Three-D Mineralogical Mapping of the Kovdor Phoscorite–Carbonatite Complex, NW Russia: I. Forsterite


 ,
,  ,
,  ,
,
Abstract
1. Introduction
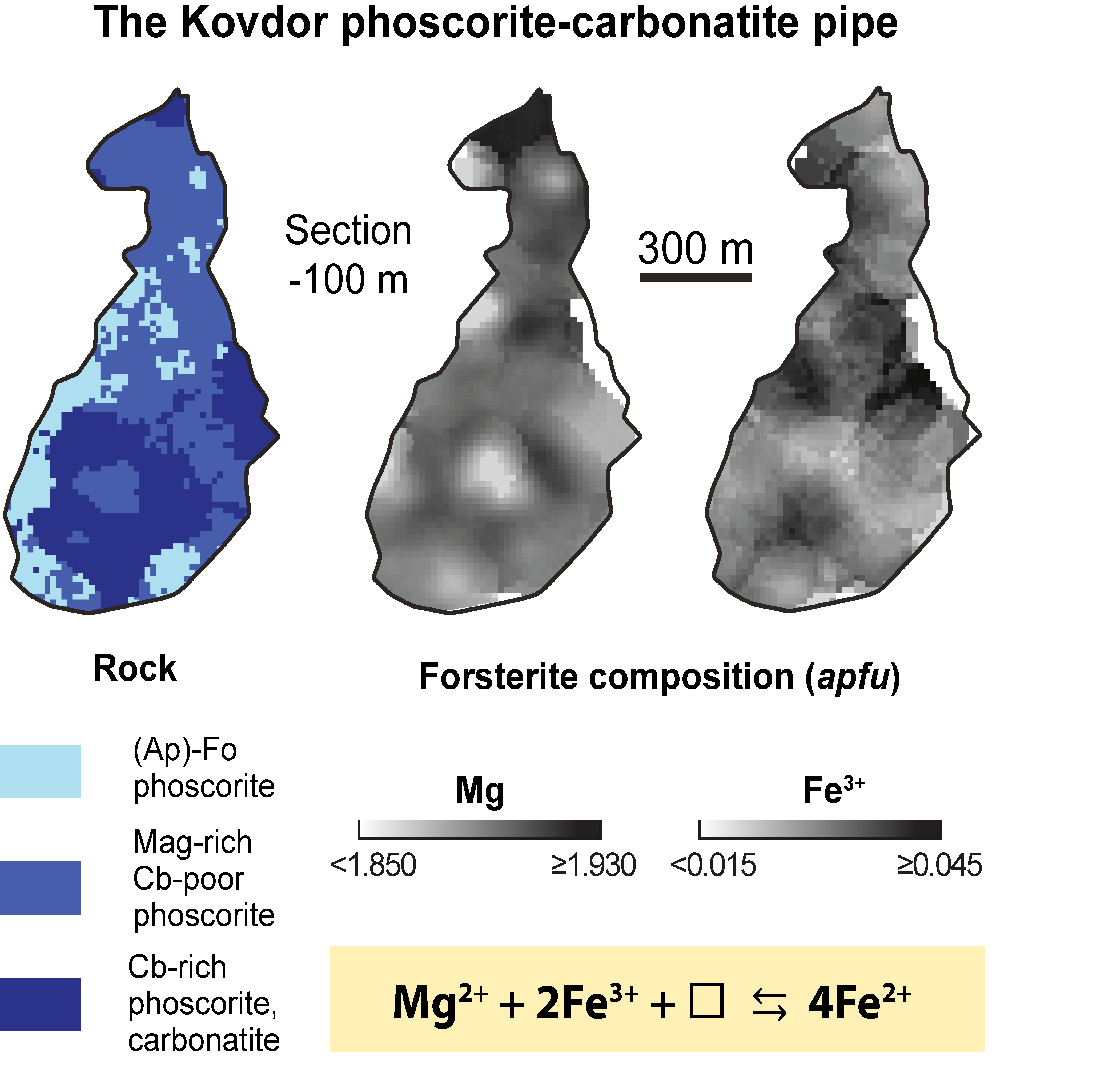
2. Geological Setting
3. Materials and Methods
4. Results
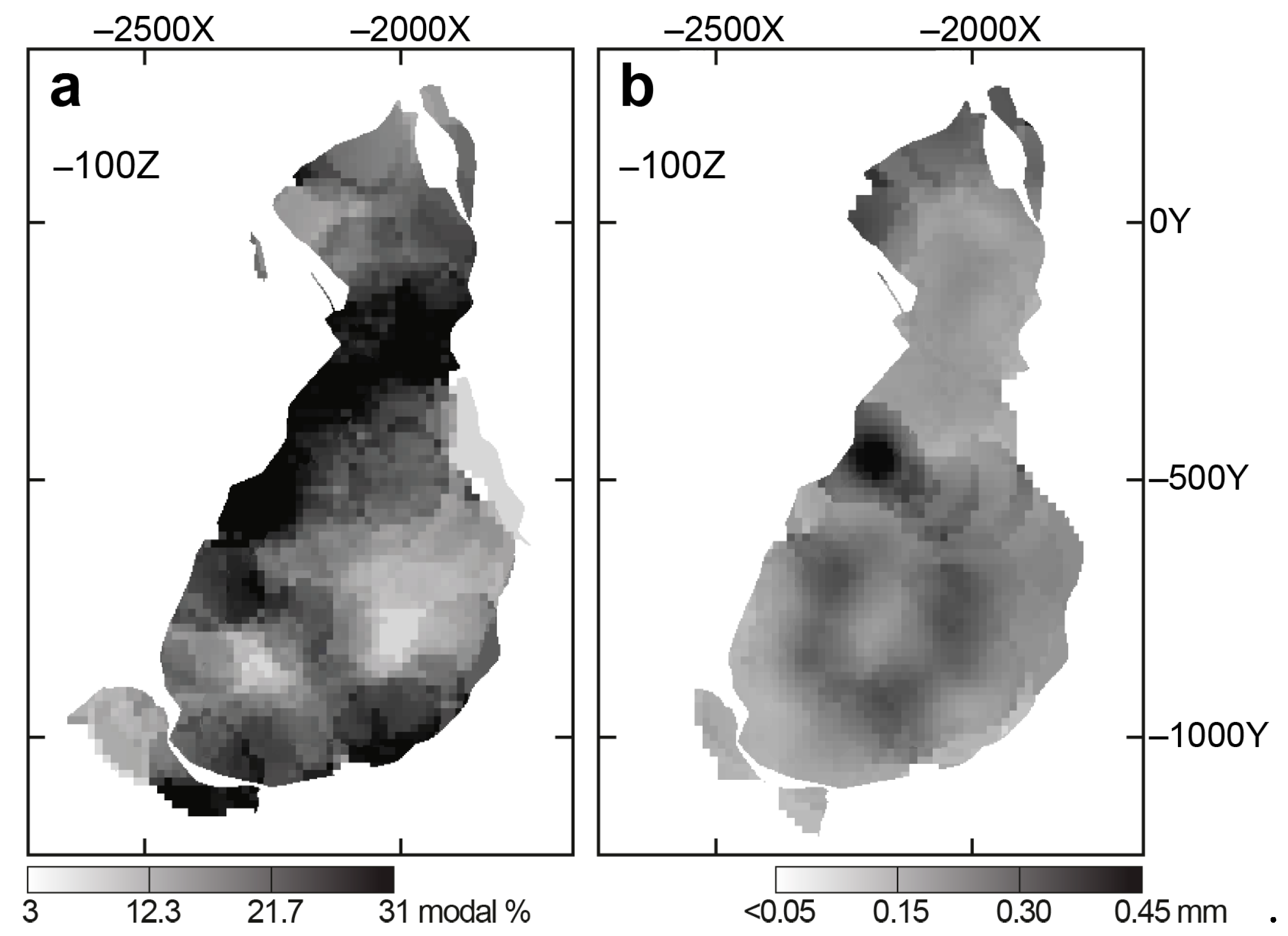
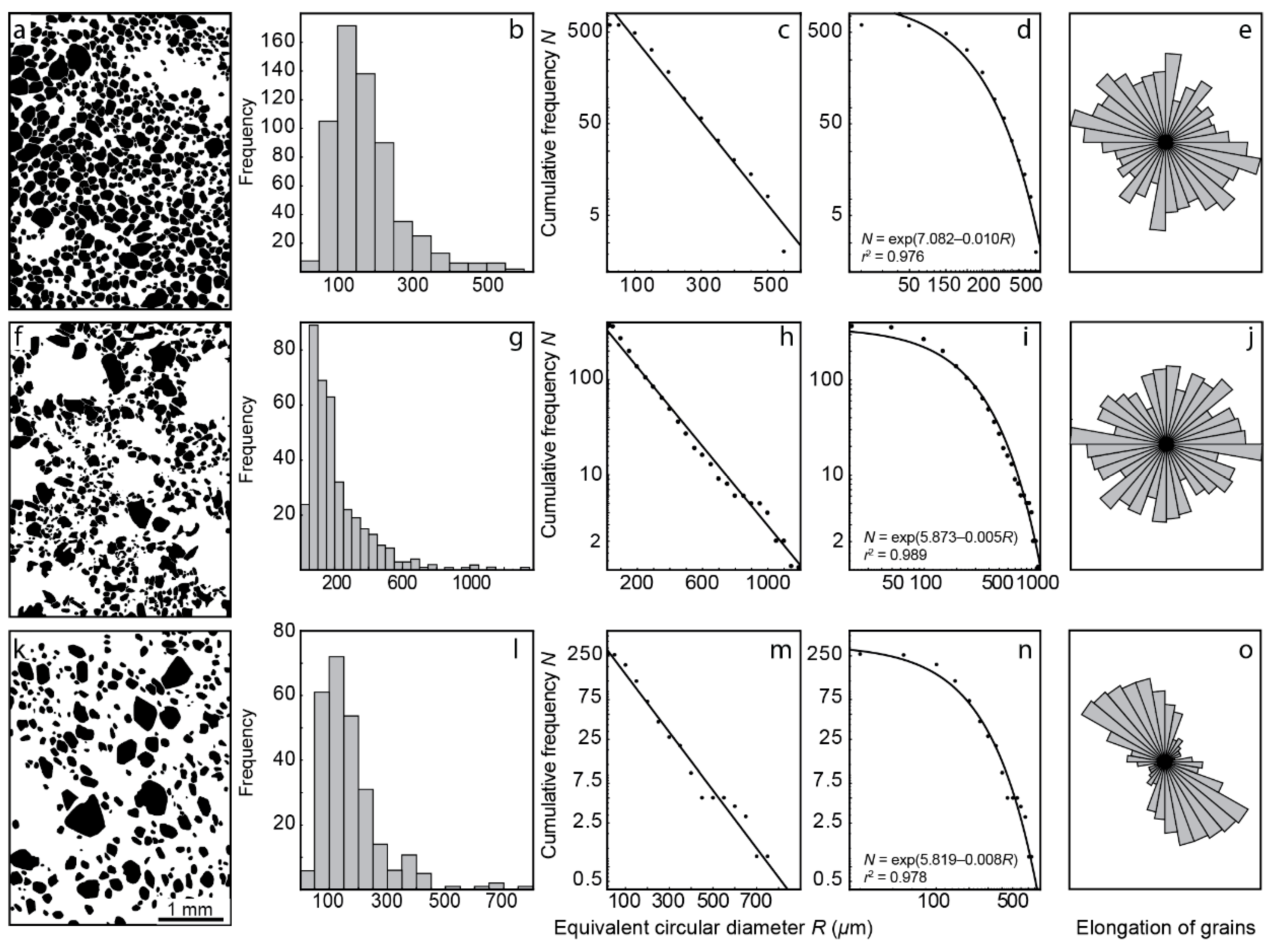
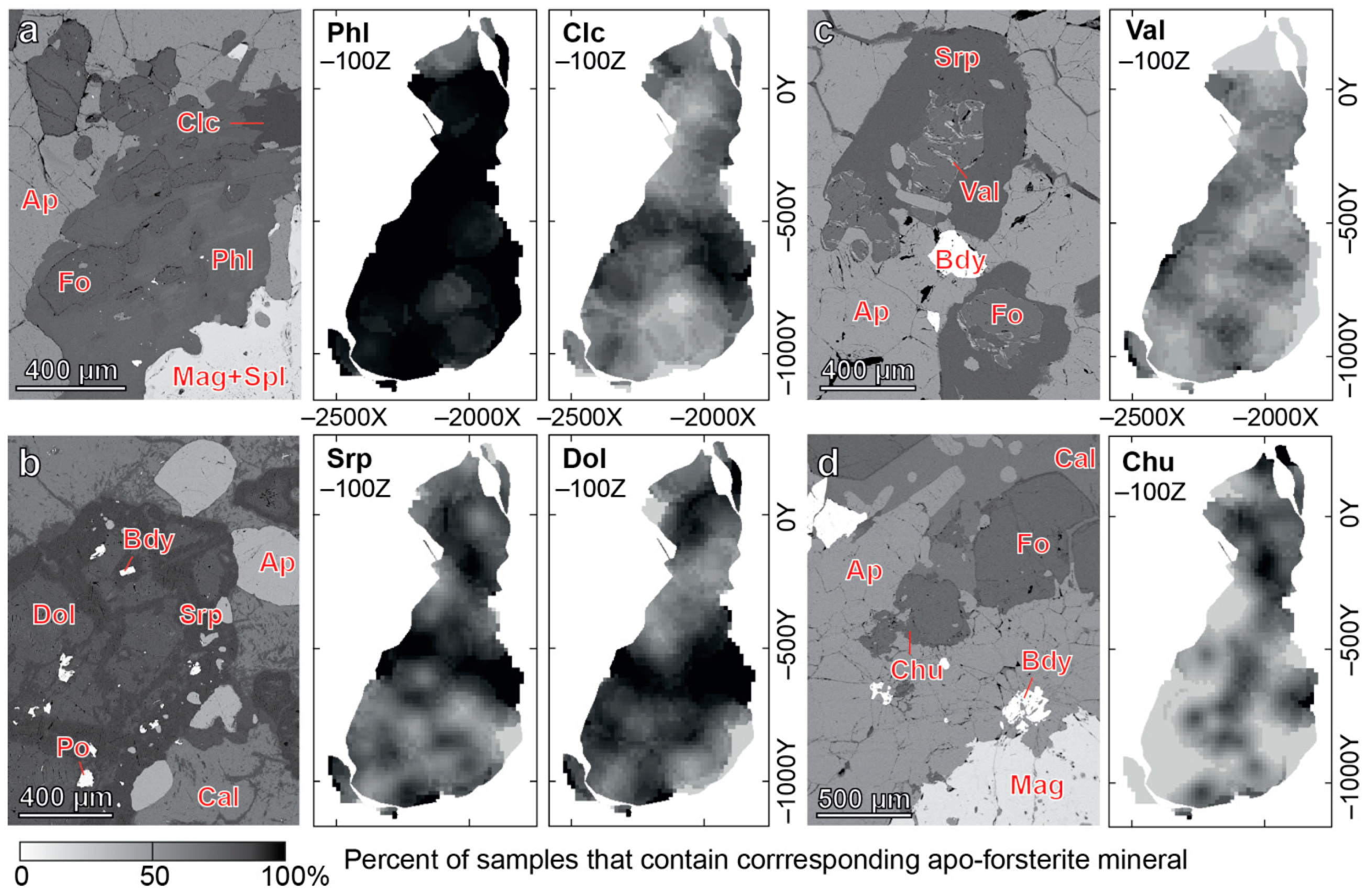
4.1. Content, Morphology and Grain Size of Forsterite
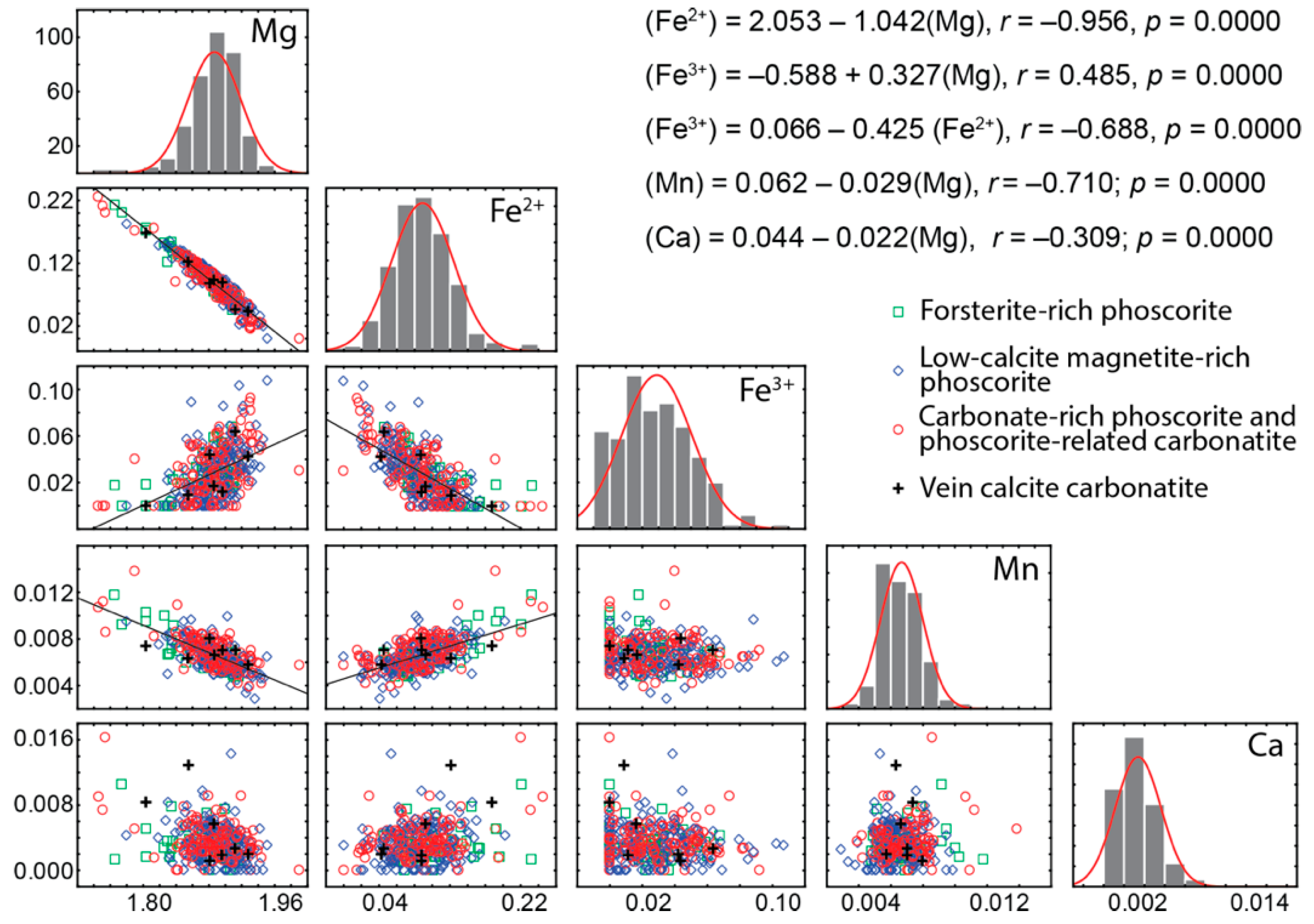
4.2. Chemical Composition
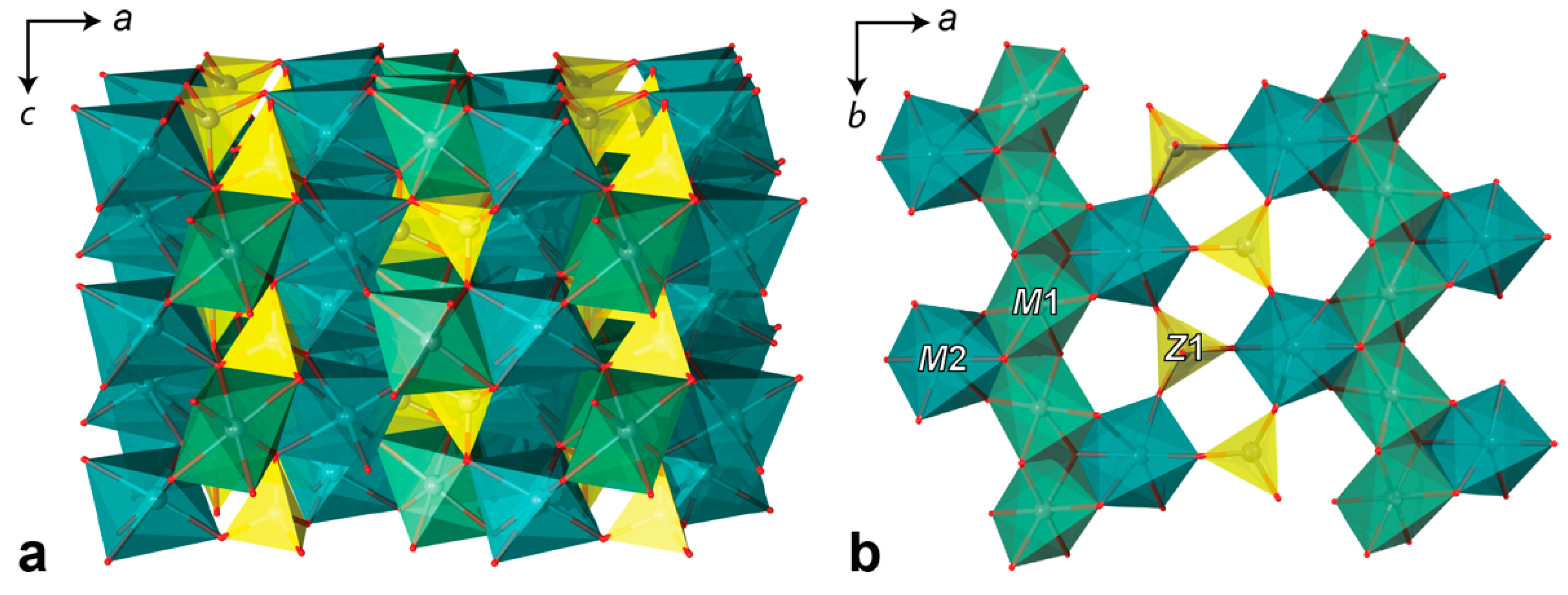
4.3. Single Crystal X-ray Diffraction
5. Discussion
6. Conclusions
- (1)
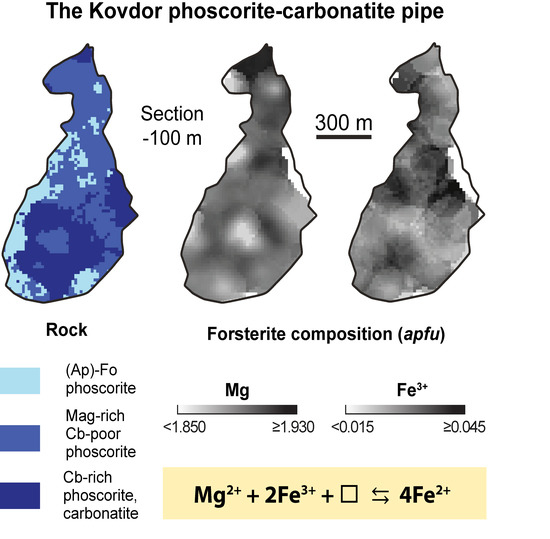
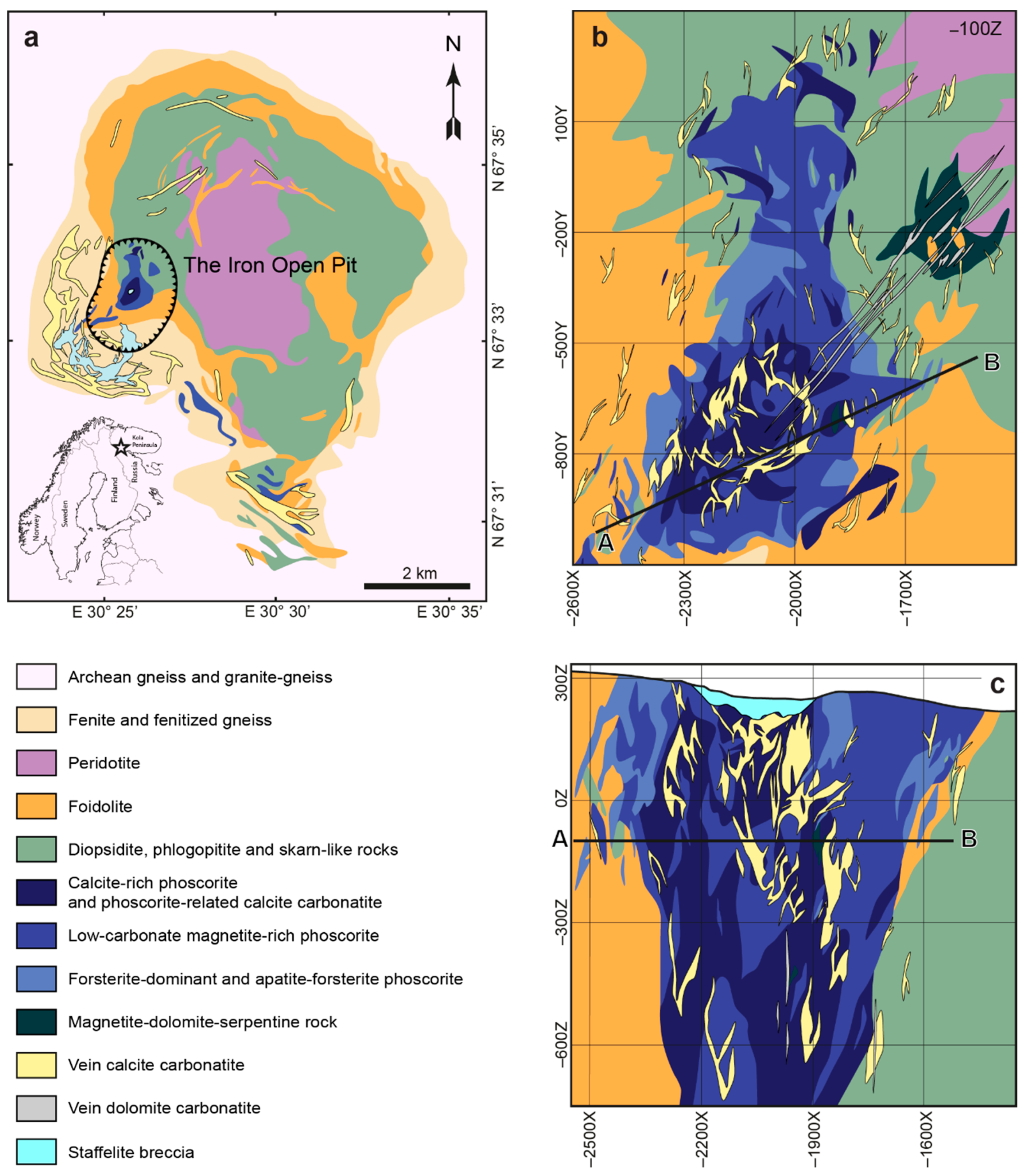
- Forsterite is the earliest mineral of both peridotite and phoscorite–carbonatite complexes, and its crystallization governed the further evolution of corresponding magmatic systems. Thus, crystallization of forsterite from the Ca-Fe-rich peridotite melt produces Si-Al-Na-K-rich residual melt-I corresponding to foidolite–melilitolite. In turn, consolidation of foidolite and melilitolite produced Fe-Ca-C-P-F-rich residual melt-II that emplaced in silicate rocks as the phoscorite–carbonatite pipe. Phoscorite crystallization started from forsterite, which launched destruction of silicate-carbonate-ferriphosphate subnetworks of the melt followed by precipitation of apatite and magnetite from the pipe wall to its axis with formation of carbonatite melt-III in the pipe axial zone;
- (2)
- Growth of forsterite grains from phoscorite–carbonatite melt was diffusion-limited, which causes constant growth rate of each grain and exponential distribution of grain size;
- (3)
- Chemical composition of forsterite in phoscorite–carbonatite pipe is determined by two schemas of isomorphism: Mg2+ + 2Fe3+ ⟷ 4(Fe, Mn)2+ and Mg2+ + 2Fe3+ ⟷ 2(Fe, Mn, Ni)2+ + Ti4+. Marginal forsterite-rich phoscorite consists of Fe2+-Mn-Ni-Ti-rich forsterite similar to olivine from peridotite, intermediate low-carbonate magnetie-rich phoscorite includes Mg-Fe3+-rich forsterite, and axial carbonate-rich phoscorite and carbonatites contain Fe2+-Mn-rich forsterite;
- (4)
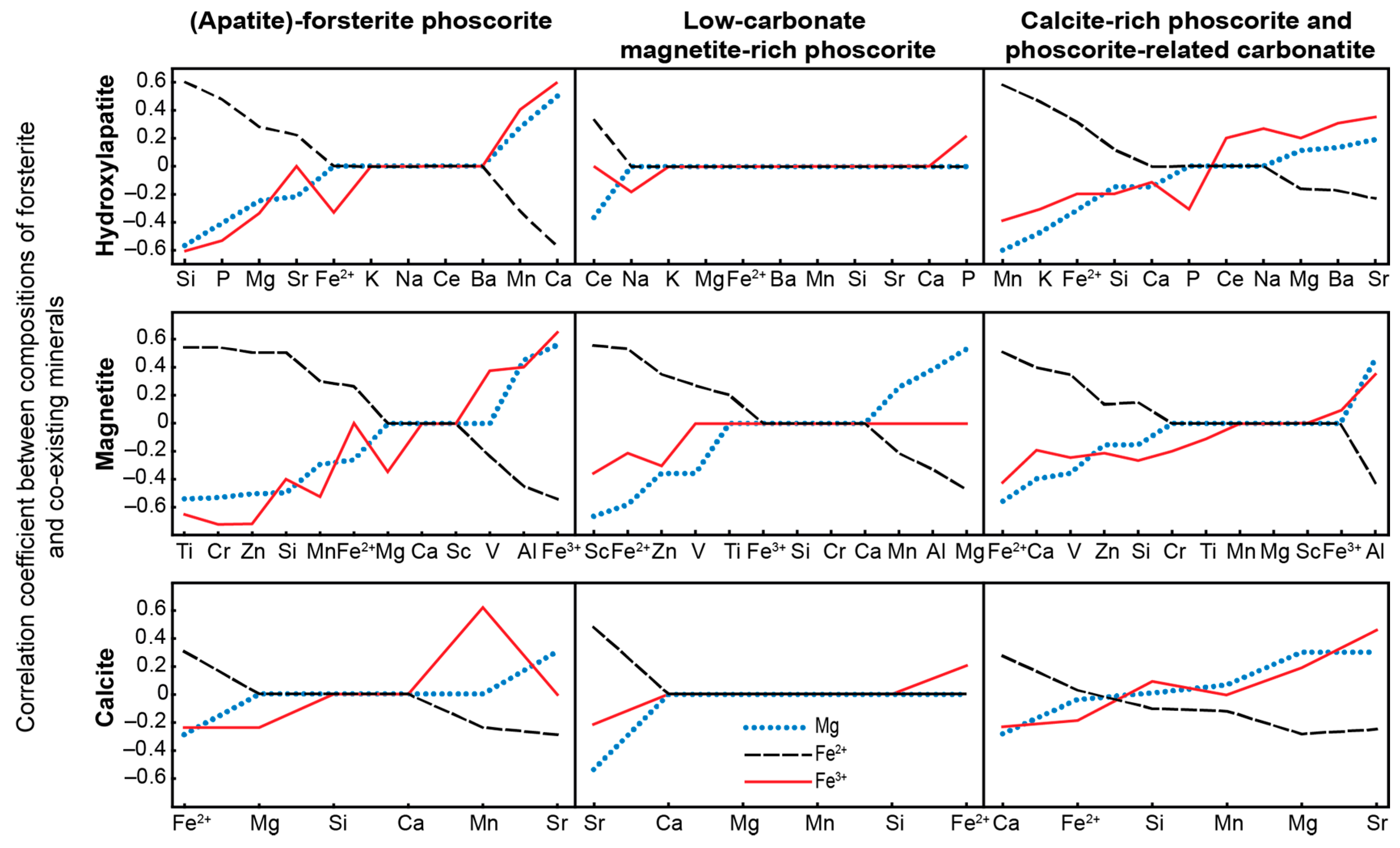
