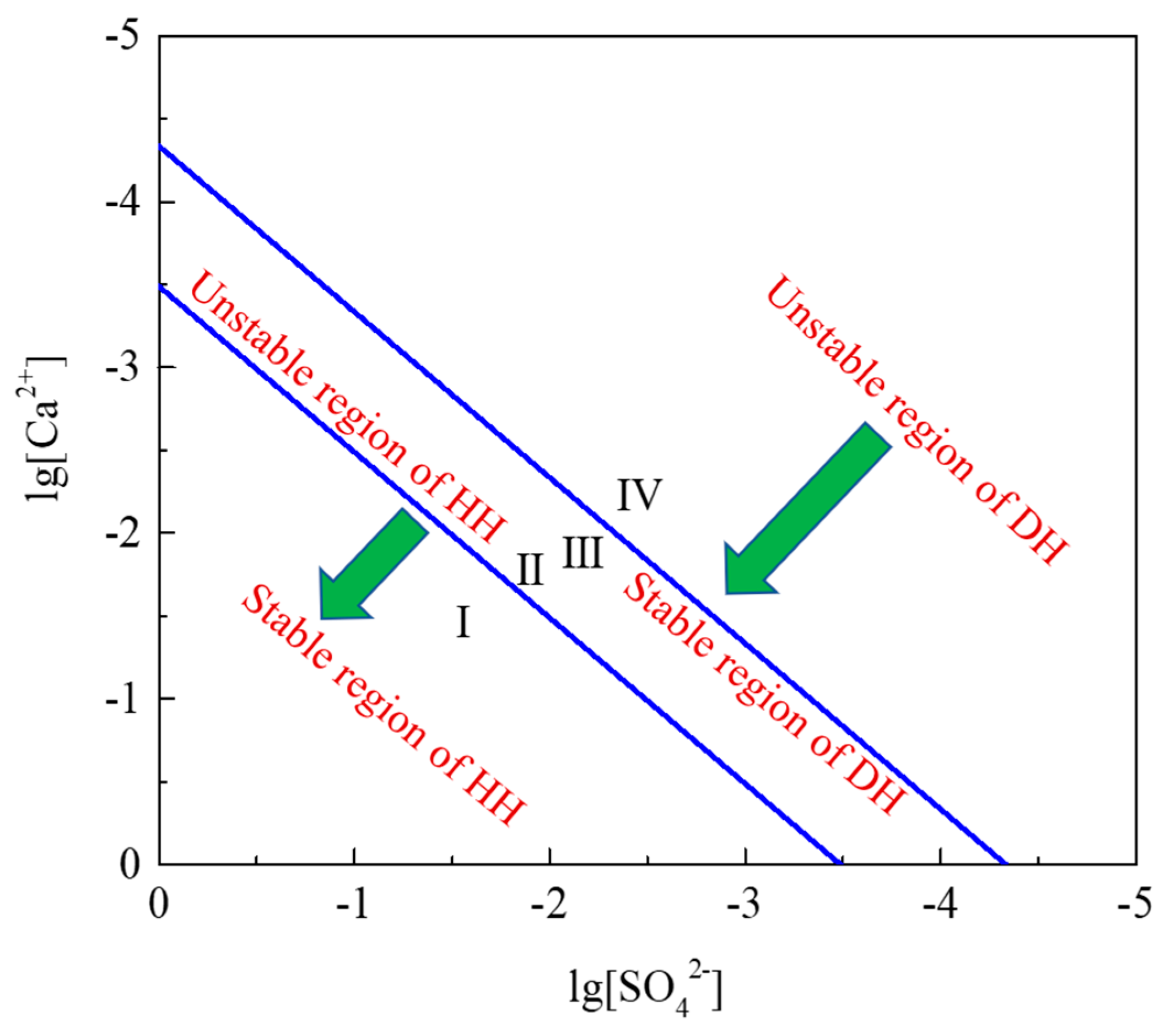
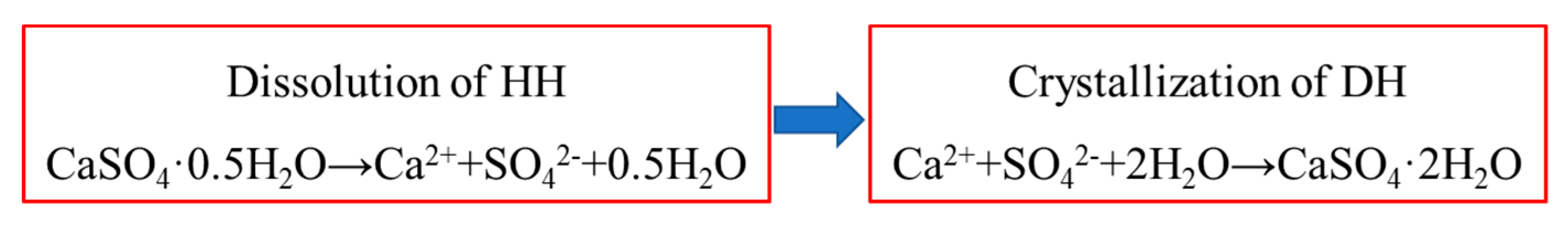
3. Thermodynamic Analysis of Hydration Process of HH
4. Results and Discussion
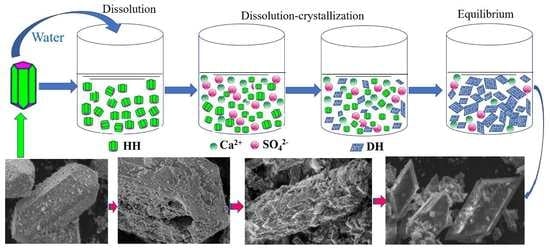
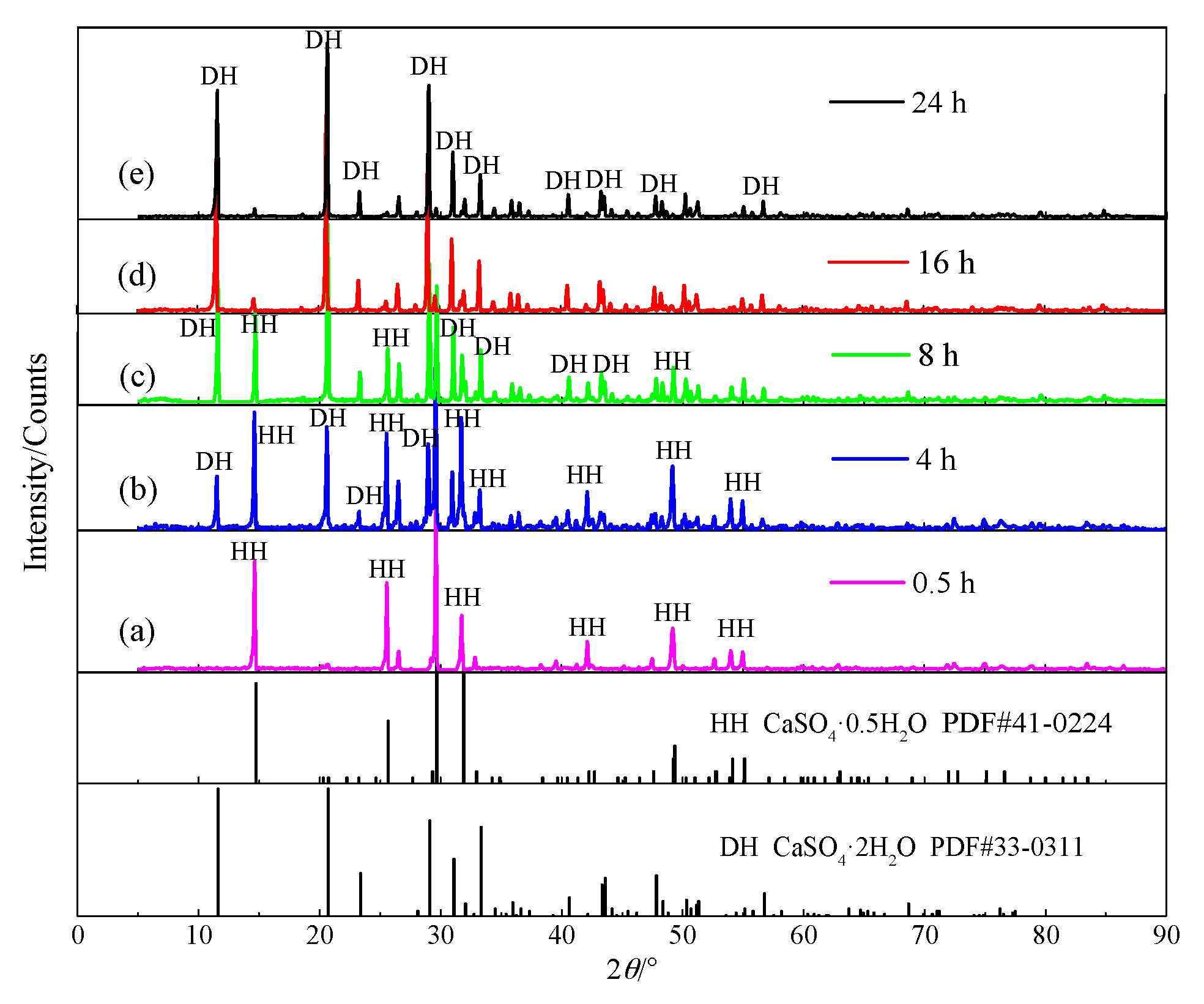
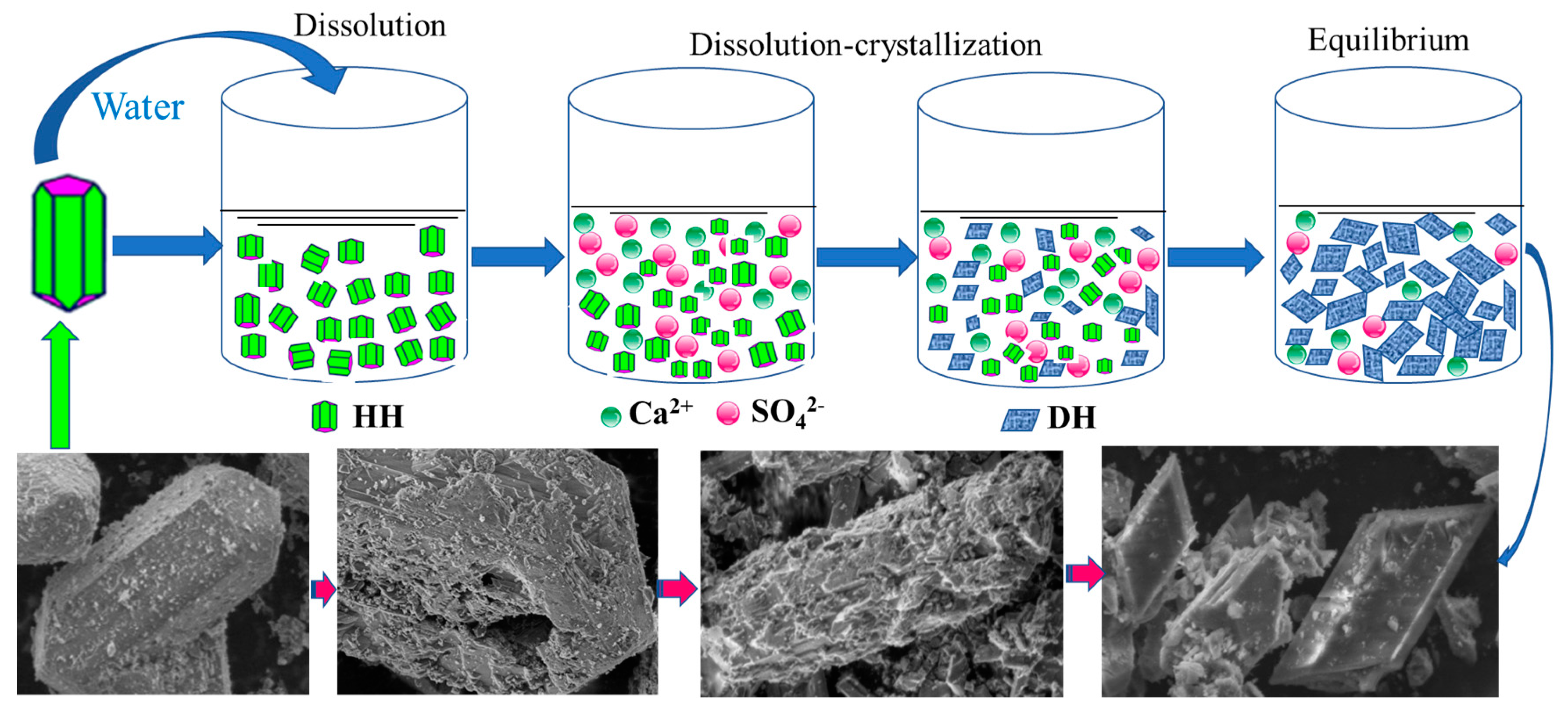
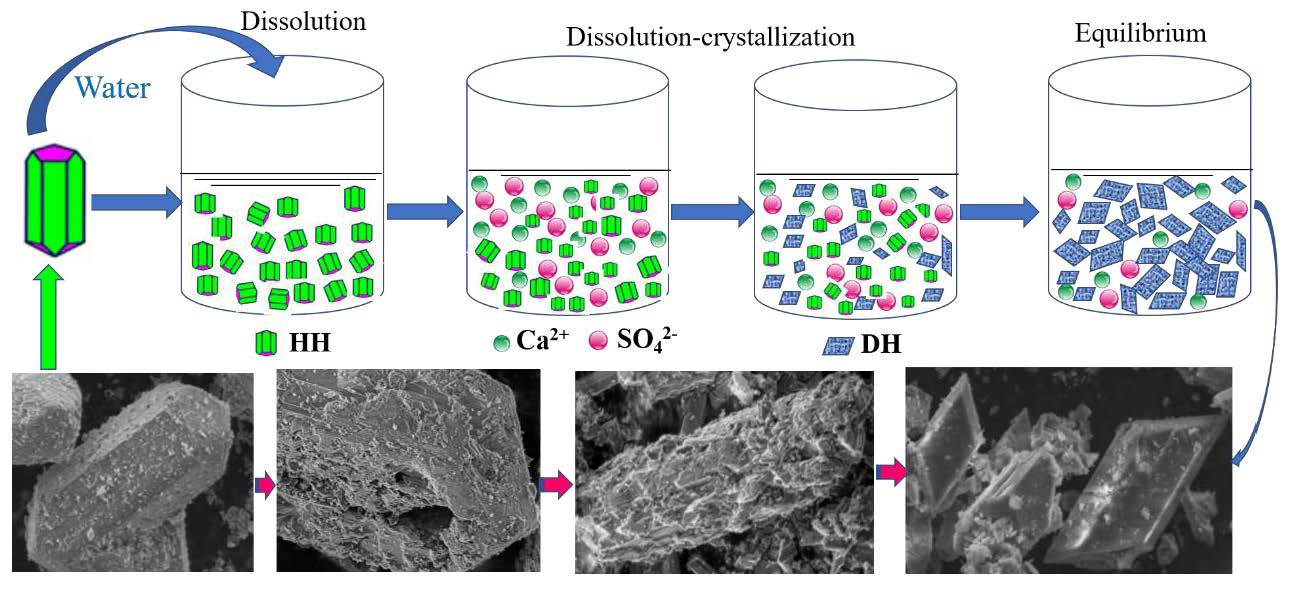
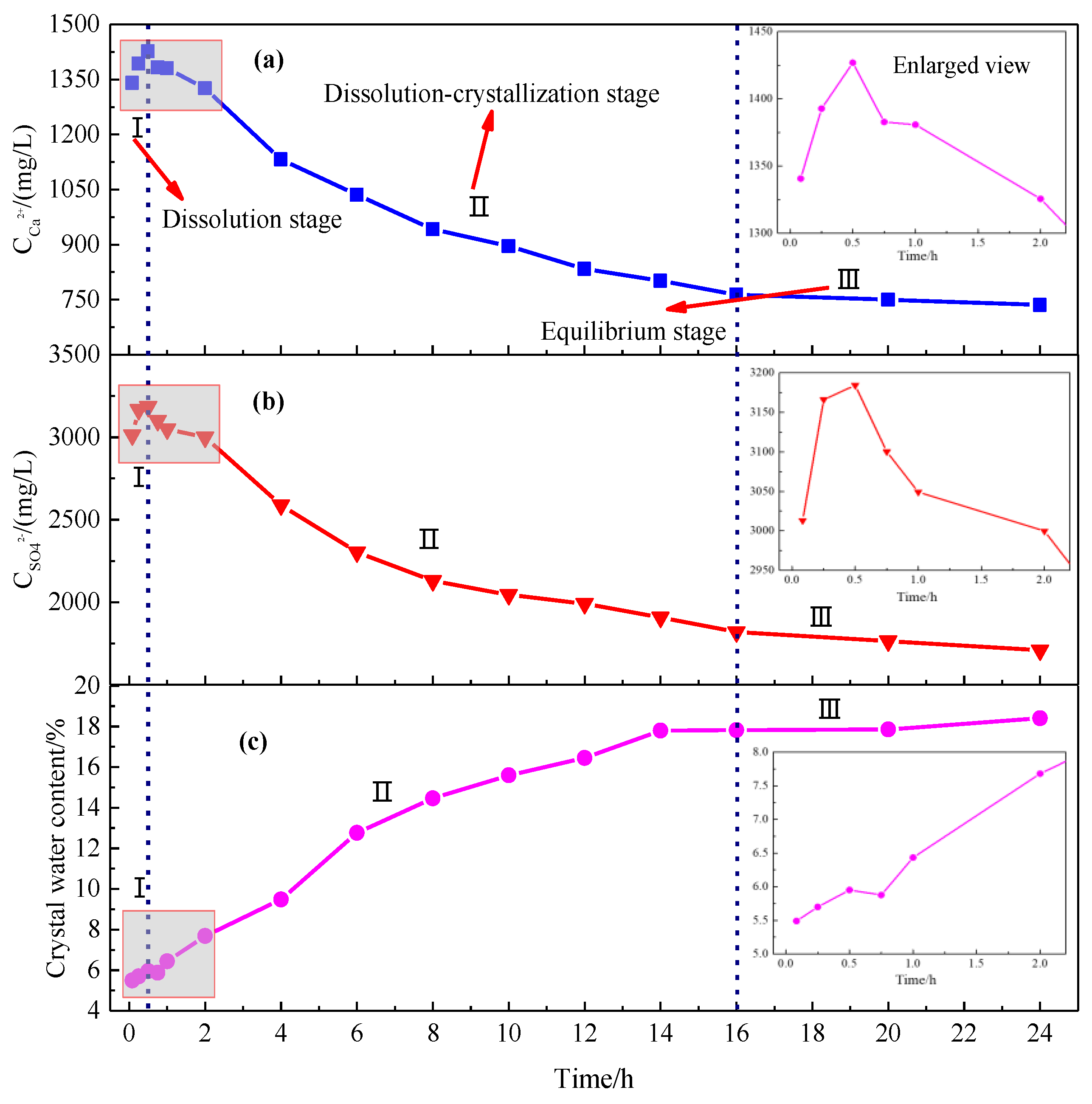
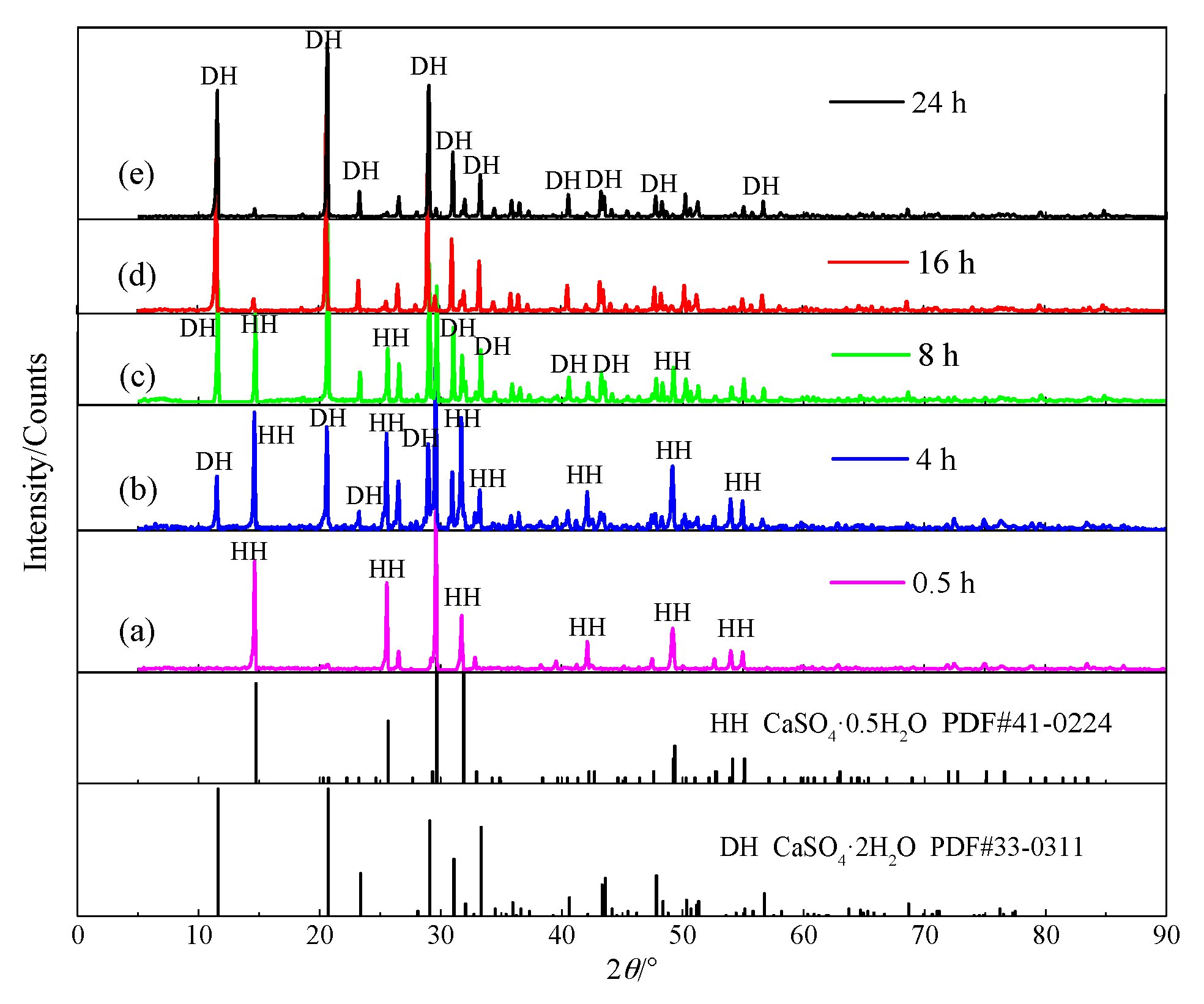
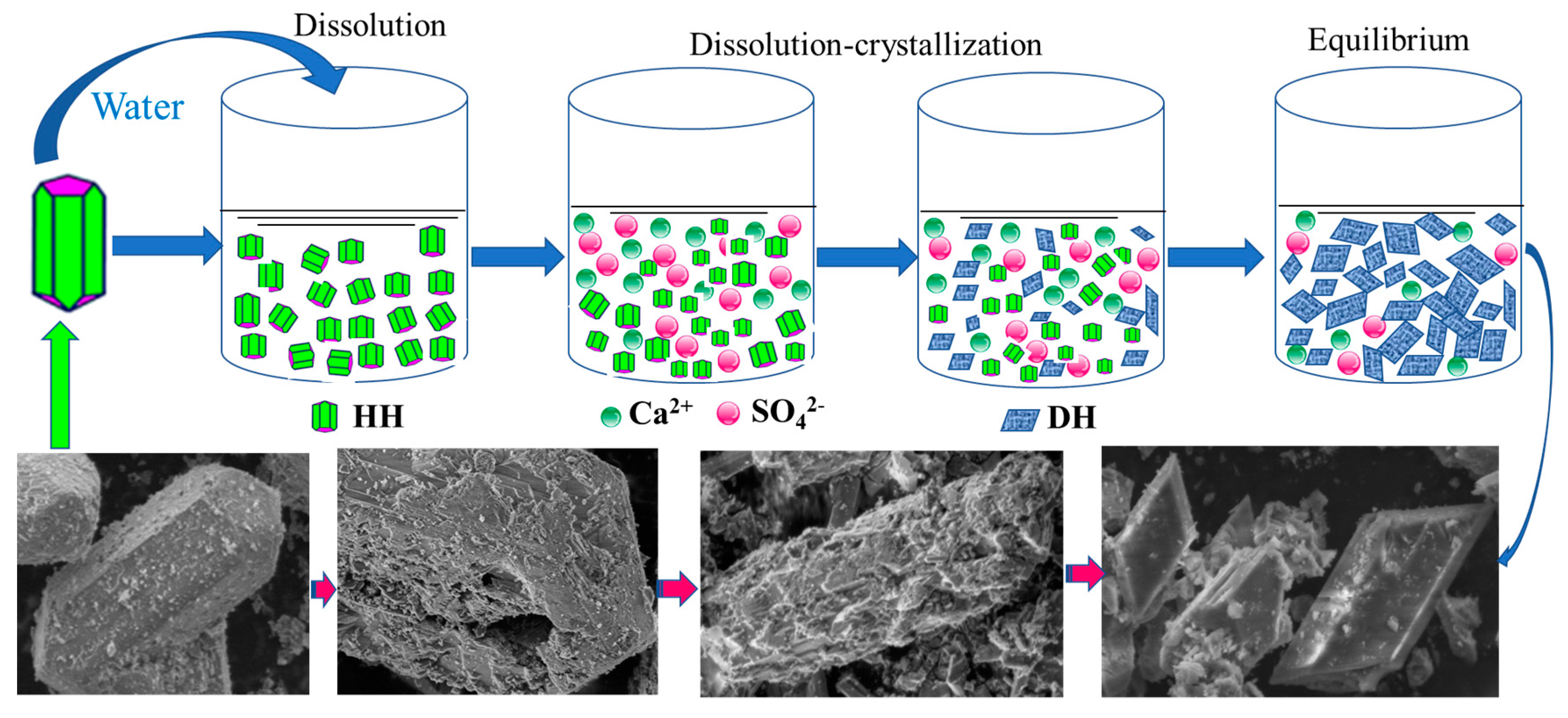
4.1. Hydration Process of HH
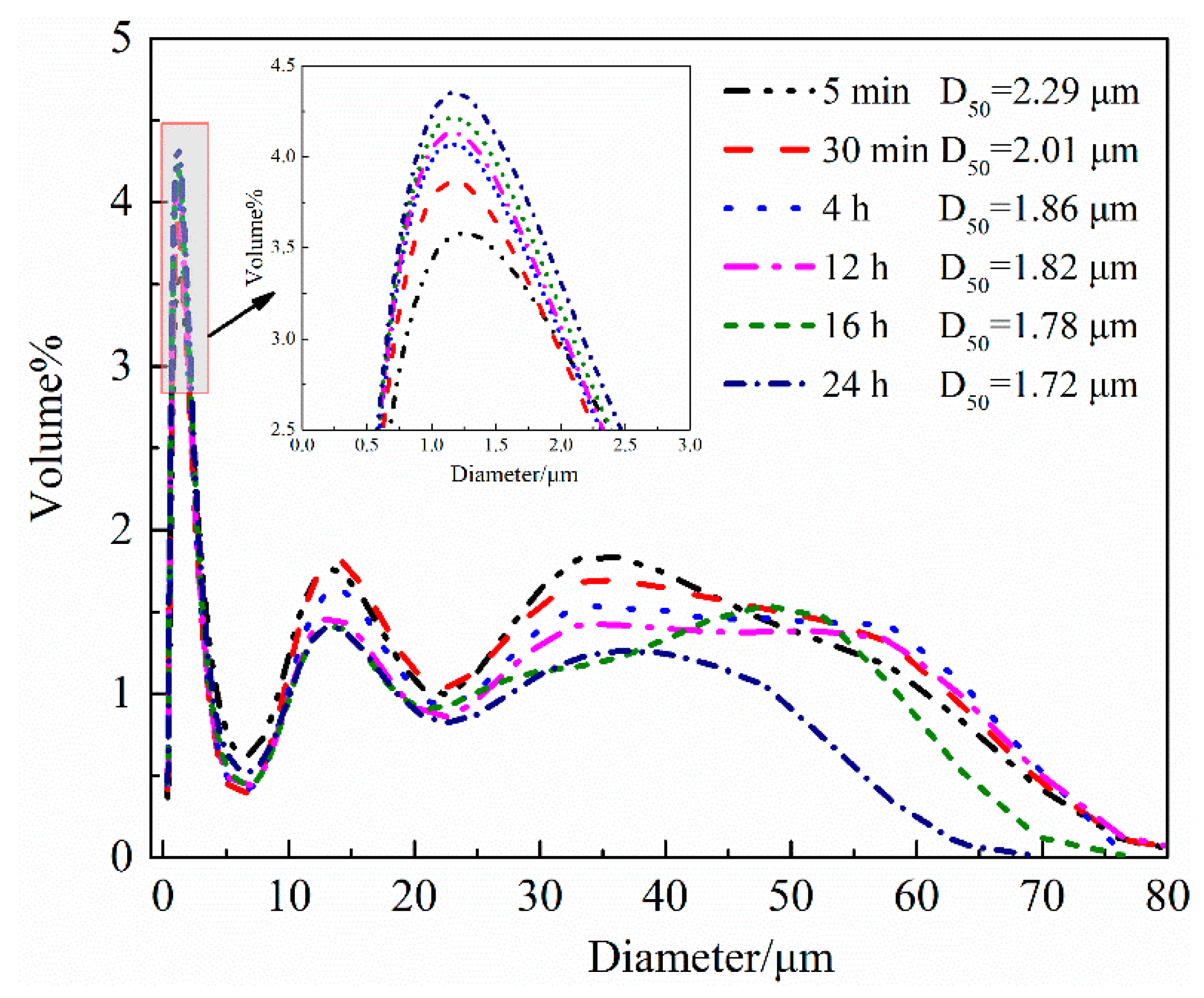
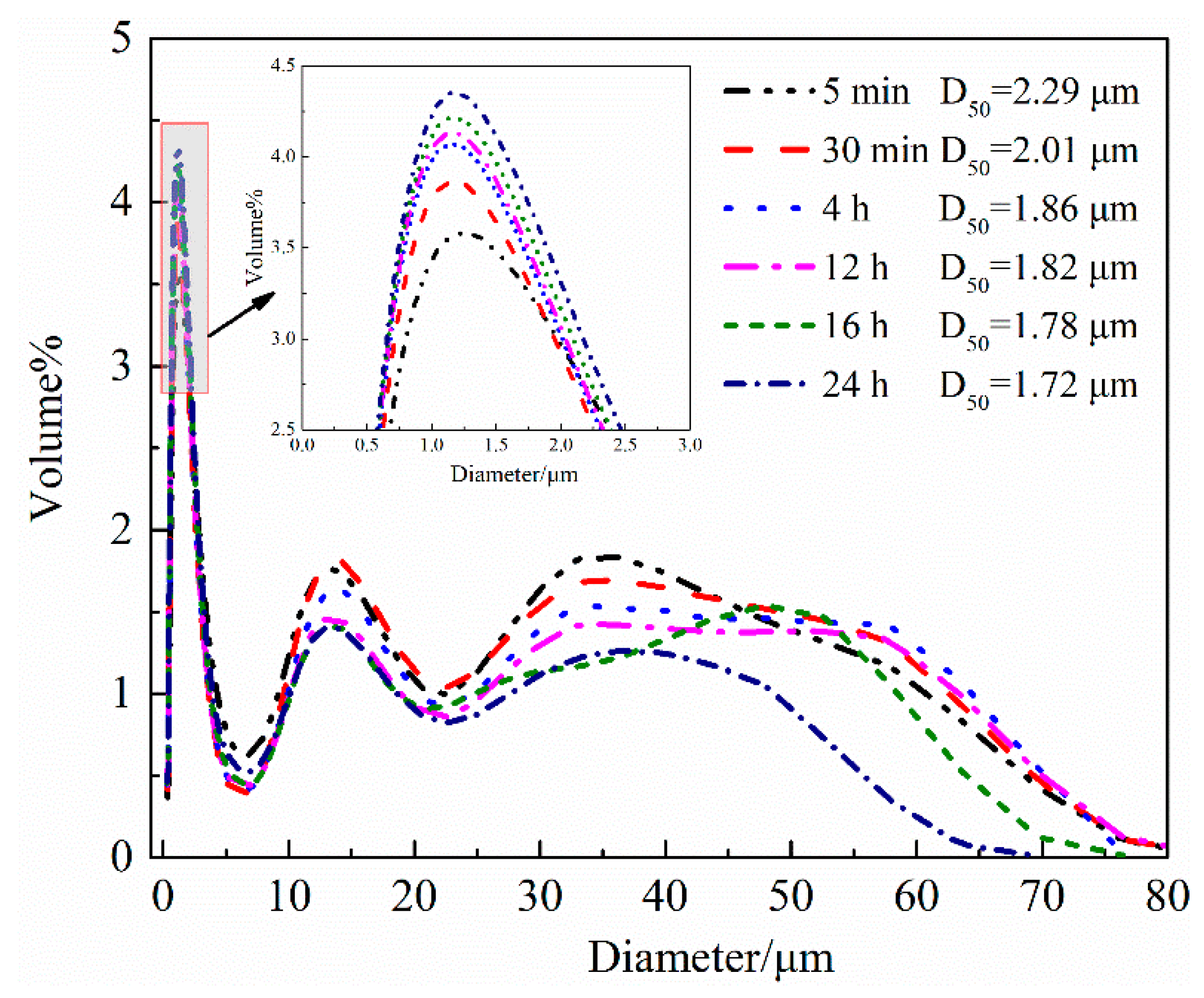
4.1.1. Hydration Process of HH Prepared in the Absence of Crystal Modifier
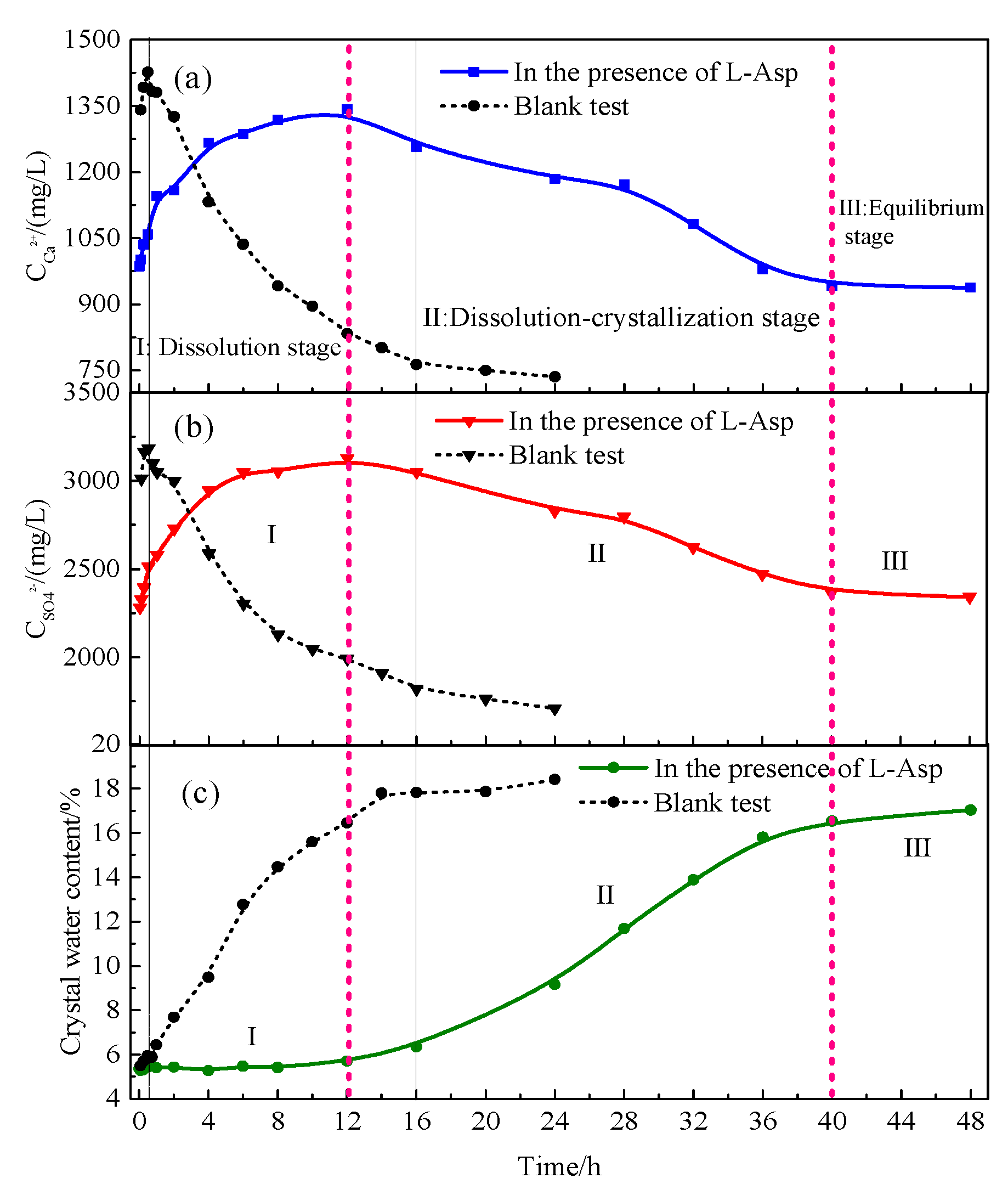
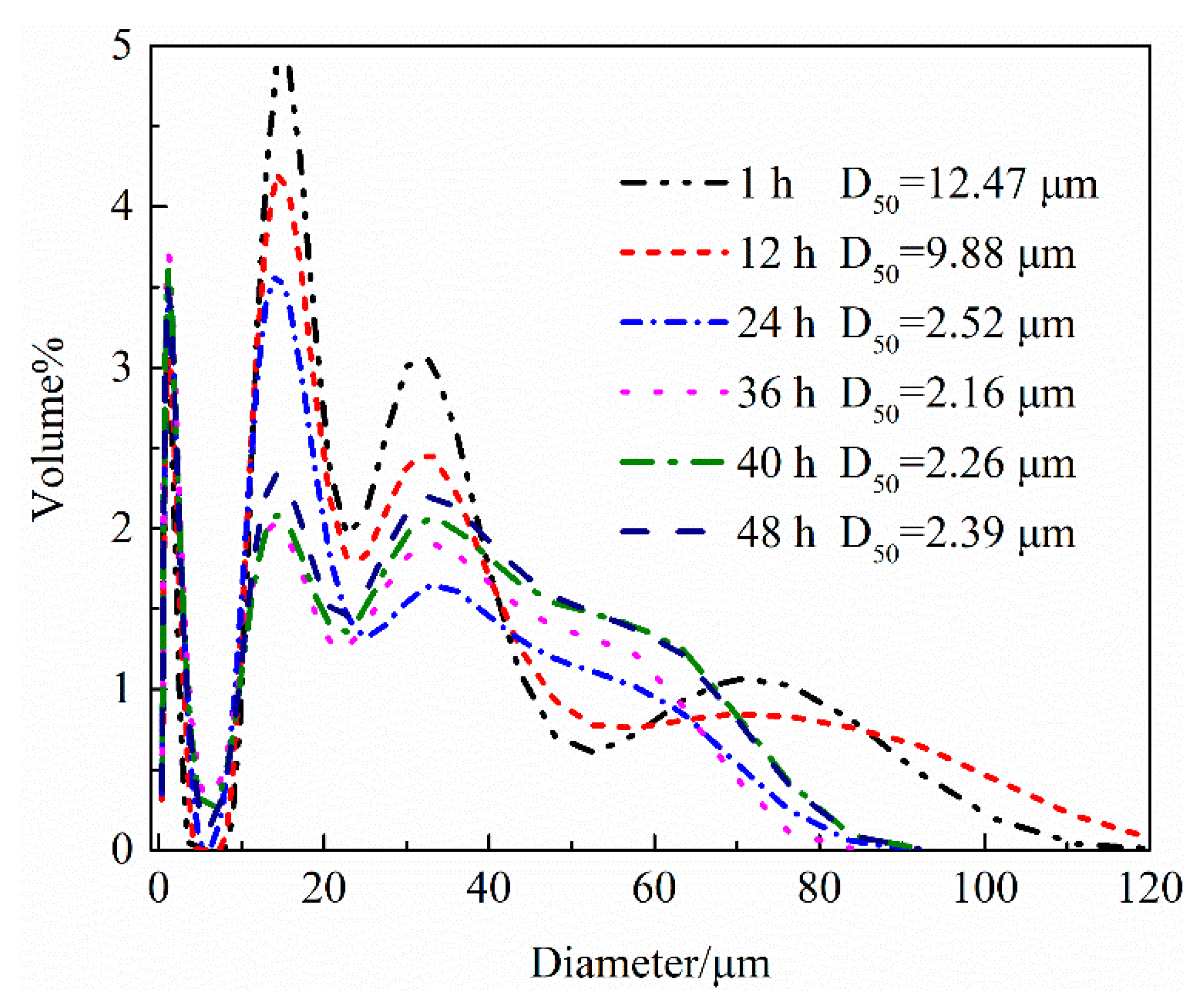
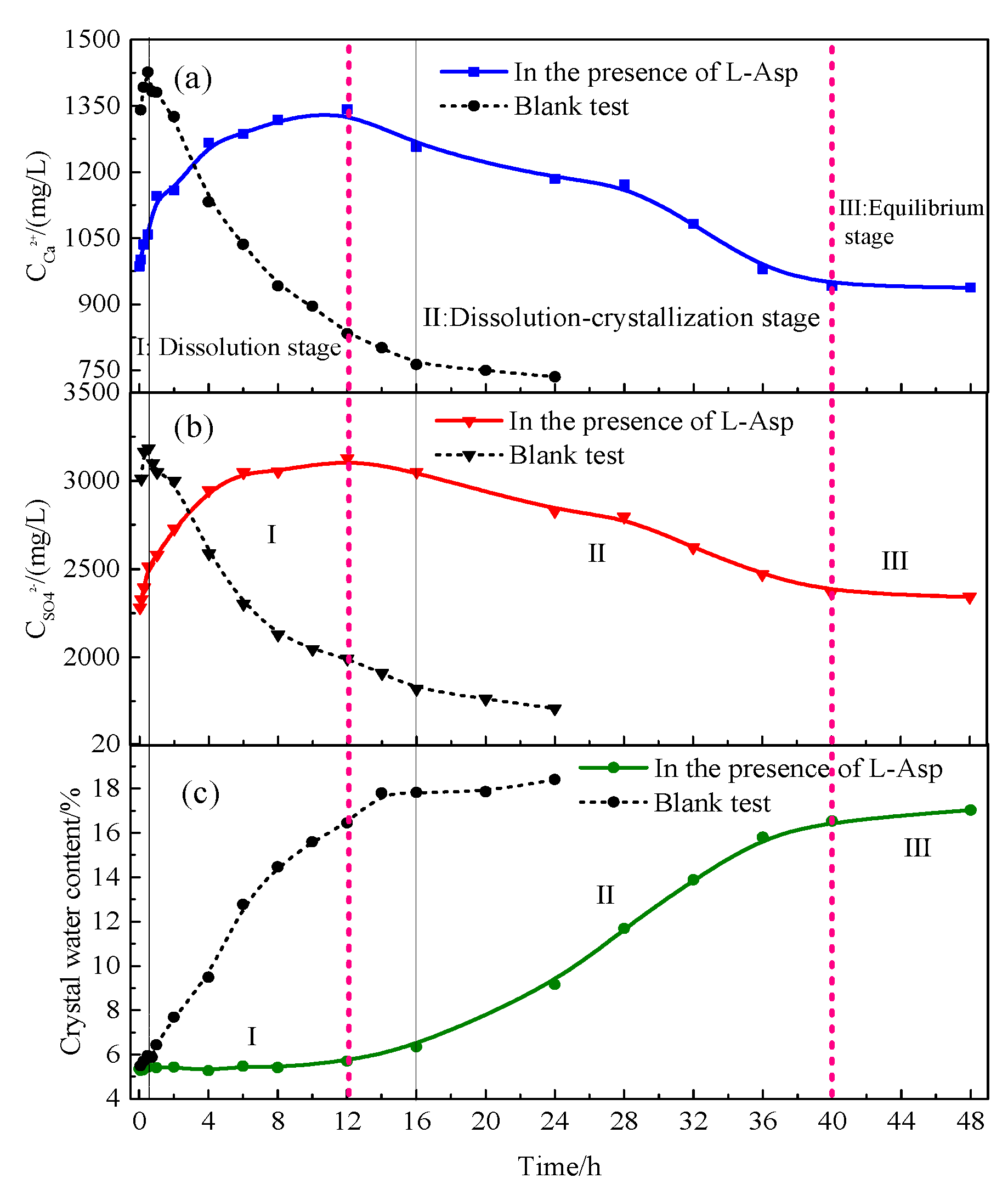
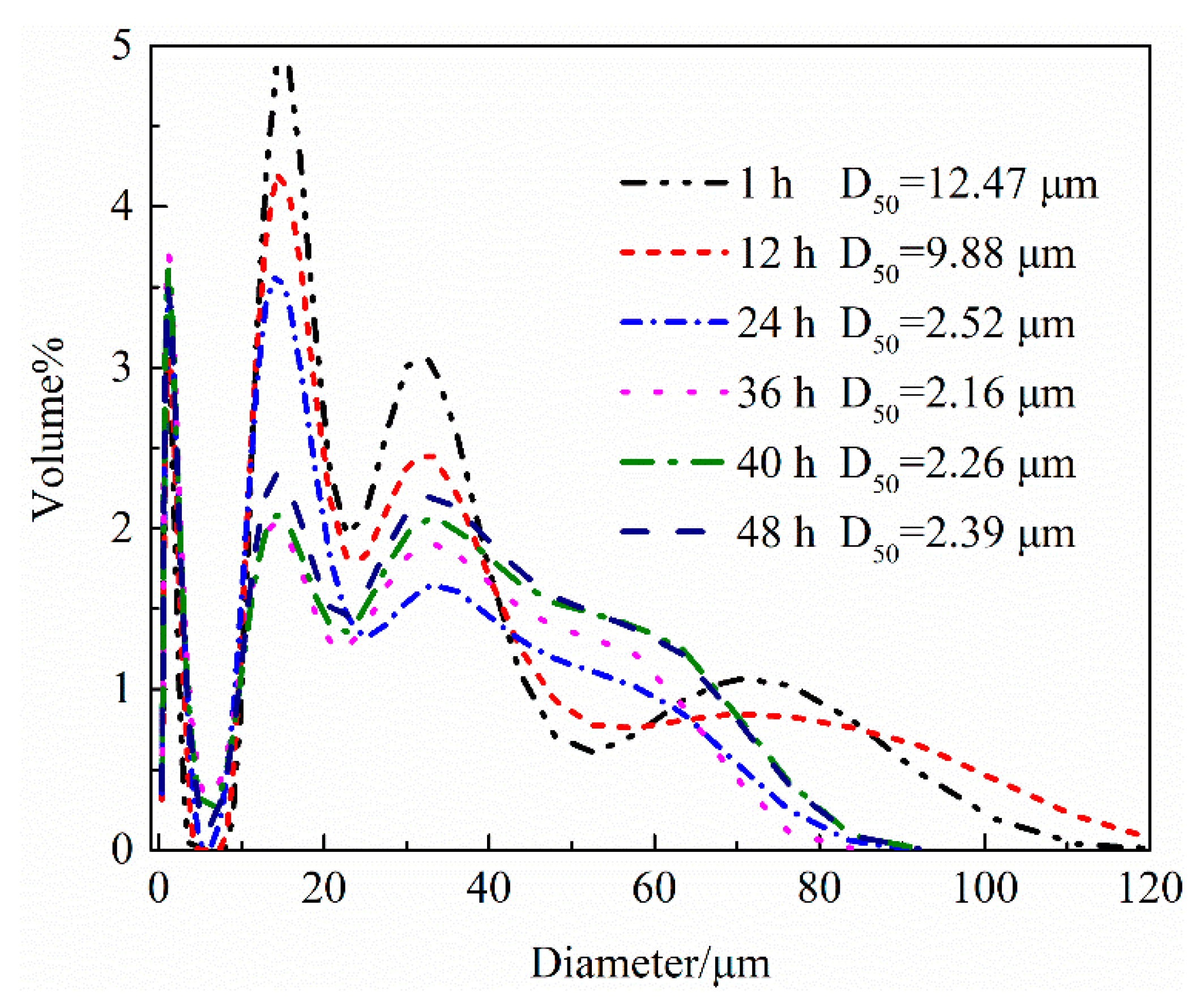
4.1.2. Hydration Process of HH Prepared in the Presence of L-Asp
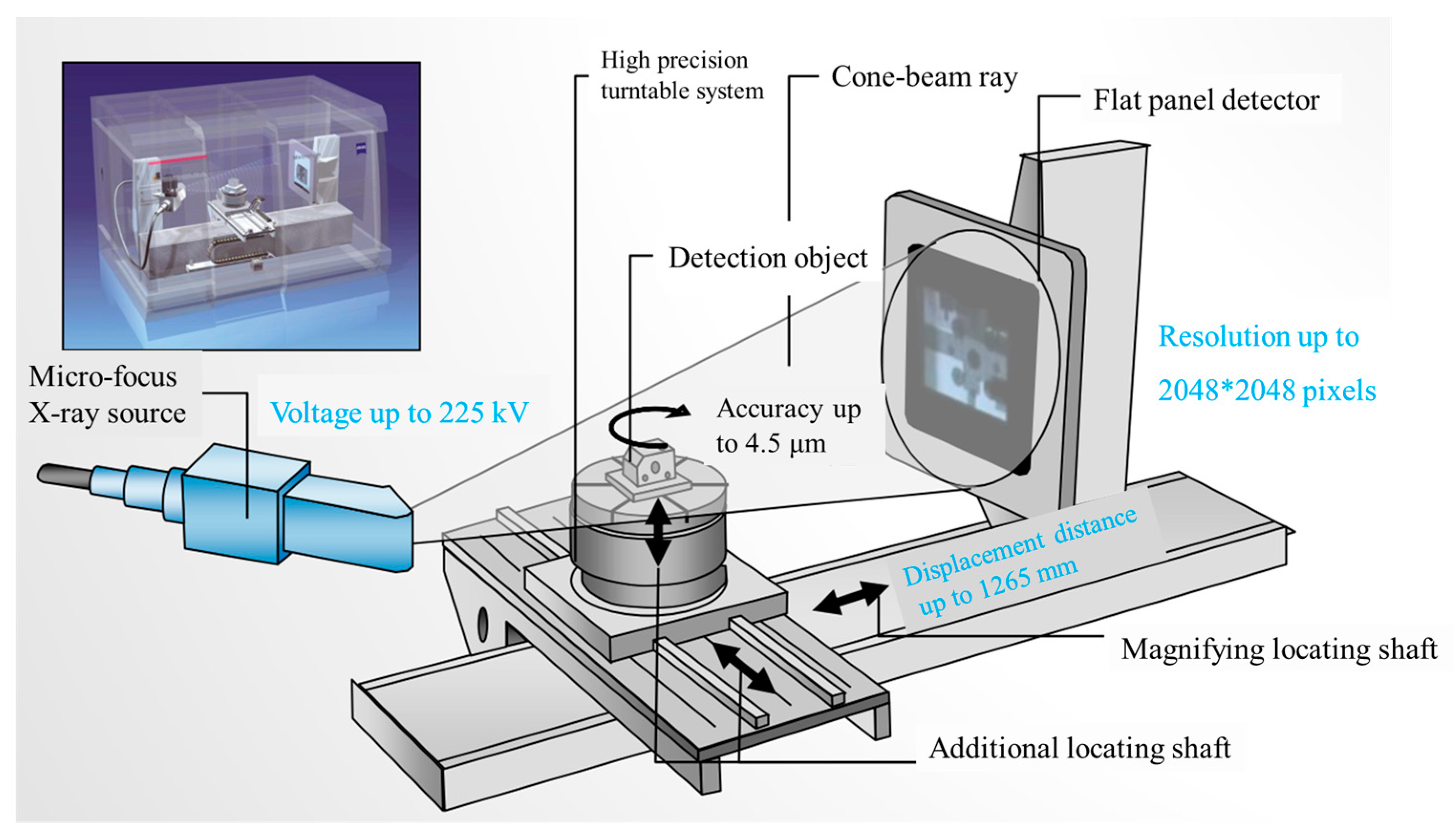
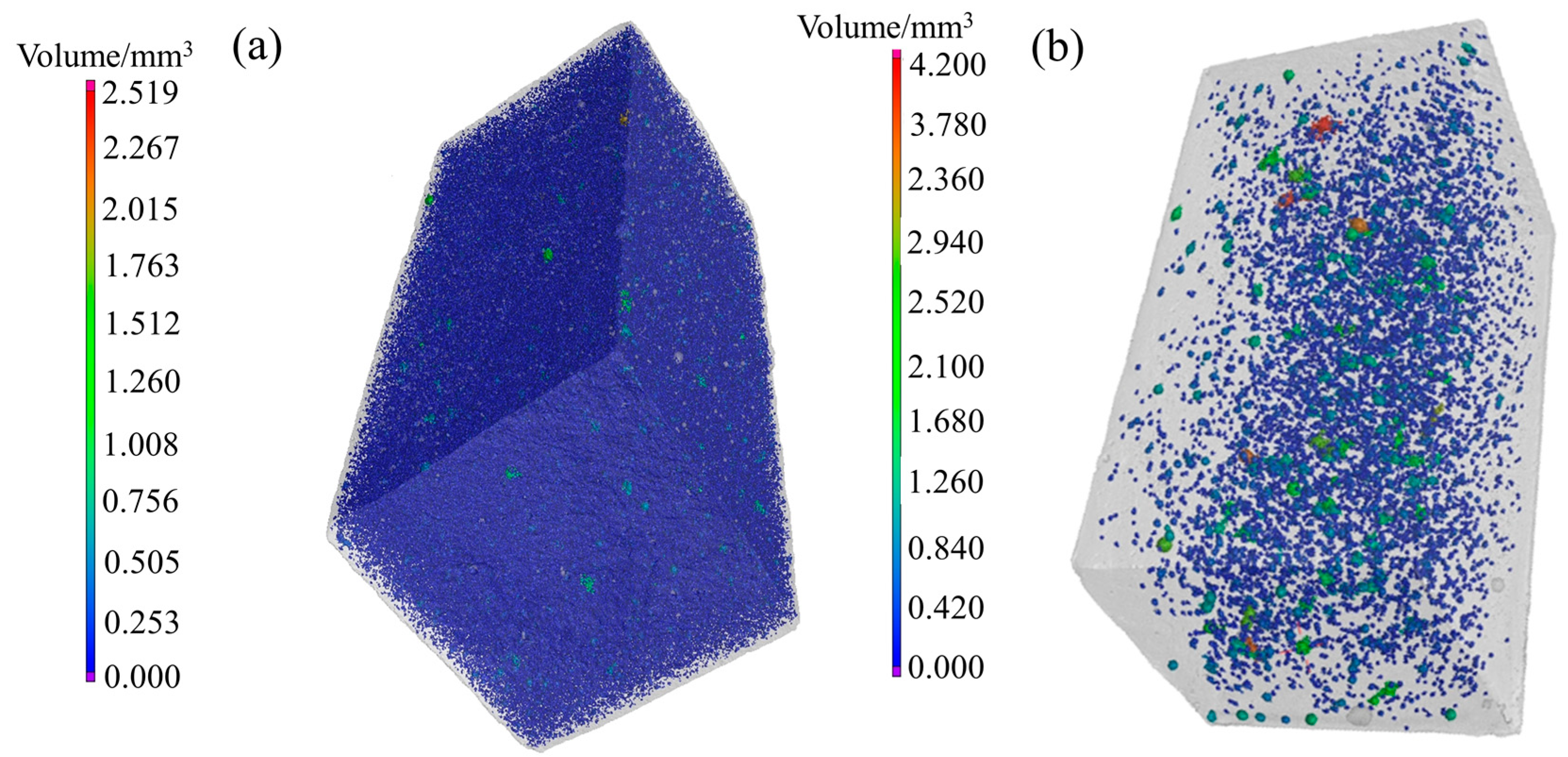
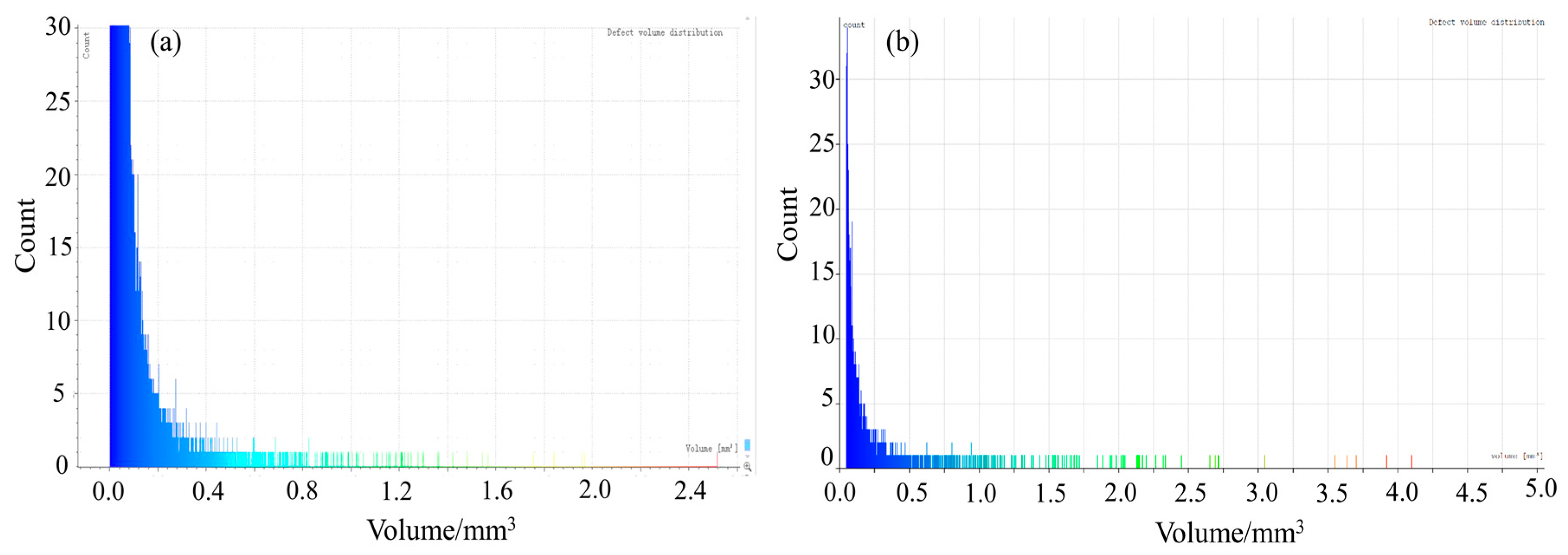
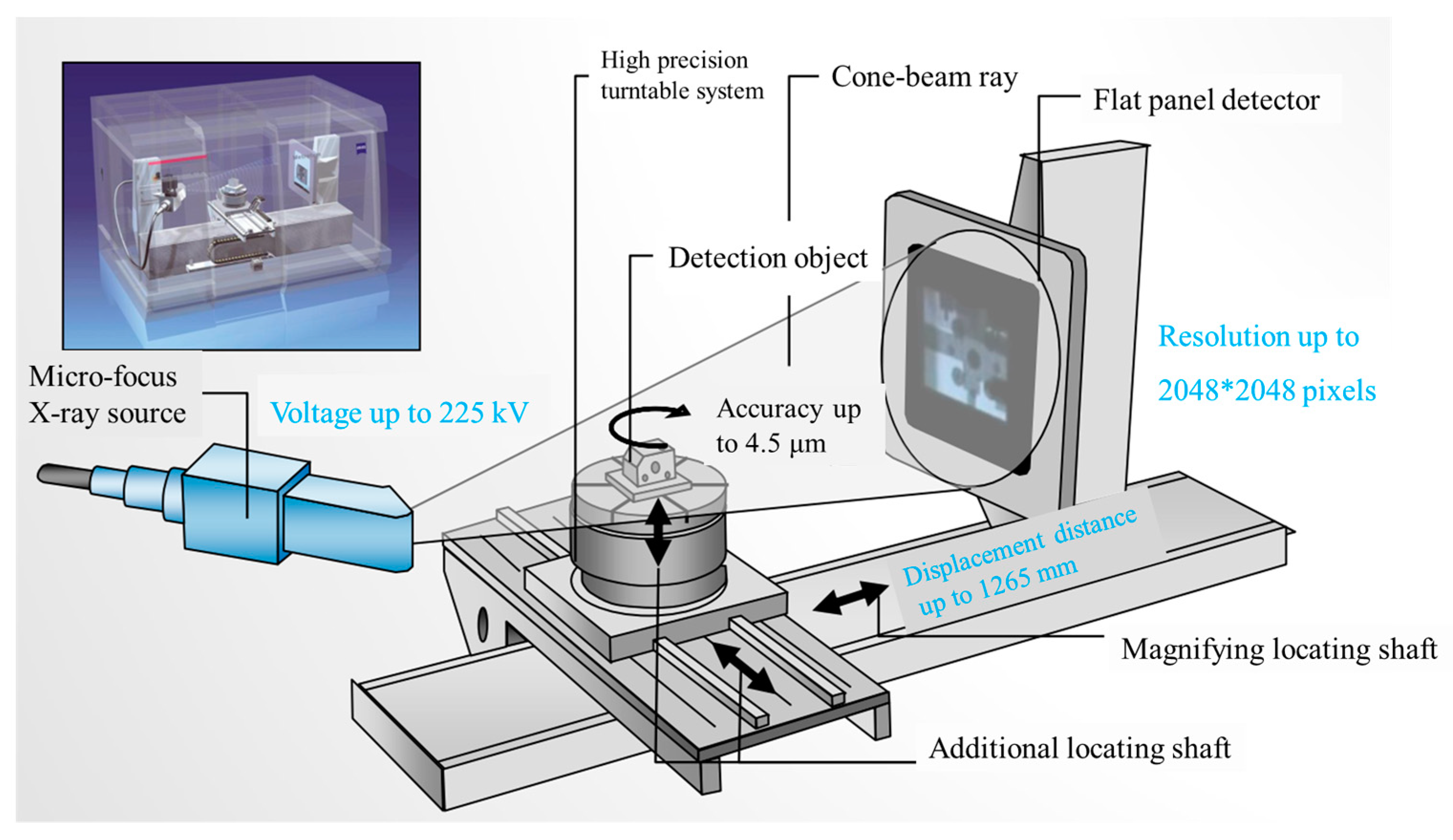
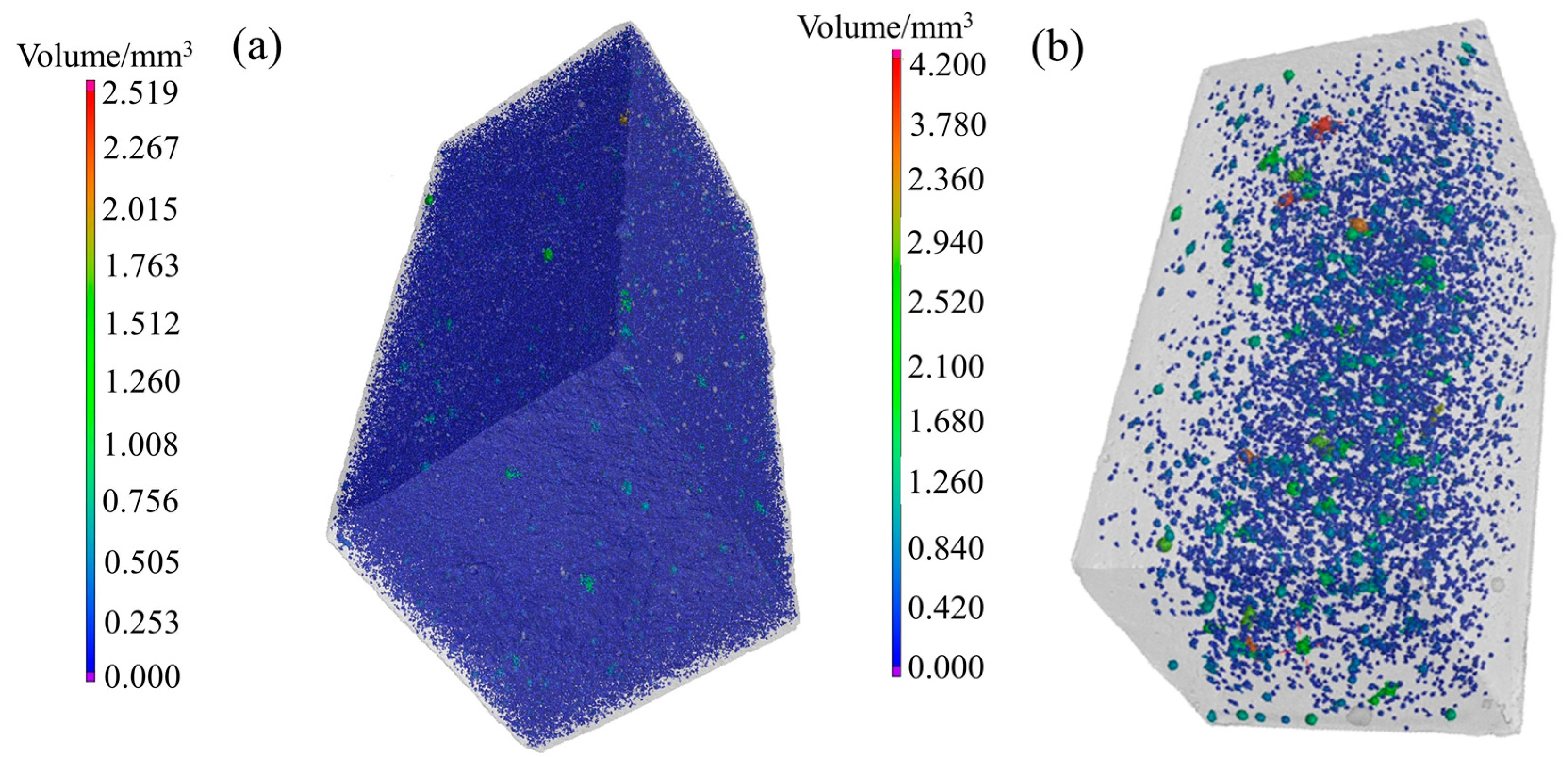
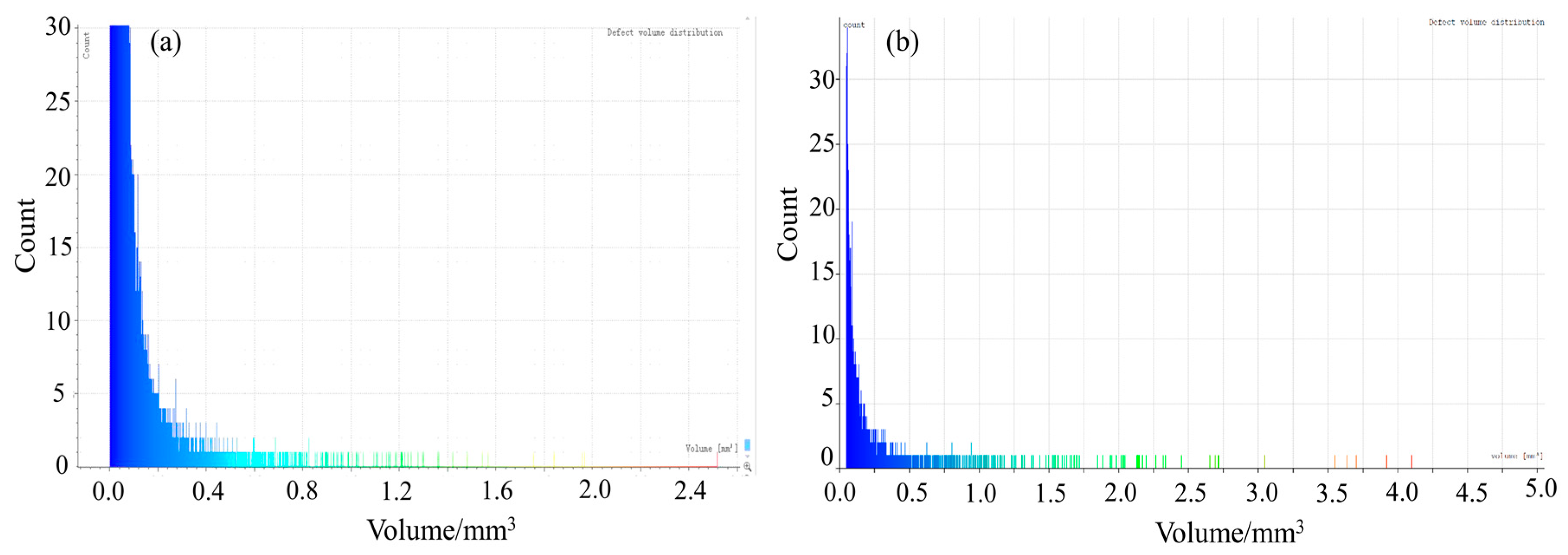
4.2. CT Scanning Analysis of Hardened Gypsum
