Interfacial Precipitation of Phosphate on Hematite and Goethite


Abstract
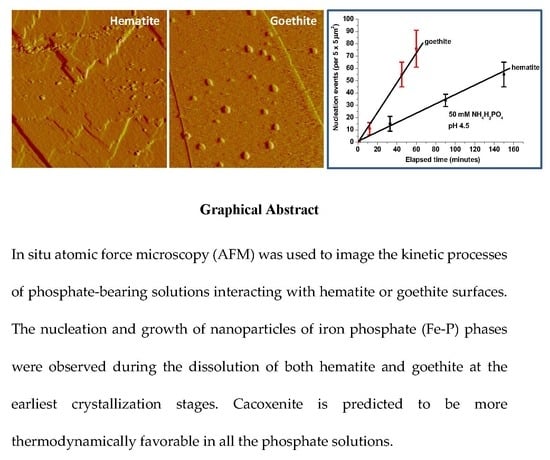
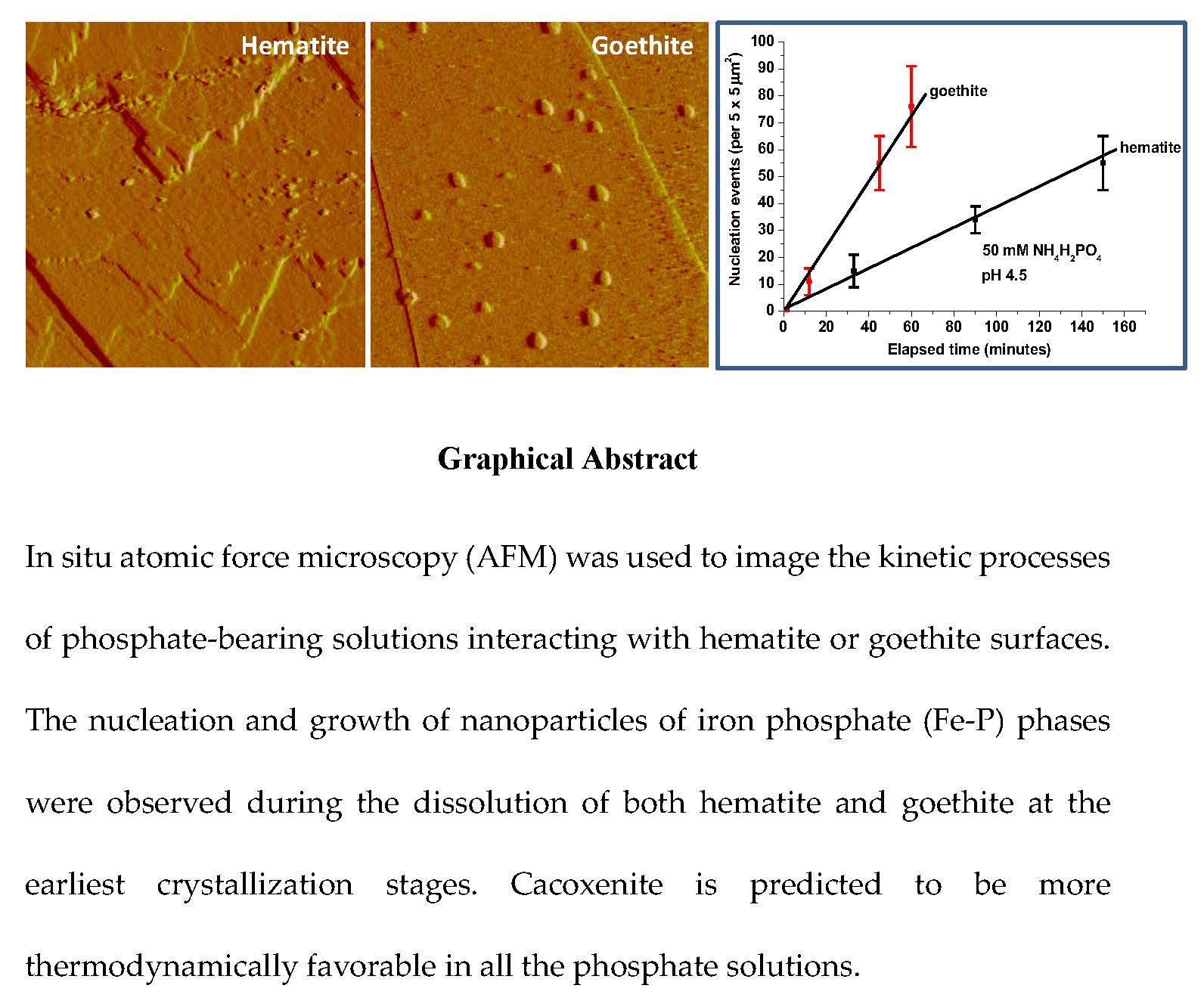{kind=link}
{kind=link}
{kind=link}
{kind=link}
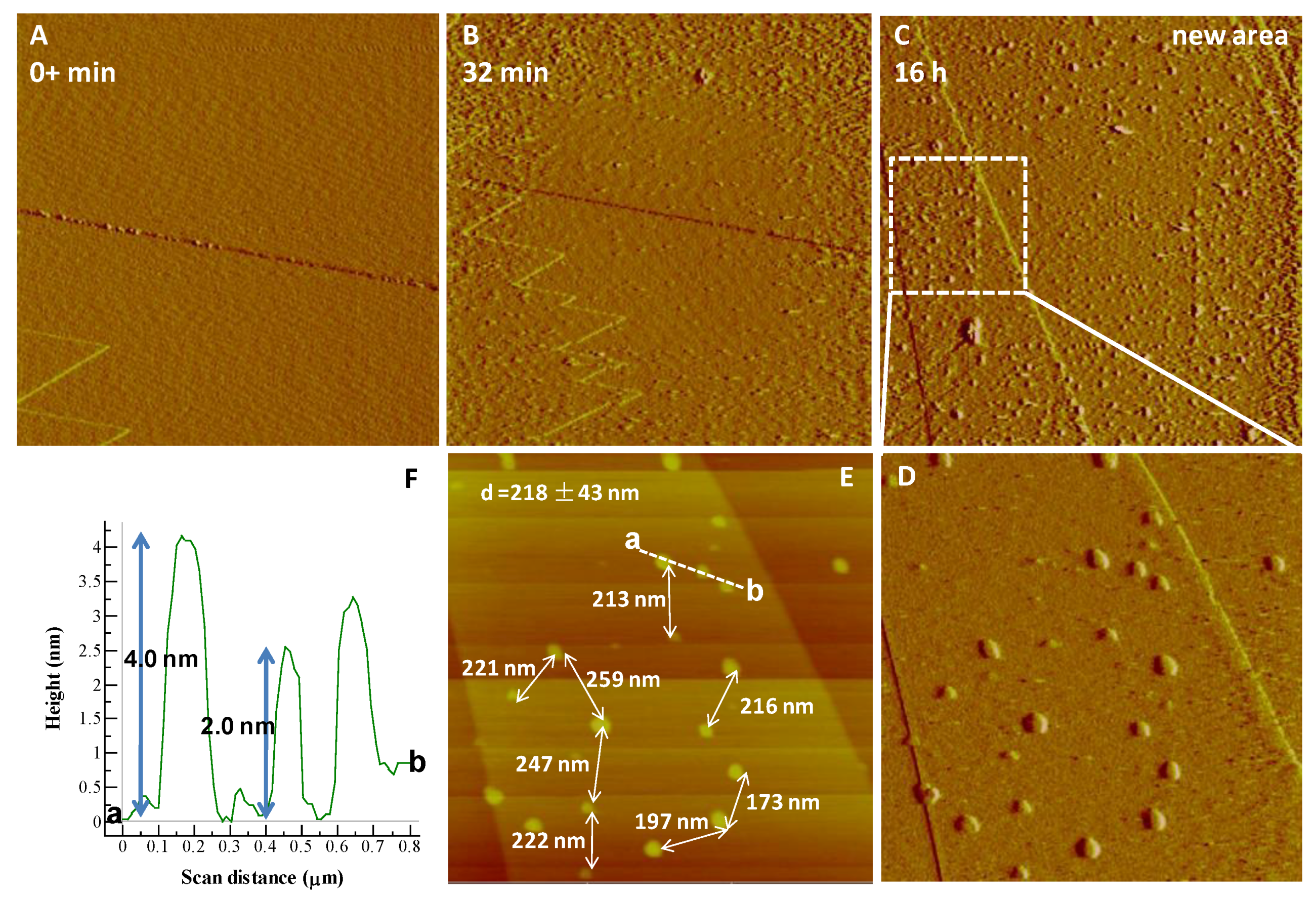{kind=link}
{kind=link}
{kind=link}
{kind=link}
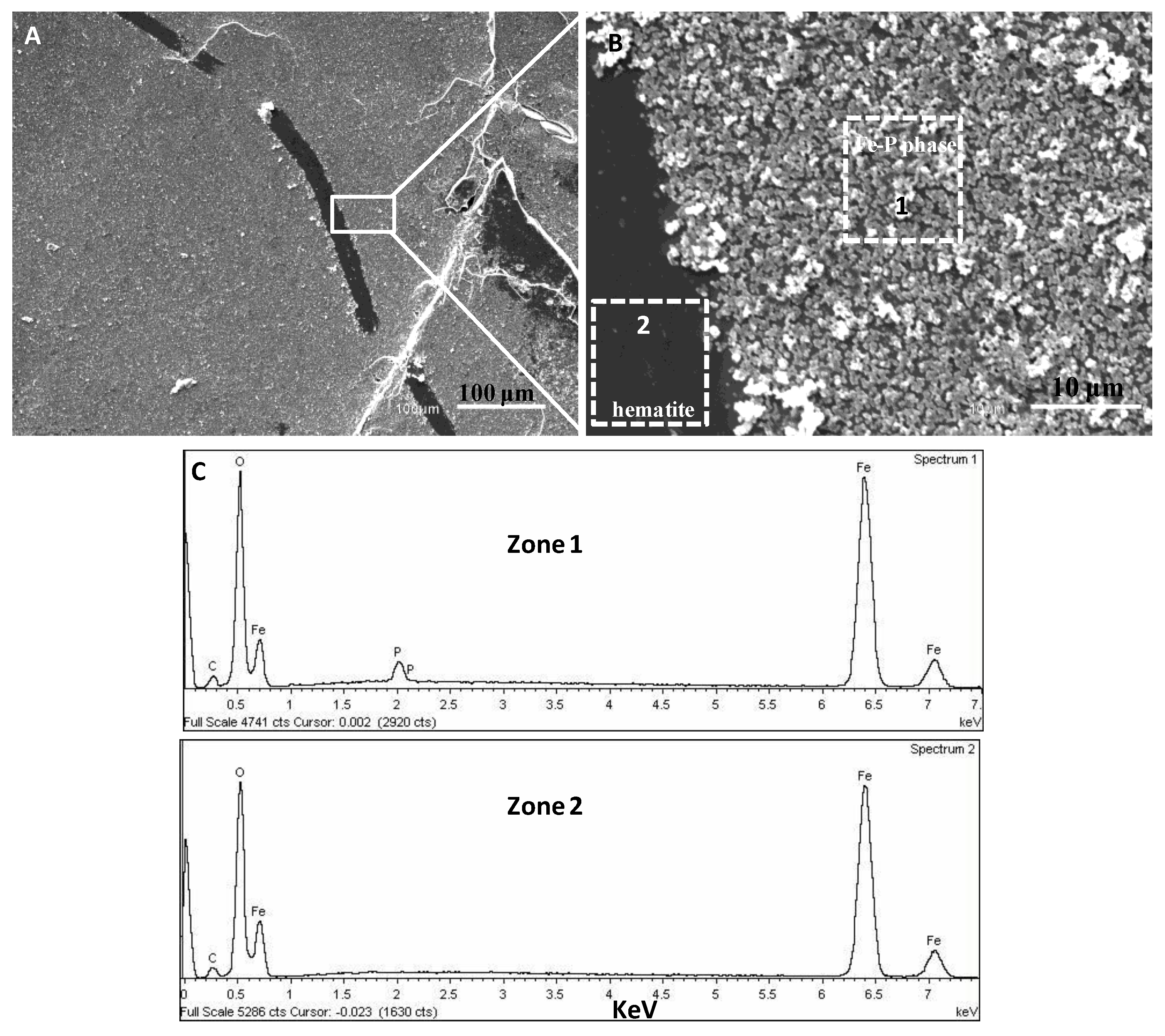{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Iron Phosphate Nucleation and Growth on Hematite and Goethite
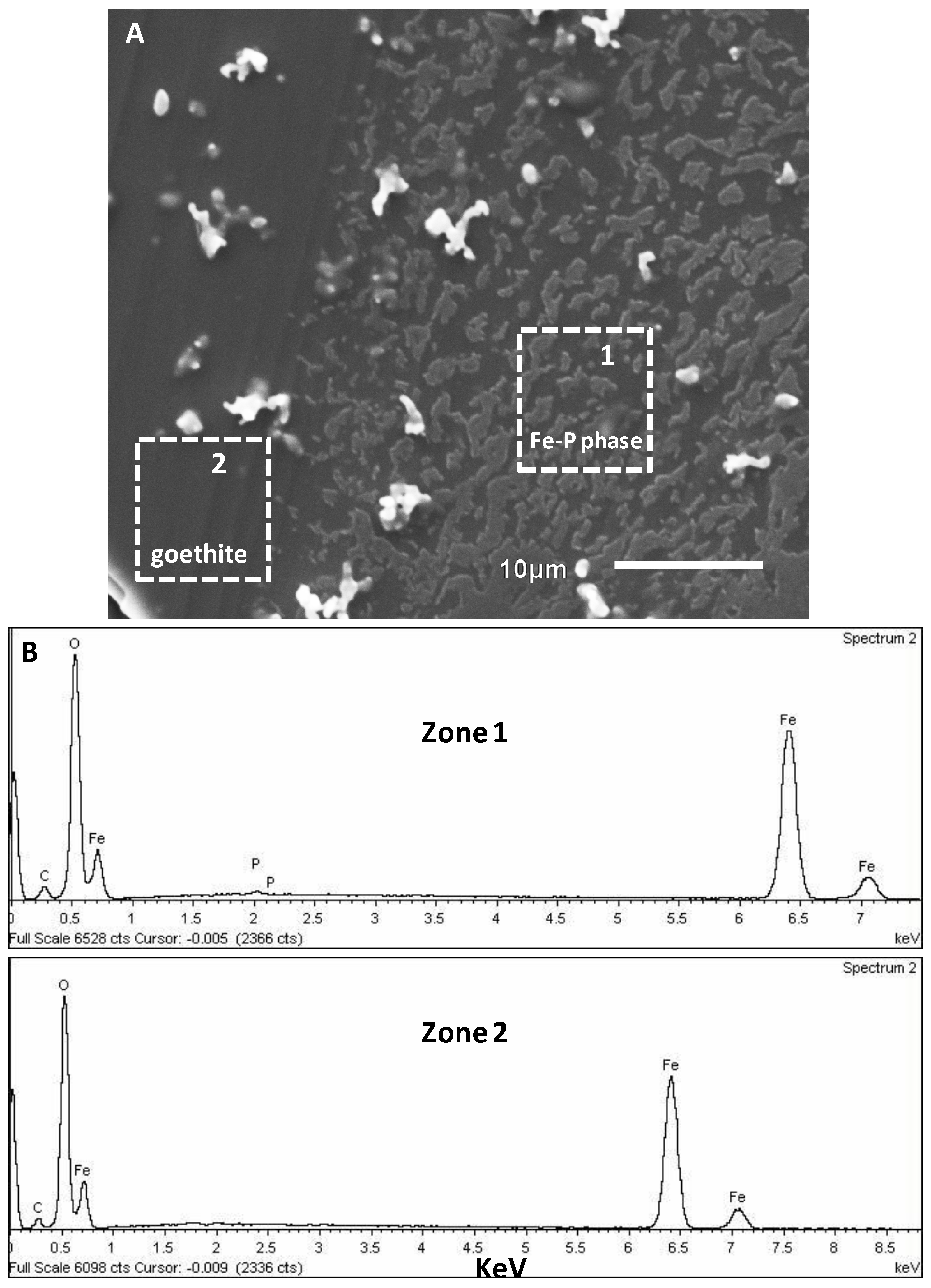
3.2. SEM-EDX Identification of Surface Precipitates on Hematite and Goethite Faces
3.3. Kinetics and Thermodynamics of Coupled Dissolution-Precipitation at the Iron Oxide-Phosphate Solution Interface
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Filippelli, G.M. The global phosphorous cycle: Past, present, and future. Elements 2008, 4, 89–95. [Google Scholar] [CrossRef]