- Trivalent iron incorporates into forsterite by scheme (3Fe2+)oct → 2Fe3+ + (□)oct that reflects redox conditions of the rock formation causing significant agreement between compositions of apatite, magnetite, calcite and forsterite;
- (5)
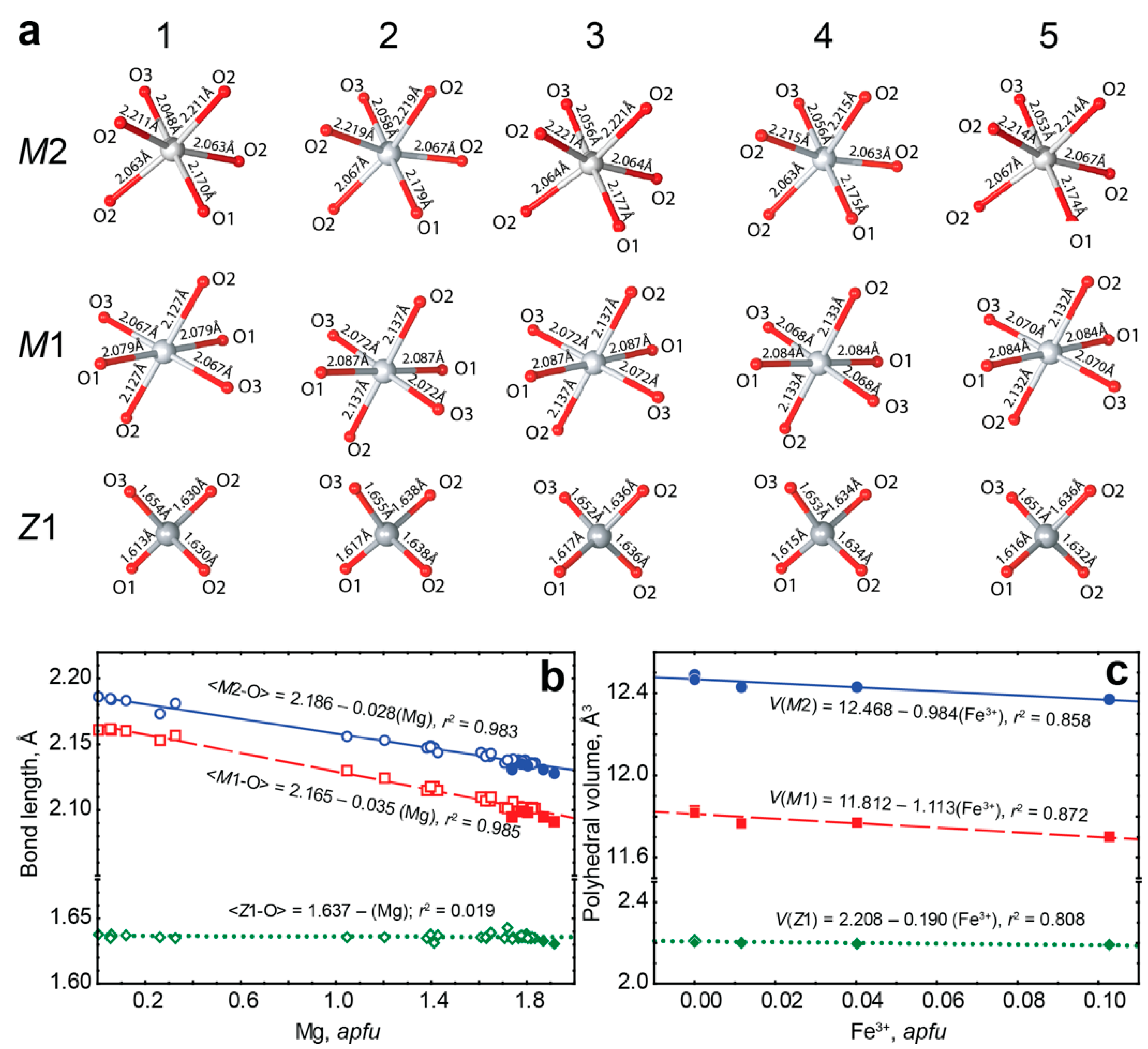
- Incorporation of trivalent iron at the octahedral M1 and M2 sites decreases the volume of these polyhedra, while volume of tetrahedral set does not change. Thus, the assumed substitution (4Fe2+)oct + (4Si4+)tet → (4Fe3+)oct + (3Si4+ + □)tet proposed by D. E. Janney and J. F. Banfield [49] was not confirmed. Our data show that laihunite-like isomorphism is more common in forsterite than it was considered to be.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Igneous Rocks. A Classification and Glossary of Terms. Recommendations of the International Union of Geological Sciences Subcommission on the Systematics of Igneous Rocks; Le Maitre, R.W., Ed.; Cambridge University Press: New York, NY, USA, 2002; ISBN 9780521662154. [Google Scholar]
- Jaireth, S.; Hoatson, D.M.; Miezitis, Y. Geological setting and resources of the major rare-earth-element deposits in Australia. Ore Geol. Rev. 2014, 62, 72–128. [Google Scholar] [CrossRef]
- Lazareva, E.V.; Zhmodik, S.M.; Dobretsov, N.L.; Tolstov, A.V.; Shcherbov, B.L.; Karmanov, N.S.; Gerasimov, E.Y.; Bryanskaya, A.V. Main minerals of abnormally high-grade ores of the Tomtor deposit (Arctic Siberia). Russ. Geol. Geophys. 2015, 56, 844–873. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Ling, M.-X.; Williams, I.S.; Yang, X.-Y.; Wang, C.Y.; Sun, W. The formation of the giant Bayan Obo REE-Nb-Fe deposit, North China, Mesoproterozoic carbonatite and overprinted Paleozoic dolomitization. Ore Geol. Rev. 2018, 92, 73–83. [Google Scholar] [CrossRef]
- Mackay, D.A.R.; Simandl, G.J. Geology, market and supply chain of niobium and tantalum—A review. Miner. Depos. 2014, 49, 1025–1047. [Google Scholar] [CrossRef]
- Mitchell, R.H. Primary and secondary niobium mineral deposits associated with carbonatites. Ore Geol. Rev. 2015, 64, 626–641. [Google Scholar] [CrossRef]