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hammas, I.; Horchani-Naifer, K.; Férid, M. Solubility study and valorization of phosphogypsum salt solution. Int. J. Miner. Process. 2013, 123, 87–93. [Google Scholar] [CrossRef]
- Romero-Hermida, I.; Morales-Flórez, V.; Santos, A.; Villena, A.; Esquivias, L. Technological proposals for recycling industrial wastes for environmental applications. Minerals 2014, 4, 746–757. [Google Scholar] [CrossRef]
- Cuadri, A.A.; Navarro, F.J.; García-Morales, M.; Bolívar, J.P. Valorization of phosphogypsum waste as asphaltic bitumen modifier. J. Hazard. Mater. 2014, 279, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gao, H.; Shu, Z.; Wang, Y.; Yan, C. Utilization of waste phosphogypsum to prepare non-fired bricks by a novel Hydration–Recrystallization process. Constr. Build. Mater. 2012, 34, 114–119. [Google Scholar] [CrossRef]
- Tian, T.; Yan, Y.; Hu, Z.H.; Xu, Y.Y.; Chen, Y.P.; Shi, J. Utilization of original phosphogypsum for the preparation of foam concrete. Constr. Build. Mater. 2016, 115, 143–152. [Google Scholar] [CrossRef]
- Guan, Q.J.; Sun, W.; Hu, Y.H.; Yin, Z.G.; Zhang, C.H.; Guan, C.P.; Zhu, X.N.; Ahmed Khoso, S. Simultaneous control of particle size and morphology of α-CaSO4·½H2O with organic additives. J. Am. Ceram. Soc. 2019, 102, 2441–2449. [Google Scholar]
