Tracing Mineral Reactions Using Confocal Raman Spectroscopy


Abstract
1. Introduction
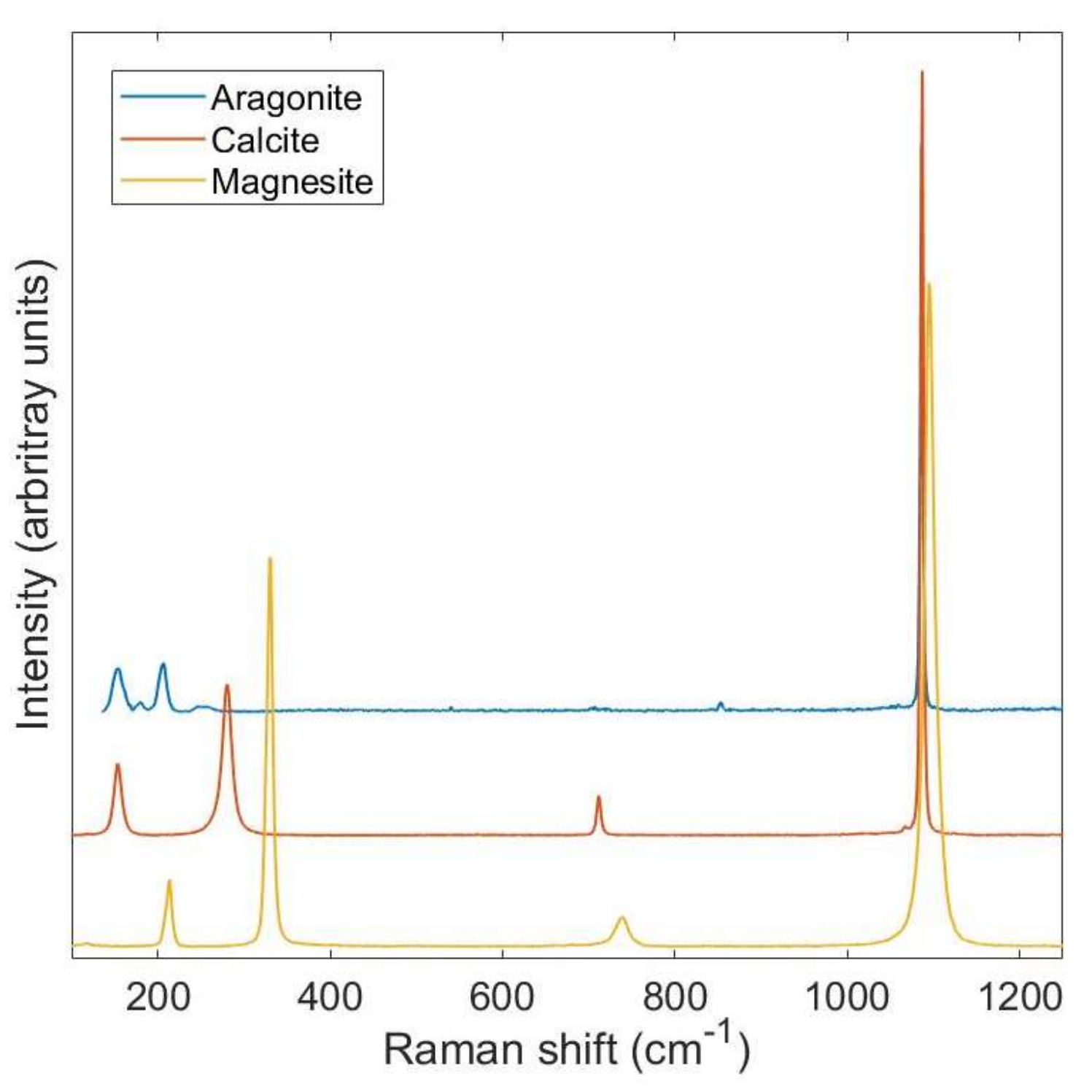
2. Background to Raman Spectroscopy
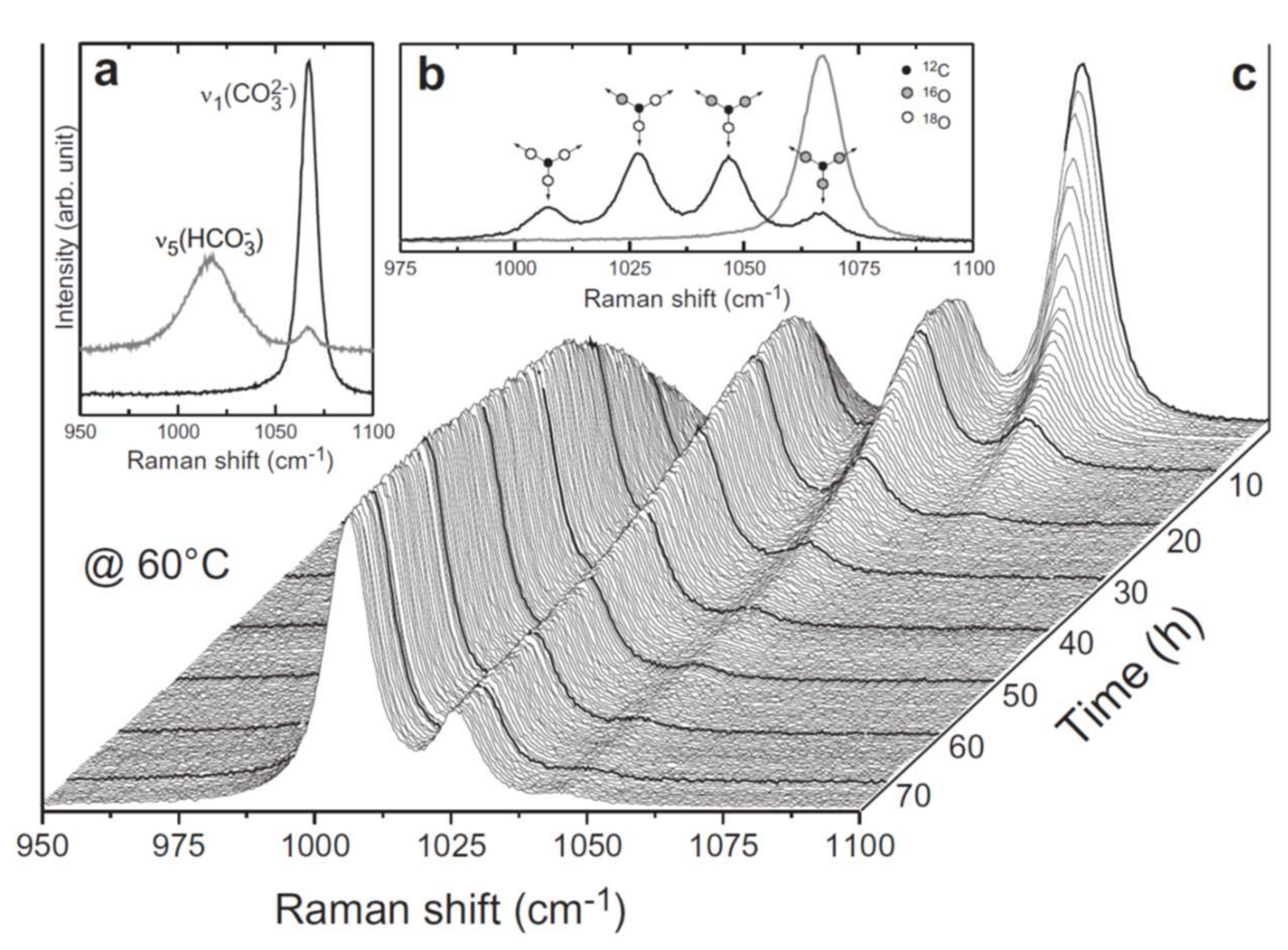
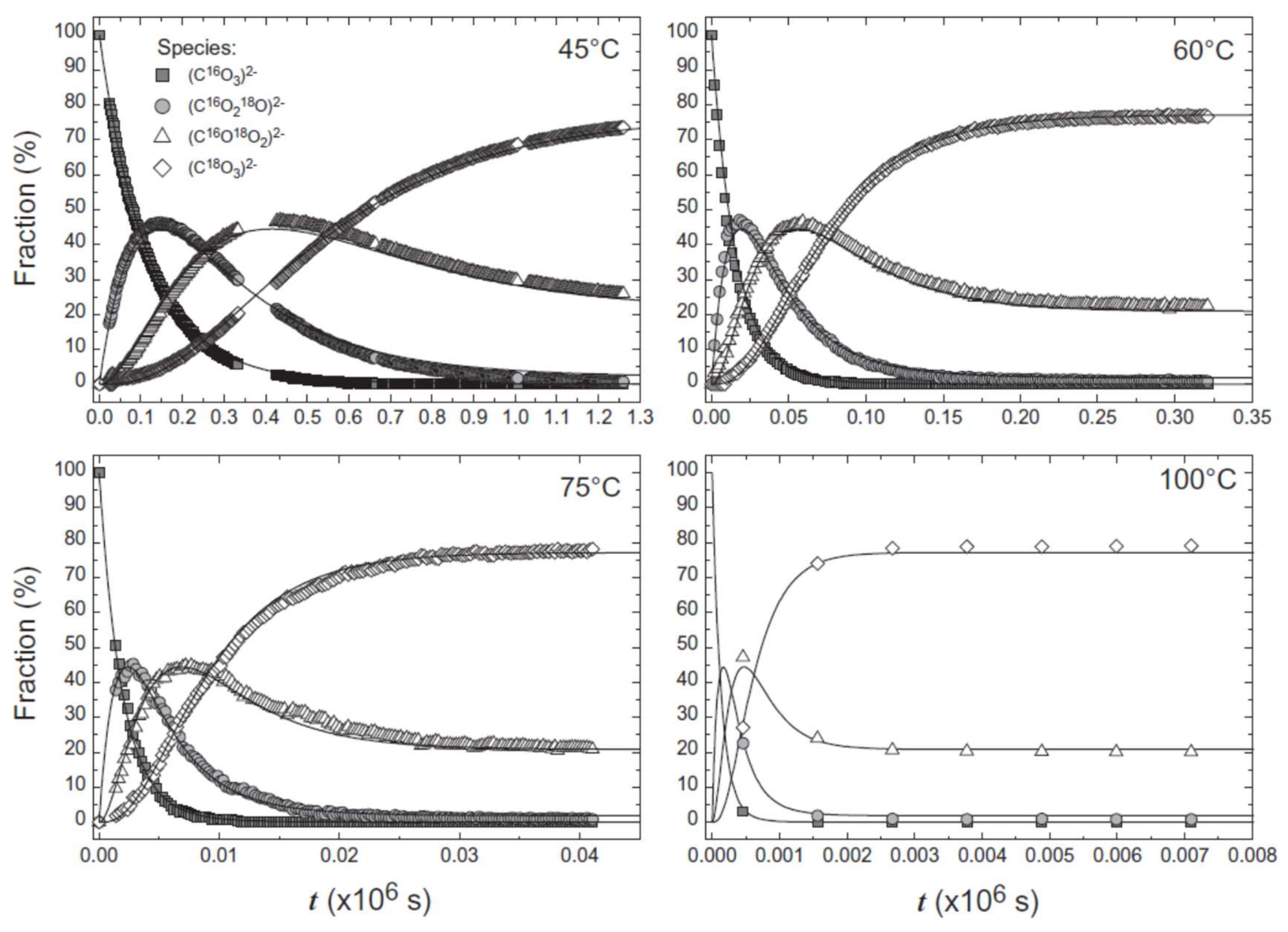
3. Isotopes in Raman Spectroscopy
4. Examining the Properties of Ions in Solution Using Raman Spectroscopy
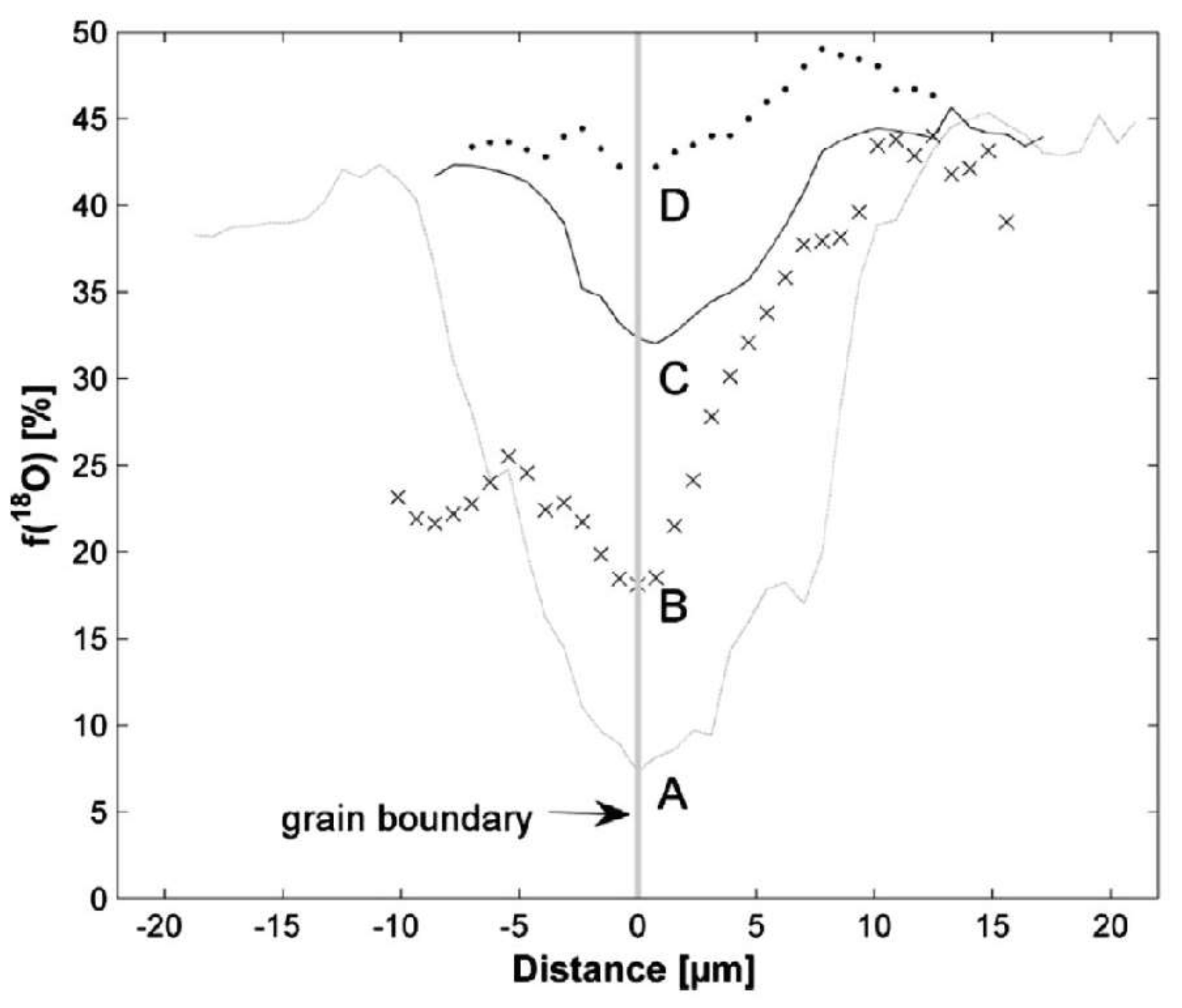
5. Isotopes and Raman Spectroscopy as a Probe for Mineral Replacement Reaction Mechanisms
6. An In Situ Stop Clock for Mineral Replacement Reactions
7. The Future for Isotopes and Raman Spectroscopy
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Landsberg, G. Eine neue Erscheinung bei der Lichtzerstreuung in Krystallen. Naturwissenschaften 1928, 16, 557–558. [Google Scholar]
- Lafuente, B.; Downs, R.T.; Yang, H.; Stone, N. The power of databases: The RRUFF project. In Highlights in Mineralogical Crystallography; Walter de Gruyter GmbH: Berlin, Germany, 2016. [Google Scholar]
- McMillan, P. Structural studies of silicate glasses and melts-applications and limitations of Raman spectroscopy. Am. Miner. 1984, 69, 622–644. [Google Scholar]
- Frezzotti, M.L.; Tecce, F.; Casagli, A. Raman spectroscopy for fluid inclusion analysis. J. Geochem. Explor. 2012, 112, 1–20. [Google Scholar] [CrossRef]
- Rosso, K.; Bodnar, R. Microthermometric and Raman spectroscopic detection limits of CO2 in fluid inclusions and the Raman spectroscopic characterization of CO2. Geochim. Cosmochim. Acta 1995, 59, 3961–3975. [Google Scholar] [CrossRef]