- Smith, M.P.; Moore, K.; Kavecsánszki, D.; Finch, A.A.; Kynicky, J.; Wall, F. From mantle to critical zone: A review of large and giant sized deposits of the rare earth elements. Geosci. Front. 2016. [Google Scholar] [CrossRef]
- Wall, F.; Zaitsev, A.N. (Eds.) Phoscorites and Carbonatites from Mantle to Mine: The Key Example of the Kola Alkaline Province; Mineralogical Society: London, UK, 2004. [Google Scholar]
- Ivanyuk, G.Y.; Yakovenchuk, V.N.; Pakhomovsky, Y.A. Kovdor; Laplandia Minerals: Apatity, Russia, 2002; ISBN 5900395413. [Google Scholar]
- Mikhailova, J.A.; Kalashnikov, A.O.; Sokharev, V.A.; Pakhomovsky, Y.A.; Konopleva, N.G.; Yakovenchuk, V.N.; Bazai, A.V.; Goryainov, P.M.; Ivanyuk, G.Y. 3D mineralogical mapping of the Kovdor phoscorite–carbonatite complex (Russia). Miner. Depos. 2016, 51, 131–149. [Google Scholar] [CrossRef]
- Ivanyuk, G.Y.; Kalashnikov, A.O.; Pakhomovsky, Y.A.; Mikhailova, J.A.; Yakovenchuk, V.N.; Konopleva, N.G.; Sokharev, V.A.; Bazai, A.V.; Goryainov, P.M. Economic minerals of the Kovdor baddeleyite-apatite-magnetite deposit, Russia: Mineralogy, spatial distribution and ore processing optimization. Ore Geol. Rev. 2016, 77, 279–311. [Google Scholar] [CrossRef]
- Ivanyuk, G.Y.; Kalashnikov, A.O.; Pakhomovsky, Y.A.; Bazai, A.V.; Goryainov, P.M.; Mikhailova, J.A.; Yakovenchuk, V.N.; Konopleva, N.G. Subsolidus Evolution of the Magnetite-Spinel-Ulvöspinel Solid Solutions in the Kovdor phoscorite–carbonatite Complex, NW Russia. Minerals 2017, 7, 215. [Google Scholar] [CrossRef]
- Kalashnikov, A.O.; Yakovenchuk, V.N.; Pakhomovsky, Y.A.; Bazai, A.V.; Sokharev, V.A.; Konopleva, N.G.; Mikhailova, J.A.; Goryainov, P.M.; Ivanyuk, G.Y. Scandium of the Kovdor baddeleyite–apatite–magnetite deposit (Murmansk Region, Russia): Mineralogy, spatial distribution, and potential resource. Ore Geol. Rev. 2016, 72, 532–537. [Google Scholar] [CrossRef]
- Bell, K.; Kjarsgaard, B.A.; Simonetti, A. Carbonatites—Into the Twenty-First Century. J. Petrol. 1998, 39, 1839–1845. [Google Scholar] [CrossRef]
- Gittins, J.; Harmer, R.E.; Barker, D.S. The bimodal composition of carbonatites: Reality or misconception? Lithos 2005, 85, 129–139. [Google Scholar] [CrossRef]