- Shen, Z.X.; Guan, B.H.; Fu, H.L.; Yang, L.C. Effect of potassium sodium tartrate and sodium citrate on the preparation of α-calcium sulfate hemihydrate from flue gas desulfurization gypsum in a concentrated electrolyte solution. J. Am. Ceram. Soc. 2009, 92, 2894–2899. [Google Scholar] [CrossRef]
- Duan, Z.Y.; Li, J.X.; Li, T.G.; Zheng, S.R.; Han, W.M.; Geng, Q.Y.; Guo, H.B. Influence of crystal modifier on the preparation of α-hemihydrate gypsum from phosphogypsum. Constr. Build. Mater. 2017, 133, 323–329. [Google Scholar] [CrossRef]
- Li, F.; Liu, J.L.; Yang, G.Y.; Pan, Z.Y.; Ni, X.; Xu, H.; Huang, Q. Effect of pH and succinic acid on the morphology of α-calcium sulfate hemihydrate synthesized by a salt solution method. J. Cryst. Growth 2013, 374, 31–36. [Google Scholar] [CrossRef]
- Guan, Q.J.; Tang, H.H.; Sun, W.; Hu, Y.H.; Yin, Z.G. Insight into influence of glycerol on preparing α-CaSO4·1/2H2O from flue gas desulfurization gypsum in glycerol–water solutions with succinic acid and NaCl. Ind. Eng. Chem. Res. 2017, 56, 9831–9838. [Google Scholar] [CrossRef]
- Guan, Q.J.; Sun, W.; Hu, Y.H.; Yin, Z.G.; Guan, C.P. Synthesis of α-CaSO4·0.5H2O from flue gas desulfurization gypsum regulated by C4H4O4Na2·6H2O and NaCl in glycerol-water solution. RSC Adv. 2017, 7, 27807–27815. [Google Scholar] [CrossRef]