- Prigiobbe, V. Estimation of nucleation and growth parameters from in situ Raman spectroscopy in carbonate systems. J. Environ. Chem. Eng. 2018, 6, 930–936. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, X.; Hu, W.; Chou, I.; Wang, X.; Chen, Y.; Xu, Z. In situ optical and Raman spectroscopic observations of the effects of pressure and fluid composition on liquid–liquid phase separation in aqueous cadmium sulfate solutions (≤400 °C, 50 MPa) with geological and geochemical implications. Geochim. Cosmochim. Acta 2017, 211, 133–152. [Google Scholar] [CrossRef]
- McMillan, P.F.; Poe, B.T.; Stanton, T.R.; Remmele, R.L. A Raman spectroscopic study of H/D isotopically substituted hydrous aluminosilicate glasses. Phys. Chem. Miner. 1993, 19, 454–459. [Google Scholar] [CrossRef]
- Sato, R.K.; McMillan, P.F. An infrared and Raman study of the isotopic species of alpha-quartz. J. Phys. Chem. 1987, 91, 3494–3498. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Jehng, J.; Wachs, I.E. In situ Raman spectroscopy of supported transition metal oxide catalysts: 18O2−16O2 Isotopic Labeling Studies. J. Phys. Chem. B 2000, 104, 7382–7387. [Google Scholar] [CrossRef]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra; Courier Corporation: New York, NY, USA, 1980. [Google Scholar]
- Edwards, H.G.M.; Villar, S.E.J.; Jehlicka, J.; Munshi, T. FT–Raman spectroscopic study of calcium-rich and magnesium-rich carbonate minerals. Spectrochim. Acta A 2005, 61, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Bhagavantam, S. Effect of crystal orientation on the Raman spectrum of calcite. Proc. Math. Sci. 1940, 11, 62–71. [Google Scholar]
- Farmer, V. Transverse and longitudinal crystal modes associated with OH stretching vibrations in single crystals of kaolinite and dickite. Spectrochim. Acta. A 2000, 56, 927–930. [Google Scholar] [CrossRef]