- Mitchell, R.H. Carbonatites and carbonatites and carbonatites. Can. Mineral. 2005, 43, 2049–2068. [Google Scholar] [CrossRef]
- Woolley, A.R.; Kjarsgaard, B.A. Paragenetic types of carbonatite as indicated by the diversity and relative abundances of associated silicate rocks: Evidence from a global database. Can. Mineral. 2008, 46, 741–752. [Google Scholar] [CrossRef]
- Woolley, A.R.; Bailey, D.K. The crucial role of lithospheric structure in the generation and release of carbonatites: Geological evidence. Mineral. Mag. 2012, 76, 259–270. [Google Scholar] [CrossRef]
- Jones, A.P.; Genge, M.; Carmody, L. Carbonate Melts and Carbonatites. Rev. Mineral. Geochem. 2013, 75, 289–322. [Google Scholar] [CrossRef]
- Russell, H.D.; Hiemstra, S.A.; Groeneveld, D. The mineralogy and petrology of the Carbonatite at Loolekop, Eastern Transvaal. S. Afr. J. Geol. 1954, 57, 197–208. [Google Scholar]
- Kukharenko, A.A.; Orlova, M.P.; Bulakh, A.G.; Bagdasarov, E.A.; Rimskaya-Korsakova, O.M.; Nefedov, E.I.; Ilyinsky, G.A.; Sergeev, A.S.; Abakumova, N.B. Caledonian Complex of Ultrabasic, Alkaline Rocks and Carbonatites of Kola Peninsula and Northern Karelia (Geology, Petrology, Mineralogy and Geochemistry) (in Russian); Nedra: Moscow, Russia, 1965. [Google Scholar]
- Yegorov, L.S. Phoscorites of the Maymecha-Kotuy ijolite-carbonatite association. Int. Geol. Rev. 1993, 35, 346–358. [Google Scholar] [CrossRef]
- Rimskaya-Korsakova, O.M. On Question about Genesis of the Kovdor Iron-Ore Deposit. In Problems of Magmatism and Metamorphism (in Russian); Leningrad State University Publishing: Leningrad, Russia, 1963; pp. 125–142. [Google Scholar]
- Kalashnikov, A.O.; Ivanyuk, G.Y.; Mikhailova, J.A.; Sokharev, V.A. Approach of automatic 3D geological mapping: The case of the Kovdor phoscorite–carbonatite complex, NW Russia. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ternovoy, V.I. Carbonatite Massifs and Their Mineral Resources (in Russian); Leningrad State University: Leningrad, Russia, 1977. [Google Scholar]
- Afanasyev, B. Mineral Resources of Alkaline-Ultrabasic Massifs of the Kola Peninsula (in Russian); Roza Vetrov Publishing: Saint-Petersburg, Russia, 2011. [Google Scholar]