- Mi, Y.; Chen, D.Y.; Wang, S.Z. Utilization of phosphogypsum for the preparation of α-calcium sulfate hemihydrate in chloride-free solution under atmospheric pressure. J. Chem. Technol. Biotechnol. 2018, 93, 2371–2379. [Google Scholar] [CrossRef]
- Shao, D.D.; Zhao, B.; Zhang, H.Q.; Wang, Z.G.; Shi, C.J.; Cao, J.L. Preparation of large-grained α-high strength gypsum with FGD gypsum. Cryst. Res. Technol. 2017, 52, 1700078. [Google Scholar] [CrossRef]
- Li, X.B.; Zhang, Q.; Ke, B.L.; Wang, X.C.; Li, L.J.; Li, X.H.; Mao, S. Insight into the effect of maleic acid on the preparation of α-hemihydrate gypsum from phosphogypsum in Na2SO4 solution. J. Cryst. Growth 2018, 493, 34–40. [Google Scholar] [CrossRef]
- Tan, H.; Dong, F. Morphological regulation of calcium sulfate hemihydrate from phosphogypsum. Mater. Werkst. 2017, 48, 1191–1196. [Google Scholar] [CrossRef]
- Li, X.B.; Zhang, Q.; Shen, Z.H.; Li, L.J.; Li, X.H.; Mao, S. L-aspartic acid: A crystal modifier for preparation of hemihydrate from phosphogypsum in CaCl2 solution. J. Cryst. Growth 2019, 511, 48–55. [Google Scholar] [CrossRef]
- Mao, J.W.; Jiang, G.M.; Chen, Q.S.; Guan, B.H. Influences of citric acid on the metastability of α-calcium sulfate hemihydrate in CaCl2 solution. Colloids Surf. A 2014, 443, 265–271. [Google Scholar] [CrossRef]