- Ishibashi, H.; Arakawa, M.; Ohi, S.; Yamamoto, J.; Miyake, A.; Kagi, H. Relationship between Raman spectral pattern and crystallographic orientation of a rock-forming mineral: A case study of Fo89Fa11 olivine. J. Raman Spectrosc. 2008, 39, 1653–1659. [Google Scholar] [CrossRef]
- Shebanova, O.N.; Lazor, P. Raman spectroscopic study of magnetite (FeFe2O4): A new assignment for the vibrational spectrum. J. Solid State Chem. 2003, 174, 424–430. [Google Scholar] [CrossRef]
- Awonusi, A.; Morris, M.D.; Tecklenburg, M.M.J. Carbonate assignment and calibration in the Raman spectrum of apatite. Calcif. Tissue Int. 2007, 81, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Alia, J.; de Mera, Y.D.; Edwards, H.G.M.; Martín, P.G.; Andres, S.L. FT-Raman and infrared spectroscopic study of aragonite-strontianite (CaxSr1− xCO3) solid solution. Spectrochim. Acta A 1997, 53, 2347–2362. [Google Scholar] [CrossRef]
- Herzberg, G. Molecular Spectra and Molecular Structure; D. Van Nostrand Company, Inc.: Princeton, NJ, USA, 1945. [Google Scholar]
- Geisler, T.; Perdikouri, C.; Kasioptas, A.; Dietzel, M. Real-time monitoring of the overall exchange of oxygen isotopes between aqueous CO32− and H2O by Raman spectroscopy. Geochim. Cosmochim. Acta 2012, 90, 1–11. [Google Scholar] [CrossRef]
- Geidel, E.; Krause, K.; Förster, H.; Bauer, F. Vibrational spectra and computer simulations of 18O-labelled NaY zeolites. J. Chem. Soc. Faraday Trans. 1997, 93, 1439–1443. [Google Scholar] [CrossRef]
- Galeener, F.L.; Mikkelsen, J., Jr. Vibrational dynamics in 18O-substituted vitreous SiO2. Phys. Rev. B 1981, 23, 5527. [Google Scholar] [CrossRef]
- Putnis, C.V.; Geisler, T.; Schmid-Beurmann, P.; Stephan, T.; Giampaolo, C. An experimental study of the replacement of leucite by analcime. Am. Miner. 2007, 92, 19–26. [Google Scholar] [CrossRef]