- Dunaev, V.A. Structure of the Kovdor deposit (in Russian). Geol. Ore Depos. 1982, 3, 28–36. [Google Scholar]
- Kapustin, Y.L. Mineralogy of Carbonatites; Amerind Publishing: New Delhi, India, 1980. [Google Scholar]
- Krasnova, N.I.; Kopylova, L.N. The Geologic Basis for Mineral-Technological Mapping at the Kovdor Ore Deposit. Int. Geol. Rev. 1988, 30, 307–319. [Google Scholar] [CrossRef]
- Krasnova, N.I.; Petrov, T.G.; Balaganskaya, E.G.; García, D.; Moutte, J.; Zaitsev, A.N.; Wall, F. Introduction to phoscorites: Occurrence, composition, nomenclature and petrogenesis. In Phoscorites and Carbonatites from Mantle to Mine: The Key Example of the Kola Alkaline Province; Zaitsev, A.N., Wall, F., Eds.; Mineralogical Society: London, UK, 2004; pp. 43–72. ISBN 0-903056-22-4. [Google Scholar]
- Bayanova, T.B.; Kirnarsky, Y.M.; Levkovich, N.V. U-Pb dating of baddeleyite from Kovdor massif (in Russian). Dokl. Earth Sci. 1997, 356, 509–511. [Google Scholar]
- Amelin, Y.; Zaitsev, A.N. Precise geochronology of phoscorites and carbonatites: The critical role of U-series disequilibrium in age interpretations. Geochim. Cosmochim. Acta 2002, 66, 2399–2419. [Google Scholar] [CrossRef]
- Rodionov, N.V.; Belyatsky, B.V.; Antonov, A.V.; Kapitonov, I.N.; Sergeev, S.A. Comparative in-situ U–Th–Pb geochronology and trace element composition of baddeleyite and low-U zircon from carbonatites of the Palaeozoic Kovdor alkaline–ultramafic complex, Kola Peninsula, Russia. Gondwana Res. 2012, 21, 728–744. [Google Scholar] [CrossRef]
- Rimskaya-Korsakova, O.M.; Krasnova, N.I. Geology of Deposits of the Kovdor Massif (in Russian); St. Petersburg University Press: Saint Petersburg, Russia, 2002. [Google Scholar]
- Shats, L.; Sorokina, I.; Kalinkin, M.; Kornyushin, A. Report on Geophysical Works Made by the Kovdor Geological Party in the Area of Kovdor in 1966 (in Russian); Archives of the Natural Reserves Department of the Murmansk region: Apatity, Russia, 1967. [Google Scholar]
- ImageJ, Open Source Image Processing Software. Available online: http://imagej.net/ (accessed on 19 June 2018).