- Saha, A.; Lee, J.; Pancera, S.M.; Bräeu, M.F.; Kempter, A.; Tripathi, A. Bose, A. New insights into the transformation of calcium sulfate hemihydrate to gypsum using time-resolved cryogenic transmission electron microscopy. Langmuir 2012, 28, 11182–11187. [Google Scholar] [CrossRef]
- Yang, L.; Cao, J.X.; Li, C.Y. Enhancing the hydration reactivity of hemi-hydrate phosphogypsum through a morphology-controlled preparation technology. Chin. J. Chem. Eng. 2016, 24, 1298–1305. [Google Scholar] [CrossRef]
- Van Driessche, A.E.; Benning, L.G.; Rodriguez-Blanco, J.D.; Ossorio, M.; Bots, P.; Garcia-Ruiz, J.M. The role and implications of bassanite as a stable precursor phase to gypsum precipitation. Science 2012, 336, 69–72. [Google Scholar] [CrossRef]
- Ossorio, M.; Stawski, T.; Rodríguez-Blanco, J.; Sleutel, M.; García-Ruiz, J.; Benning, L.; Van Driessche, A. Physicochemical and additive controls on the multistep precipitation pathway of gypsum. Minerals 2017, 7, 140. [Google Scholar] [CrossRef]
- Liu, C.B.; Gao, J.M.; Tang, Y.B.; Zhao, Y.S. Early hydration and microstructure of gypsum plaster revealed by environment scanning electron microscope. Mater. Lett. 2019, 234, 49–52. [Google Scholar] [CrossRef]