- Gillet, P.; McMillan, P.; Schott, J.; Badro, J.; Grzechnik, A. Thermodynamic properties and isotopic fractionation of calcite from vibrational spectroscopy of 18O-substituted calcite. Geochim. Cosmochim. Acta 1996, 60, 3471–3485. [Google Scholar] [CrossRef]
- Crawford, B., Jr. Vibrational intensities. II. The use of isotopes. J. Chem. Phys. 1952, 20, 977–981. [Google Scholar] [CrossRef]
- Petreanu, E.; Pinchas, S.; Samuel, D. The infra-red absorption of 75% 18O-barium phosphate. J. Inorg. Nucl. Chem. 1965, 27, 2519–2523. [Google Scholar] [CrossRef]
- Corno, M.; Busco, C.; Civalleri, B.; Ugliengo, P. Periodic ab initio study of structural and vibrational features of hexagonal hydroxyapatite Ca10(PO4)6(OH)2. Phys. Chem. Chem. Phys. 2006, 8, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Montero, S.; Schmölz, R.; Haussühl, S. Raman spectra of orthorhombic sulfate single crystals I: K2SO4, Rb2SO4, Cs2SO4 and Tl2SO4. J. Raman Spectrosc. 1974, 2, 101–113. [Google Scholar] [CrossRef]
- Pinchas, S.; Shamir, J. The anomalous behaviour of 18O-labelled compounds. Part IV. Vibrational spectra of isotopic vanadate ions. Isr. J. Chem. 1969, 7, 805–811. [Google Scholar] [CrossRef]
- Arakawa, M.; Yamamoto, J.; Kagi, H. Developing micro-Raman mass spectrometry for measuring carbon isotopic composition of carbon dioxide. Appl. Spectrosc. 2007, 61, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Menneken, M.; Geisler, T. An evaluation of the potential of using Raman spectroscopy to determine the carbon isotope composition of CO2 inclusions. J. Geochem. Explor. 2009, 101, 70. [Google Scholar] [CrossRef]
- Menneken, M.; Geisler, T.; Nemchin, A.A.; Strauss, H. Raman spectroscopic determination of the isotope composition of CO2 inclusions. Geochim. Cosmochim. Acta Suppl. 2009, 73, A869. [Google Scholar]