- Shen, J.B.; Yuan, L.; Zhang, J.L.; Li, H.; Bai, Z.; Chen, X.; Zhang, W.; Zhang, F.S. Phosphorus dynamics: From soil to plant. Plant Physiol. 2011, 156, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N. The disappearing nutrient. Nature 2009, 461, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Ruiz-Agudo, E.; Putnis, C.V.; Menneken, M.; Putnis, A. Kinetics of calcium phosphate nucleation and growth on calcite: Implications for predicting the fate of dissolved phosphate species in alkaline soils. Environ. Sci. Technol. 2012, 46, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Putnis, C.V.; Ruiz-Agudo, E.; Hovelmann, J.; Putnis, A. In situ imaging of interfacial precipitation of phosphate on goethite. Environ. Sci. Technol. 2015, 49, 4184–4192. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.D.; Kubicki, J.D. Molecular orbital theory study on surface complex structures of phosphates to iron hydroxides: Calculation of vibrational frequencies and adsorption energies. Langmuir 2004, 20, 9249–9254. [Google Scholar] [CrossRef] [PubMed]
- Khare, N.; Hesterberg, D.; Martin, J.D. XANES investigation of phosphate sorption in single and binary systems of iron and aluminum oxide minerals. Environ. Sci. Technol. 2005, 39, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Rahnemaie, R.; Hiemstra, T.; van Riemsdijk, W.H. Geometry, charge distribution, and surface speciation of phosphate on goethite. Langmuir 2007, 23, 3680–3689. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.P.; Vega, F.A.; Van Riemsdijk, W.H. Competitive and synergistic effects in pH dependent phosphate adsorption in soils: LCD modeling. Environ. Sci. Technol. 2011, 45, 8420–8428. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Li, W.; Philips, B.L.; Grey, C.P. Phosphate adsorption on the iron oxyhydroxides goethite, akaganeite, and lepidocrocite: A 31P NMR study. Energy Environ. Sci. 2011, 4, 4298–4305. [Google Scholar] [CrossRef]
- Liu, H.; Chen, T.; Frost, R.L. An overview of the role of goethite surfaces in the environment. Chemosphere 2014, 103, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, R.G.; Martin, R.R.; Giuliacci, M.E.; Tazaki, K. Surface reactions of goethite with phosphate. J. Chem. Soc. Faraday Trans. 1988, 1, 2311–2315. [Google Scholar] [CrossRef]
- Li, L.; Stanforth, R. Distinguishing adsorption and surface precipitation of phosphate on goethite (a-FeOOH). J. Colloid Interface Sci. 2000, 230, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ler, A.; Stanforth, R. Evidence for surface precipitation of phosphate on goethite. Environ. Sci. Technol. 2003, 37, 2694–2700. [Google Scholar] [CrossRef] [PubMed]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrence and Uses, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Torrent, J. Interactions between phosphate and iron oxide. Adv. Geoecol. 1997, 30, 321–344. [Google Scholar]
- Parkhurst, D.L.; Appelo, C.A.J. Users Guide to PHREEQC (Version 2): A Computer Program for Speciation, Batch Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Water-Resources Investigations Report 99-4259; US Geological Survey: Reston, VA, USA, 1999; 312p.