- Zhao, H.; Hu, G.H.; Ye, G.B.; Ren, X.M.; Zhang, Q.C.; Jiang, T.; Zhao, H.; Hu, G.H.; Ye, G.B.; Ren, X.M. Effects of superplasticisers on hydration process, structure and properties of α -hemihydrate calcium sulfate. Adv. Cem. Res. 2017, 30, 1–8. [Google Scholar] [CrossRef]
- Eve, S.; Gomina, M.; Jernot, J.P.; Ozouf, J.C.; Orange, G. Microstructure characterization of polyamide fibre/latex-filled plaster composites. J. Eur. Ceram. Soc. 2007, 27, 3517–3525. [Google Scholar] [CrossRef]
- Gypsum Plasters–Determination of Water of Crystallization Content; GB/T 17669.2-1999; The State Bureau of Quality Technical Supervision: Beijing, China, 1999.
- Water Quality–Determination of Calcium–EDTA Titrimetric Method; The State Department of Environmental Conservation: Beijing, China, 1987; GB 7476-87.
- Water Quality–Determination of Sulfate–Gravimetric Method; The State Department of Environmental Conservation: Beijing, China, 1989; GB 11899-89.
- Yue, W.H.; Zhao, Q.N. Study of the thermodynamics during the process of hydration and hardening of the anhydrite. J. Wuhan Univ. Technol. 1988, 3, 283–289. [Google Scholar]