- Carey, D.M.; Korenowski, G.M. Measurement of the Raman spectrum of liquid water. J. Chem. Phys. 1998, 108, 2669–2675. [Google Scholar] [CrossRef]
- Pye, C.C.; Rudolph, W.W. An ab initio and Raman investigation of magnesium (II) hydration. J. Phys. Chem. A 1998, 102, 9933–9943. [Google Scholar] [CrossRef]
- Bulmer, J.T.; Irish, D.E.; Ödberg, L. The temperature dependence of Raman band parameters for aquated Mg(II) and Zn(II). Can. J. Chem. 1975, 53, 3806–3811. [Google Scholar] [CrossRef]
- Martinez, I.; Sanchez-Valle, C.; Daniel, I.; Reynard, B. High-pressure and high-temperature Raman spectroscopy of carbonate ions in aqueous solution. Chem. Geol. 2004, 207, 47–58. [Google Scholar] [CrossRef]
- Pye, C.C.; Rudolph, W.W. An ab initio, infrared, and Raman investigation of phosphate ion hydration. J. Phys. Chem. A 2003, 107, 8746–8755. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Irmer, G.; Hefter, G.T. Raman spectroscopic investigation of speciation in MgSO4(aq). Phys. Chem. Chem. Phys. 2003, 5, 5253–5261. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Chou, I.; Hu, W.; Wan, Y.; Li, Z. Properties of lithium under hydrothermal conditions revealed by in situ Raman spectroscopic characterization of Li2O-SO3-H2O (D2O) systems at temperatures up to 420 °C. Chem. Geol. 2017, 451, 104–115. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Brooker, M.H.; Tremaine, P.R. Raman spectroscopic investigation of aqueous FeSO4 in neutral and acidic solutions from 25 °C to 303 °C: Inner-and outer-sphere complexes. J. Solut. Chem. 1997, 26, 757–777. [Google Scholar] [CrossRef]