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Agilent CrysAlis PRO. 2014. Available online: https://www.rigaku.com/en/products/smc/crysalis (accessed on 19 June 2018).
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. Diamond–Crystal and Molecular Structure Visualization; Crystal Impact GbR: Bonn, Germany, 2012. [Google Scholar]
- Dolivo-Dobrovolsky, D.D. MINAL, Free Software. Available online: http://www.dimadd.ru (accessed on 8 July 2013).
- StatSoft Inc, Statistica 8. Available online: www.statsoft.ru (accessed on 19 June 2018).
- TableCurve 2D. Available online: www.sigmaplot.co.uk/products/tablecurve2d/tablecurve2d.php (accessed on 19 June 2018).
- Micromine Pty Ltd. Micromine 16.1. Available online: https://www.micromine.com/ (accessed on 19 June 2018).
- Veksler, I.V.; Nielsen, T.F.D.; Sokolov, S.V. Mineralogy of Crystallized Melt Inclusions from Gardiner and Kovdor Ultramafic Alkaline Complexes: Implications for Carbonatite Genesis. J. Petrol. 1998, 39, 2015–2031. [Google Scholar] [CrossRef]
- Tarasenko, Y.; Litsarev, M.A.; Tretyakova, L.I.; Vokhmentsev, A.Y. Chrysolite of the Kovdor phlogopite deposit (in Russian). Izv. AN SSSR Seriya Geol. 1986, 9, 67–80. [Google Scholar]
- Zeira, S.; Hafner, S.S. The location of Fe3+ ions in forsterite (Mg2SiO4). Earth Planet. Sci. Lett. 1974, 21, 201–208. [Google Scholar] [CrossRef]
- Janney, D.E.; Banfield, J.F. Distribution of cations and vacancies and the structure of defects in oxidized intermediate olivine by atomic-resolution TEM and image simulation. Am. Mineral. 1998, 83, 799–810. [Google Scholar] [CrossRef]
- Bragg, W.L.; Brown, G.B. XXX. Die Struktur des Olivins. Z. Krist. Cryst. Mater. 1926, 63, 538–556. [Google Scholar] [CrossRef]
- Brown, G.E. Crystal Chemistry of the Olivines; Virginia Polytechnic Institute and State University: Blacksburg, VA, USA, 1970. [Google Scholar]
- Huggins, F.E. Cation order in olivines: Evidence from vibrational spectra. Chem. Geol. 1973, 11, 99–108. [Google Scholar] [CrossRef]
- Nover, G.; Will, G. Structure refinements of seven natural olivine crystals and the influence of the oxygen partial pressure on the cation distribution. Z. Krist. Cryst. Mater. 1981, 155. [Google Scholar] [CrossRef]
- Francis, C.A. New data on the forsterite-tephroite series. Am. Mineral. 1985, 70, 568–575. [Google Scholar]
- Seifert, F.A.; Virgo, D. Kinetics of the Fe2+-Mg, Order-Disorder Reaction in Anthophyllites: Quantitative Cooling Rates. Science 1975, 188, 1107–1109. [Google Scholar] [CrossRef] [PubMed]
- Seifert, F.; Virgo, D. Temperature dependence of intracrystalline Fe2+-Mg distribution in a natural anthophyllite. In Carnegie Institute of Washington Year Book; Carnegie Institute of Washington: Washington, DC, USA, 1974; pp. 405–411. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Riekel, C.; Weiss, A. Cation-Ordering in Synthetic Mg2−xFexSiO4-Olivines. Z. Naturforsch. B 1978, 33. [Google Scholar] [CrossRef]
- Redfern, S.A.T.; Henderson, C.M.B.; Knight, K.S.; Wood, B.J. High-temperature order-disorder in (Fe0.5Mn0.5)2SiO4 and (Mg0.5Mn0.5)2SiO4 olivines: An in situ neutron diffraction study. Eur. J. Mineral. 1997, 9, 287–300. [Google Scholar] [CrossRef]
- Shen, B.; Tamada, O.; Kitamura, M.; Morimoto, N. Superstructure of laihunite-3M (□0.40Fe2+0.80Fe3+0.80SiO4). Am. Mineral. 1986, 71, 1455–1460. [Google Scholar]
- Brown, G.E.; Prewitt, C.T. High-temperature crystal chemistry of hortonolite. Am. Mineral. 1973, 58, 577–587. [Google Scholar]
- Princivalle, F.; Secco, L. Crystal structure refinement of 13 olivines in the forsterite-fayalite series from volcanic rocks and ultramafic nodules. TMPM 1985, 34, 105–115. [Google Scholar] [CrossRef]
- Motoyama, T.; Matsumoto, T. The crystal structures and the cation distributions of Mg and Fe of natural olivines. Miner. J. 1989, 14, 338–350. [Google Scholar] [CrossRef]
- Princivalle, F. Influence of temperature and composition on Mg-Fe2+ intracrystalline distribution in olivines. Mineral. Petrol. 1990, 43, 121–129. [Google Scholar] [CrossRef]
- Heinemann, R.; Kroll, H.; Kirfel, A.; Barbier, B. Order and anti-order in olivine III: Variation of the cation distribution in the Fe,Mg olivine solid solution series with temperature and composition. Eur. J. Mineral. 2007, 19, 15–27. [Google Scholar] [CrossRef]
- Kroll, H.; Kirfel, A.; Heinemann, R.; Barbier, B. Volume thermal expansion and related thermophysical parameters in the Mg, Fe olivine solid-solution series. Eur. J. Mineral. 2012, 24, 935–956. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, F.; Yang, J.; Gao, Y. Exsolutions of Diopside and Magnetite in Olivine from Mantle Dunite, Luobusa Ophiolite, Tibet, China. Acta Geol. Sin. Engl. Ed. 2010, 82, 377–384. [Google Scholar] [CrossRef]
- Markl, G.; Marks, M.A.W.; Wirth, R. The influence of T, aSiO2, and fO2 on exsolution textures in Fe-Mg olivine: An example from augite syenites of the Ilimaussaq Intrusion, South Greenland. Am. Mineral. 2001, 86, 36–46. [Google Scholar] [CrossRef]
- Libourel, G. Systematics of calcium partitioning between olivine and silicate melt: Implications for melt structure and calcium content of magmatic olivines. Contrib. Mineral. Petrol. 1999, 136, 63–80. [Google Scholar] [CrossRef]
- Carmichael, I.S.E.; Nicholls, J. Iron-titanium oxides and oxygen fugacities in volcanic rocks. J. Geophys. Res. 1967, 72, 4665–4687. [Google Scholar] [CrossRef]
- Mysen, B.O.; Richet, P. Silicate Glasses and Melts. Properties and Structure; Elsevier: New York, NY, USA, 2005; ISBN 0-444-52011-2. [Google Scholar]
- Moseley, D. Symplectic exsolution in olivine. Am. Mineral. 1984, 69, 139–153. [Google Scholar]
- Wyllie, P.J.; Baker, M.B.; White, B.S. Experimental boundaries for the origin and evolution of carbonatites. Lithos 1990, 26, 3–19. [Google Scholar] [CrossRef]
- Veksler, I.V.; Petibon, C.; Jenner, G.A.; Dorfman, A.M.; Dingwell, D.B. Trace Element Partitioning in Immiscible Silicate-Carbonate Liquid Systems: An Initial Experimental Study Using a Centrifuge Autoclave. J. Petrol. 1998, 39, 2095–2104. [Google Scholar] [CrossRef]
- Wyllie, P.J.; Tuttle, O.F. The System CaO-CO2-H2O and the Origin of Carbonatites. J. Petrol. 1960, 1, 1–46. [Google Scholar] [CrossRef]
- Koster van Groos, A.F.; Wyllie, P.J. Liquid immiscibility in the system Na2O-Al2O3-SiO2-CO2 at pressures to 1 kilobar. Am. J. Sci. 1966, 264, 234–255. [Google Scholar] [CrossRef]
- Koster van Groos, A.F.; Wyllie, P.J. Liquid immiscibility in the join NaAlSi3O8-Na2CO3-H2O and its bearing on the genesis of carbonatites. Am. J. Sci. 1968, 266, 932–967. [Google Scholar] [CrossRef]