- Bellotto, M.; Artioli, G.; Dalconi, M.C.; Corso, R. On the preparation of concentrated gypsum slurry to reuse sulfate-process TiO2 byproduct stream. J. Clean. Prod. 2018, 195, 1468–1475. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, L.M. Effect of amino acid retarders on the hydration of flue gas desulfurization gypsum. New Build. Mater. 2017, 44, 21–23. [Google Scholar]
- Harouaka, K.; Kubicki, J.D.; Fantle, M.S. Effect of amino acids on the precipitation kinetics and Ca isotopic composition of gypsum. Geochim. Cosmochim. Acta 2017, 218, 343–364. [Google Scholar] [CrossRef]
- Ding, Y.; Fang, Y.C.; Fang, H.; Zhang, Q.C.; Zhang, F.J.; Oh, W. Study on the retarding mechanism and strength loss of gypsum from hydrolyzed wheat protein retarder. J. Korean Ceram. Soc. 2015, 52, 28–32. [Google Scholar] [CrossRef]
- Singh, N.B.; Vellmer, C.; Middendorf, B. Effect of carboxylic acids on the morphology, physical characteristics and hydration of α-hemihydrate plaster. Indian J. Eng. Mater. Sci. 2005, 12, 337–344. [Google Scholar]
- Badens, E.; Veesler, S.; Boistelle, R. Crystallization of gypsum from hemihydrate in presence of additives. J. Cryst. Growth 1999, 198, 704–709. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydration Time/h | 0.08 | 0.5 | 2 | 6 | 10 | 16 | 24 |
|---|---|---|---|---|---|---|---|