- Preston, C.M.; Adams, W.A. A laser Raman spectroscopic study of aqueous orthophosphate salts. J. Phys. Chem. 1979, 83, 814–821. [Google Scholar] [CrossRef]
- Zotov, N.; Keppler, H. Silica speciation in aqueous fluids at high pressures and high temperatures. Chem. Geol. 2002, 184, 71–82. [Google Scholar] [CrossRef]
- King, H.E.; Satoh, H.; Tsukamoto, K.; Putnis, A. Nanoscale observations of magnesite growth in chloride-and sulfate-rich solutions. Environ. Sci. Technol. 2013, 47, 8684–8691. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chou, I.; Hu, W.; Burruss, R.C. In situ observations of liquid–liquid phase separation in aqueous MgSO4 solutions: Geological and geochemical implications. Geochim. Cosmochim. Acta 2013, 103, 1–10. [Google Scholar] [CrossRef]
- Wang, X.; Wan, Y.; Hu, W.; Chou, I.; Cao, J.; Wang, X.; Wang, M.; Li, Z. In situ observations of liquid–liquid phase separation in aqueous ZnSO4 solutions at temperatures up to 400 °C: Implications for Zn2+–SO42− association and evolution of submarine hydrothermal fluids. Geochim. Cosmochim. Acta 2016, 181, 126–143. [Google Scholar] [CrossRef]
- Gebauer, D.; Cölfen, H. Prenucleation clusters and non-classical nucleation. Nano Today 2011, 6, 564–584. [Google Scholar] [CrossRef]
- Geisler, T.; Kasioptas, A.; Menneken, M.; Perdikouri, C.; Putnis, A. A preliminary in situ Raman spectroscopic study of the oxygen isotope exchange kinetics between H2O and (PO4) aq. J. Geochem. Explor. 2009, 101, 37. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Irmer, G.; Königsberger, E. Speciation studies in aqueous HCO3−–CO32− solutions. A combined Raman spectroscopic and thermodynamic study. Dalton Trans. 2008, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, S.; Anbalagan, G.; Pandi, S. Raman and infrared spectra of carbonates of calcite structure. J. Raman Spectrosc. 2006, 37, 892–899. [Google Scholar] [CrossRef]