- Nriagu, J.O.; Dell, C.I. Diagenetic formation of iron phosphates in recent lake sediments. Am. Mineralogist 1974, 59, 934–946. [Google Scholar]
- Vieillard, P.; Tardy, Y. Stability fields of clays and aluminum phosphates: Parageneses in lateritic weathering of argillaceous phosphatic sediments. Am. Mineral. 1979, 64, 626–634. [Google Scholar]
- Putnis, A.; Putnis, C.V. The mechanism of reequilibration of solids in the presence of a fluid phase. J. Solid State Chem. 2007, 180, 1783–1786. [Google Scholar] [CrossRef]
- Putnis, A. Why mineral interfaces matter. Science 2014, 343, 1441–1442. [Google Scholar] [CrossRef] [PubMed]
- Giuffre, A.J.; Hamm, L.M.; Han, N.; De Yoreo, J.J.; Dove, P.M. Polysaccharide chemistry regulates kinetics of calcite nucleation through competition of interfacial energies. Proc. Natl. Acad. Sci. USA 2013, 110, 9261–9266. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Agudo, E.; Kowacz, M.; Putnis, C.V.; Putnis, A. The role of background electrolytes on the kinetics and mechanism of calcite dissolution. Geochim. Cosmochim. Acta 2010, 74, 1256–1267. [Google Scholar] [CrossRef]
- Stack, A.G. Molecular dynamics simulations of solvation and kink site formation at the {001} barite-water interface. J. Phys. Chem. C 2009, 113, 2104–2110. [Google Scholar] [CrossRef]
- Qin, L.H.; Zhang, W.J.; Lu, J.W.; Stack, A.G.; Wang, L.J. Direct imaging of nanoscale dissolution of dicalcium phosphate dihydrate by an organic ligand: Concentration matters. Environ. Sci. Technol. 2013, 47, 13365–13374. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.H.; Wang, L.J.; Wang, B.S. Role of alcoholic hydroxyls of dicarboxylic acids in regulating nanoscale dissolution kinetics of dicalcium phosphate dihydrate. ACS Sustain. Chem. Eng. 2017, 5, 3920–3928. [Google Scholar] [CrossRef]
- Matzapetakis, M.; Raptopoulou, C.P.; Tsohos, A.; Papaefthymiou, V.; Moon, S.N.; Salifoglou, A. Synthesis, spectroscopic and structural characterization of the first mononuclear, water soluble iron-citrate complex, (NH4)5Fe(C6H4O7)2·2H2O. J. Am. Chem. Soc. 1998, 120, 13266–13267. [Google Scholar] [CrossRef]
- Fischer, C.; Karius, V.; Weidler, P.G.; Lüttge, A. Relationship between micrometer to submicrometer surface roughness and topography variations of natural iron oxides and trance element concentration. Langmuir 2008, 24, 3250–3266. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Putnis, C.V.; Hövelmann, J.; Putnis, A. Interfacial Precipitation of Phosphate on Hematite and Goethite. Minerals 2018, 8, 207. https://doi.org/10.3390/min8050207
Wang L, Putnis CV, Hövelmann J, Putnis A. Interfacial Precipitation of Phosphate on Hematite and Goethite. Minerals. 2018; 8(5):207. https://doi.org/10.3390/min8050207
Chicago/Turabian StyleWang, Lijun, Christine V. Putnis, Jörn Hövelmann, and Andrew Putnis. 2018. "Interfacial Precipitation of Phosphate on Hematite and Goethite" Minerals 8, no. 5: 207. https://doi.org/10.3390/min8050207
APA StyleWang, L., Putnis, C. V., Hövelmann, J., & Putnis, A. (2018). Interfacial Precipitation of Phosphate on Hematite and Goethite. Minerals, 8(5), 207. https://doi.org/10.3390/min8050207