| zeta potential/mV | −1.49 | −1.51 | −1.64 | −1.54 | −1.40 | −0.92 | −1.22 |
| Crystal Modifier | CSO42−,max/(mg/L) | CSO42−,equ/(mg/L) | CDH,max/(mg/L) | CDH,equ/(mg/L) | Smax |
|---|---|---|---|---|---|
| None | 3184.14 | 1708.96 | 5707.50 | 3063.28 | 0.86 |
| L-Asp | 3128.16 | 2342.00 | 5604.62 | 4196.08 | 0.34 |
| Crystal Modifier | Defect Area/mm2 | Specific Surface Area of Defect /(cm2/g) | Pores Volume /mm3 | Porosity/% | Compressive Strength/MPa |
|---|---|---|---|---|---|
| None | 291,235.47 | 17.28 | 7141.95 | 6.07 | 9.7 |
| L-Asp | 20,861.44 | 0.99 | 1023.81 | 0.80 | 28.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhang, Q. Hydration Mechanism and Hardening Property of α-Hemihydrate Phosphogypsum. Minerals 2019, 9, 733. https://doi.org/10.3390/min9120733
Li X, Zhang Q. Hydration Mechanism and Hardening Property of α-Hemihydrate Phosphogypsum. Minerals. 2019; 9(12):733. https://doi.org/10.3390/min9120733
Chicago/Turabian StyleLi, Xianbo, and Qin Zhang. 2019. "Hydration Mechanism and Hardening Property of α-Hemihydrate Phosphogypsum" Minerals 9, no. 12: 733. https://doi.org/10.3390/min9120733
APA StyleLi, X., & Zhang, Q. (2019). Hydration Mechanism and Hardening Property of α-Hemihydrate Phosphogypsum. Minerals, 9(12), 733. https://doi.org/10.3390/min9120733