- Pinchas, S.; Sadeh, D. Fundamental vibration frequencies of the main isotopic PO43− ions in aqueous solutions. J. Inorg. Nucl. Chem. 1968, 30, 1785–1789. [Google Scholar] [CrossRef]
- Campbell, N.J.; Flanagan, J.; Griffith, W.P. Vibrational spectra of [B(18OH)4]−, [V18O4]3−, and [P18O4]3−: An anomaly resolved. J. Chem. Phys. 1985, 83, 3712–3713. [Google Scholar] [CrossRef]
- Putnis, A.; John, T. Replacement Processes in the Earth's Crust. Elements 2010, 6, 159–164. [Google Scholar] [CrossRef]
- Nahon, D.; Merino, E. Pseudomorphic replacement in tropical weathering: Evidence, geochemical consequences, and kinetic-rheological origin. Am. J. Sci. 1997, 297, 393–417. [Google Scholar] [CrossRef]
- Henao, D.M.O.; Godoy, M.A.M. Jarosite pseudomorph formation from arsenopyrite oxidation using Acidithiobacillus ferrooxidans. Hydrometallurgy 2010, 104, 162–168. [Google Scholar] [CrossRef]
- Laves, F. Artificial preparation of microcline. J. Geol. 1951, 59, 511–512. [Google Scholar] [CrossRef]
- Fiebig, J.; Hoefs, J. Hydrothermal alteration of biotite and plagioclase as inferred from intragranular oxygen isotope-and cation-distribution patterns. Eur. J. Miner. 2002, 14, 49–60. [Google Scholar] [CrossRef]
- Labotka, T.C.; Cole, D.R.; Fayek, M.; Riciputi, L.R.; Stadermann, F.J. Coupled cation and oxygen-isotope exchange between alkali feldspar and aqueous chloride solution. Am. Miner. 2004, 89, 1822–1825. [Google Scholar] [CrossRef]
- Niedermeier, D.R.D.; Putnis, A.; Geisler, T.; Golla-Schindler, U.; Putnis, C.V. The mechanism of cation and oxygen isotope exchange in alkali feldspars under hydrothermal conditions. Contrib. Miner. Petrol. 2009, 157, 65–76. [Google Scholar] [CrossRef]