- Freestone, I.C.; Hamilton, D.L. The role of liquid immiscibility in the genesis of carbonatites? An experimental study. Contrib. Mineral. Petrol. 1980, 73, 105–117. [Google Scholar] [CrossRef]
- Kjarsgaard, B.A.; Hamilton, D.L. Liquid immiscibility and the origin of alkali-poor carbonatites. Mineral. Mag. 1988, 52, 43–55. [Google Scholar] [CrossRef]
- Kjarsgaard, B.A.; Hamilton, D.L. The genesis of carbonatites by liquid immiscibility. In Carbonatites: Genesis and Evolution; Bell, K.E., Ed.; Unwin Hyman: London, UK, 1989; pp. 388–404. [Google Scholar]
- Watkinson, D.H.; Wyllie, P.J. Experimental Study of the Composition Join NaAlSiO4-CaCO3-H2O and the Genesis of Alkalic Rock—Carbonatite Complexes. J. Petrol. 1971, 12, 357–378. [Google Scholar] [CrossRef]
- Franz, G.W. Melting Relationships in the System CaO-MgO-SiO2-CO2-H2O: A Study of Synthetic Kimberlites; The Pennsylvania State University: State College, PA, USA, 1965. [Google Scholar]
- Franz, G.W.; Wyllie, P.J. Experimental Studies in the system CaO-MgO-SiO2-CO2-H2O. In Ultramafic and Related Rocks; Wyllie, P.J., Ed.; John Wiley and Sons: New York, NY, USA, 1967; pp. 323–326. [Google Scholar]
- Toplis, M.J.; Libourel, G.; Carroll, M.R. The role of phosphorus in crystallisation processes of basalt: An experimental study. Geochim. Cosmochim. Acta 1994, 58, 797–810. [Google Scholar] [CrossRef]
- Mysen, B.O. Iron and phosphorus in calcium silicate quenched melts. Chem. Geol. 1992, 98, 175–202. [Google Scholar] [CrossRef]
- Mysen, B.O.; Ryerson, F.J.; Virgo, D. The structural role of phosphorus in silicate melts. Am. Mineral. 1981, 66, 106–117. [Google Scholar]
- Mikhailova, J.A.; Krasnova, N.I.; Krezer, Y.L.; Wall, F. Inclusions in minerals of the Kovdor intrusion of ultrabasic, alkaline rocks and carbonatites as indicators of the endogenic evolution processes. In Deep-Seated Magmatism, Magmatic Sources and the Problem of Plumes (in Russian); Vladykin, N.V., Ed.; Siberian Branch of the Russian Academy of Sciences: Irkutsk/Valdivostok, Russia, 2002; pp. 296–320. [Google Scholar]
- Biggar, G.M. High Pressure High Temperature Phase Equilibrium Studies in the System CaO-CaF2-P2O5-H2O-CO2 with Special Reference to the Apatites; University of Leeds: Leeds, UK, 1962. [Google Scholar]
- Wyllie, P.J.; Biggar, G.M. Fractional Crystallization in the “Carbonatite Systems” CaO-MgO-CO2-H2O and CaO-CaF2-P2O5-CO2-H2O. In Papers and Proceedings of the Fourth General Meeting. International Mineralogical; International Mineralogical Association: Gauteng, South Africa, 1966; pp. 92–105. [Google Scholar]
- Wyllie, P.; Haas, J. The system CaO-SiO2-CO2-H2O: 1. Melting relationships with excess vapor at 1 kilobar pressure. Geochim. Cosmochim. Acta 1965, 29, 871–892. [Google Scholar] [CrossRef]
- Moussallam, Y.; Florian, P.; Corradini, D.; Morizet, Y.; Sator, N.; Vuilleumier, R.; Guillot, B.; Iacono-Marziano, G.; Schmidt, B.C.; Gaillard, F. The molecular structure of melts along the carbonatite-kimberlite-basalt compositional joint: CO2 and polymerisation. Earth Planet. Sci. Lett. 2016, 434, 129–140. [Google Scholar] [CrossRef]
- Klemme, S. Experimental constraints on the evolution of iron and phosphorus-rich melts: Experiments in the system CaO-MgO-Fe2O3-P2O5-SiO2-H2O-CO2. J. Mineral. Petrol. Sci. 2010, 105, 1–8. [Google Scholar] [CrossRef]
- Eberl, D.D.; Kile, D.E.; Drits, V.A. On geological interpretations of crystal size distributions: Constant vs. proportionate growth. Am. Mineral. 2002, 87, 1235–1241. [Google Scholar] [CrossRef]
- Minarik, W.G.; Watson, E.B. Interconnectivity of carbonate melt at low melt fraction. Earth Planet. Sci. Lett. 1995, 133, 423–437. [Google Scholar] [CrossRef]
- Minarik, W.G. Complications to Carbonate Melt Mobility due to the Presence of an Immiscible Silicate Melt. J. Petrol. 1998, 39, 1965–1973. [Google Scholar] [CrossRef]
- Keppler, H. Water solubility in carbonatite melts. Am. Mineral. 2003, 88, 1822–1824. [Google Scholar] [CrossRef]
- Krasnova, N.I. Geology, mineralogy and problems of genesis of apatite-francolite rocks of the Kovdor massif (in Russian). In Composition of Phosphorites; Nauka: Novosibirsk, Russia, 1979; pp. 164–172. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Rock | Rock | Mineral Content, Modal % | |||
|---|---|---|---|---|---|
| Fo | Ap | Mag | Cal | ||
| Forsterite-rich phoscorite (Cal < 10 modal %, Mag < 10 modal %) | Forsteritite (F) | 85–90 | 0–5 | 1–8 | – |
| Apatite-forsterite phoscorite (AF) | 10–85 | 10–80 | 0–8 | 0–5 | |
| Low-carbonate magnetite-rich phoscorite (Cal < 10 modal %, Mag > 10 modal %) | Magnetite-forsterite phoscorite (MF) | 10–70 | 0–5 | 15–85 | 0–5 |
| Magnetite-apatite-forsterite phoscorite (MAF) | 10–70 | 10–70 | 10–70 | 0–8 | |
| Magnetite-apatite phoscorite (MA) | 0–5 | 5–50 | 40–85 | 0–5 | |
| Magnetitite (M) | 0–8 | 0–5 | 80–95 | 0–5 | |
| Calcite-rich phoscorite (10 modal % < Cal < 50 modal %) | Calcite-magnetite-apatite-forsterite phoscorite (CMAF) | 10–60 | 10–60 | 10–55 | 10–40 |
| Calcite-magnetite-forsterite phoscorite (CMF) | 10–70 | 0–5 | 15–60 | 10–45 | |
| Calcite-apatite-forsterite phoscorite (CAF) | 10–45 | 20–55 | 2–8 | 20–40 | |
| Calcite-magnetite-apatite phoscorite (CMA) | 0–6 | 10–63 | 11–79 | 10–45 | |
| Calcite-apatite phoscorite (CA) | 2–6 | 50–65 | 1–6 | 27–41 | |
| Calcite-magnetite phoscorite (CM) | 0–5 | 0–5 | 70–84 | 16–20 | |
| Phoscorite-related carbonatite (Cal > 50 modal %) | Calcite carbonatite (C) | 0–35 | 2–40 | 1–35 | 50–82 |
| Element | Type of Crystal | Standards | DL, wt % |
|---|---|---|---|
| Mg | KAP | Forsterite | 0.1 |
| Al | KAP | Pyrope | 0.05 |
| Si | KAP | Forsterite | 0.05 |
| Ca | PET | Diopside | 0.03 |
| Sc | PET | Thortveitite | 0.02 |
| Ti | PET | Lorenzenite | 0.02 |
| Cr | Quartz | Chromite | 0.02 |
| Mn | Quartz | Synthetic MnCO3 | 0.01 |
| Fe | Quartz | Hematite | 0.01 |
| Ni | LiF | Metal nickel | 0.01 |
| Rock | Peridotite | Phoscorite | Carbonatites | |||
|---|---|---|---|---|---|---|
| (Ap)-Fo | Low-Cb Mag-Rich | Cal-Rich | Phoscorite-Related | Vein | ||
| n | 7 | 39 | 176 | 117 | 20 | 7 |
| SiO2, wt % | 41.1 ± 0.9 40.37–42.63 | 40.8 ± 0.6 39.73–42.55 | 40.9 ± 0.6 38.48–42.21 | 40.8 ± 0.6 39.33–43.98 | 40.8 ± 0.6 39.60–42.01 | 40.8 ± 0.7 39.65–41.68 |
| MgO | 47 ± 2 44.12–50.26 | 52 ± 1 47.73–53.87 | 53 ± 1 47.10–55.25 | 52 ± 1 46.65–55.93 | 53 ± 1 48.51–54.43 | 52 ± 2 48.53–53.69 |
| FeO | 10 ± 1 8.68–12.11 | 7 ± 1 4.43–11.10 | 6 ± 1 3.48–8.82 | 6 ± 1 1.53–10.89 | 6 ± 1 3.73–10.22 | 6 ± 1 4.22–8.04 |
| MnO | 0.4 ± 0.2 0.10–0.55 | 0.34 ± 0.07 0.23–0.56 | 0.3 ± 0.3 0.14–0.49 | 0.33 ± 0.06 0.19–0.53 | 0.34 ± 0.09 0.25–0.66 | 0.33 ± 0.04 0.28–0.39 |
| CaO | 0.3 ± 0.1 0.13–0.36 | 0.13 ± 0.08 <0.03–0.40 | 0.12 ± 0.08 <0.03–0.55 | 0.13 ± 0.08 <0.03–0.60 | 0.17 ± 0.06 0.09–0.31 | 0.19 ± 0.16 0.05–0.49 |
| TiO2 | <0.02 <0.02–0.02 | <0.02 <0.02–0.07 | <0.02 <0.02–0.05 | <0.02 <0.02–0.04 | <0.02 <0.02–0.03 | <0.02 <0.02–0.03 |
| Al2O3 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 |
| Cr2O3 | <0.02 <0.02–0.03 | <0.02 | <0.02 | ≤0.02 | <0.02 <0.02–0.04 | <0.02 |
| NiO | 0.10 ± 0.05 <0.02–0.17 | <0.02 <0.01–0.06 | <0.01 <0.01–0.03 | <0.01 <0.01–0.03 | <0.01 | <0.01 |
| Sc2O3 | <0.02 | <0.02 | <0.02 <0.02–0.03 | <0.02 <0.02–0.11 | <0.02 | <0.02 |
| Mg, apfu | 1.75 ± 0.06 1.66–1.82 | 1.87 ± 0.04 1.77–1.92 | 1.89 ± 0.03 1.74–1.95 | 1.89 ± 0.04 1.75–1.99 | 1.89 ± 0.03 1.79–1.93 | 1.88 ± 0.04 1.8–1.93 |
| Fe2+ | 0.21 ± 0.03 0.15–0.24 | 0.11 ± 0.04 0.05–0.21 | 0.09 ± 0.03 0.00–0.18 | 0.08 ± 0.04 0.00–0.23 | 0.09 ± 0.03 0.03–0.17 | 0.09 ± 0.04 0.04–0.17 |
| Fe3+ | 0.01 ± 0.01 0.00–0.03 | 0.02 ± 0.02 0.00–0.07 | 0.03 ± 0.02 0.00–0.11 | 0.03 ± 0.02 0.00–0.09 | 0.03 ± 0.02 0.00–0.05 | 0.03 ± 0.02 0.00–0.06 |
| Mn | 0.01 0.00–0.01 | 0.01 0.00–0.01 | 0.01+0.01 0.00–0.01 | 0.01 0.00–0.01 | 0.01 0.00–0.01 | 0.01 0.00–0.01 |
| Ca | 0.01 0.00–0.01 | 0.00 0.00–0.01 | 0.00 0.00–0.01 | 0.00 0.00–0.02 | 0.00 0.00–0.01 | 0.00 0.00–0.01 |
| Si | 1.02 ± 0.03 0.98–1.08 | 0.99 ± 0.01 0.97–1.04 | 0.99 ± 0.01 0.93–1.03 | 0.99 ± 0.01 0.95–1.06 | 0.98 ± 0.01 0.97–1.00 | 0.99 ± 0.01 0.97–1.01 |
| Variables | Factor Loadings | |
|---|---|---|
| Factor 1 | Factor 2 | |
| Mg | −0.927 | −0.248 |
| Fe2+ | 0.961 | 0.179 |
| Fe3+ | −0.680 | −0.021 |
| Mn | 0.676 | 0.219 |
| Ca | 0.357 | 0.014 |
| Ti | 0.164 | 0.844 |
| Ni | 0.090 | 0.861 |
| Explained variance | 2.863 | 1.596 |
| % of total variance | 40.9 | 22.8 |
| Sample | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Drill hole | 919 | 924 | 924 | 966 | 987 |
| Depth, m | 18.5 | 26.7 | 169.1 | 62.9 | 67.2 |
| Phoscorite | Mag-Ap-Fo | Cal-Mag-Ap-Fo | Ap-Fo | Mag-Ap-Fo | Cal-Mag-Ap-Fo |
| SiO2, wt % | 39.37 | 40.69 | 40.71 | 40.16 | 40.74 |
| TiO2 | bd | 0.01 | 0.07 | bd | bd |
| FeO | 6.20 | 8.96 | 8.51 | 8.42 | 6.89 |
| MnO | 0.30 | 0.46 | 0.49 | 4.27 | 0.39 |
| MgO | 53.37 | 47.79 | 48.96 | 47.10 | 52.18 |
| CaO | 0.13 | 0.34 | 0.06 | 0.14 | 0.09 |
| NiO | bd | bd | 0.06 | bd | bd |
| Total | 99.37 | 98.25 | 98.86 | 100.09 | 100.29 |
| Mg, apfu | 1.917 | 1.778 | 1.804 | 1.738 | 1.871 |
| Fe2+ | 0.022 | 0.187 | 0.176 | 0.163 | 0.098 |
| Fe3+ | 0.103 | – | – | 0.012 | 0.040 |
| Mn2+ | 0.006 | 0.010 | 0.010 | 0.090 | 0.008 |
| Ca2+ | 0.003 | 0.009 | 0.002 | 0.004 | 0.002 |
| Ni2+ | – | – | 0.001 | – | – |
| Ti4+ | – | – | 0.001 | – | – |
| Si4+ | 0.949 | 1.016 | 1.006 | 0.994 | 0.980 |
| Sample | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Refined formula | Mg1.94Fe0.06SiO4 | Mg1.87Fe0.13SiO4 | Mg1.84Fe0.16SiO4 | Mg1.89Fe0.11SiO4 | Fe0.10Mg1.90SiO4 |
| Temperature/K | 293(2) | ||||
| Crystal system | orthorhombic | ||||
| Space group | Pnma | ||||
| a, (Å) | 10.1899(6) | 10.2165(4) | 10.2097(4) | 10.2027(4) | 10.1980(4) |
| b, (Å) | 5.9730(4) | 5.9911(2) | 5.9876(3) | 5.9775(3) | 5.9810(2) |
| c, (Å) | 4.7403(3) | 4.76168(14) | 4.7600(2) | 4.7541(2) | 4.75403(16) |
| α = β = γ, (°) | 90 | 90 | 90 | 90 | 90 |
| Volume, (Å3) | 288.51(3) | 291.453(18) | 290.99(2) | 289.94(2) | 289.970(18) |
| Z | 4 | 4 | 4 | 4 | 4 |
| ρcalc, (g/cm3) | 3.279 | 3.298 | 3.331 | 3.299 | 3.299 |
| μ/mm−1 | 1.321 | 1.639 | 1.813 | 1.544 | 1.544 |
| Crystal size, (mm3) | 0.23 × 0.18 × 0.16 | 0.27 × 0.21 × 0.18 | 0.29 × 0.25 × 0.16 | 0.18 × 0.15 × 0.14 | 0.19 × 0.17 × 0.16 |
| Radiation | MoKα (λ = 0.71073) | ||||
| 2Θ range for data collection, (°) | 7.99–54.916 | 7.978–54.914 | 7.984–54.942 | 7.99–54.844 | 7.992–54.86 |
| Index ranges | −13 ≤ h ≤ 11, −7 ≤ k ≤ 4, −6 ≤ l ≤ 5 | −13 ≤ h ≤ 5, −4 ≤ k ≤ 7, −6 ≤ l ≤ 5 | −13 ≤ h ≤ 3, −6 ≤ k ≤ 7, −6 ≤ l ≤ 3 | −13 ≤ h ≤ 9, −6 ≤ k ≤ 7, −5 ≤ l ≤ 6 | −6 ≤ h ≤ 13, −5 ≤ k ≤ 7, −6 ≤ l ≤ 3 |
| Reflections collected | 725 | 755 | 794 | 753 | 738 |
| Independent reflections | 361 [Rint = 0.0222, Rsigma = 0.0333] | 364 [Rint = 0.0161, Rsigma = 0.0228] | 364 [Rint = 0.0291, Rsigm = 0.0414] | 363 [Rint = 0.0212, Rsigma = 0.0290] | 363 [Rint = 0.0149, Rsigma = 0.0214] |
| Data/restraints/parameters | 361/0/42 | 364/0/36 | 364/0/42 | 363/0/42 | 363/0/42 |
| Goodness-of-fit on F2 | 1.040 | 1.146 | 1.077 | 1.149 | 1.253 |
| Final R indexes [I >= 2σ (I)] | R1 = 0.0334, wR2 = 0.0852 | R1 = 0.0274, wR2 = 0.0713 | R1 = 0.0260, wR2 = 0.0593 | R1 = 0.0239, wR2 = 0.0602 | R1 = 0.0258, wR2 = 0.0683 |
| Final R indexes [all data] | R1 = 0.0359, wR2 = 0.0873 | R1 = 0.0283, wR2 = 0.0722 | R1 = 0.0336, wR2 = 0.0634 | R1 = 0.0261, wR2 = 0.0619 | R1 = 0.0273, wR2 = 0.0690 |
| Largest diff. peak/hole, (e/Å−3) | 0.57/−0.55 | 0.53/−0.69 | 0.50/−0.48 | 0.47/−0.60 | 0.46/−0.65 |
| Sample | M1 | M2 |
|---|---|---|
| 1 | 0.020 | 0.035 |
| 2 | 0.057 | 0.070 |
| 3 | 0.080 | 0.084 |
| 4 | 0.047 | 0.057 |
| 5 | 0.04 | 0.065 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailova, J.A.; Ivanyuk, G.Y.; Kalashnikov, A.O.; Pakhomovsky, Y.A.; Bazai, A.V.; Panikorovskii, T.L.; Yakovenchuk, V.N.; Konopleva, N.G.; Goryainov, P.M. Three-D Mineralogical Mapping of the Kovdor Phoscorite–Carbonatite Complex, NW Russia: I. Forsterite. Minerals 2018, 8, 260. https://doi.org/10.3390/min8060260
Mikhailova JA, Ivanyuk GY, Kalashnikov AO, Pakhomovsky YA, Bazai AV, Panikorovskii TL, Yakovenchuk VN, Konopleva NG, Goryainov PM. Three-D Mineralogical Mapping of the Kovdor Phoscorite–Carbonatite Complex, NW Russia: I. Forsterite. Minerals. 2018; 8(6):260. https://doi.org/10.3390/min8060260
Chicago/Turabian StyleMikhailova, Julia A., Gregory Yu. Ivanyuk, Andrey O. Kalashnikov, Yakov A. Pakhomovsky, Ayya V. Bazai, Taras L. Panikorovskii, Victor N. Yakovenchuk, Nataly G. Konopleva, and Pavel M. Goryainov. 2018. "Three-D Mineralogical Mapping of the Kovdor Phoscorite–Carbonatite Complex, NW Russia: I. Forsterite" Minerals 8, no. 6: 260. https://doi.org/10.3390/min8060260
APA StyleMikhailova, J. A., Ivanyuk, G. Y., Kalashnikov, A. O., Pakhomovsky, Y. A., Bazai, A. V., Panikorovskii, T. L., Yakovenchuk, V. N., Konopleva, N. G., & Goryainov, P. M. (2018). Three-D Mineralogical Mapping of the Kovdor Phoscorite–Carbonatite Complex, NW Russia: I. Forsterite. Minerals, 8(6), 260. https://doi.org/10.3390/min8060260