- Hövelmann, J.; Putnis, A.; Geisler, T.; Schmidt, B.C.; Golla-Schindler, U. The replacement of plagioclase feldspars by albite: Observations from hydrothermal experiments. Contrib. Miner. Petrol. 2010, 159, 43–59. [Google Scholar] [CrossRef]
- Velbel, M.A. Formation of protective surface layers during silicate-mineral weathering under well-leached, oxidizing conditions. Am. Miner. 1993, 78, 405–414. [Google Scholar]
- Liu, Y.; Olsen, A.A.; Rimstidt, D. Mechanism for the dissolution of olivine series minerals in acidic solutions. Am. Miner. 2006, 91, 455–458. [Google Scholar] [CrossRef]
- Zakaznova-Herzog, V.P.; Nesbitt, H.W.; Bancroft, G.M.; Tse, J.S. Characterization of leached layers on olivine and pyroxenes using high-resolution XPS and density functional calculations. Geochim. Cosmochim. Acta 2008, 72, 69–86. [Google Scholar] [CrossRef]
- Schott, J.; Pokrovsky, O.S.; Spalla, O.; Devreux, F.; Gloter, A.; Mielczarski, J.A. Formation, growth and transformation of leached layers during silicate minerals dissolution: The example of wollastonite. Geochim. Cosmochim. Acta 2012, 98, 259–281. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Schott, J. Forsterite surface composition in aqueous solutions: A combined potentiometric, electrokinetic, and spectroscopic approach. Geochim. Cosmochim. Acta 2000, 64, 3299–3312. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; King, H.E.; Patiño-López, L.D.; Putnis, C.V.; Geisler, T.; Rodriguez-Navarro, C.; Putnis, A. Control of silicate weathering by interface-coupled dissolution-precipitation processes at the mineral-solution interface. Geology 2016, 44, 567–570. [Google Scholar] [CrossRef]
- King, H.E.; Plümper, O.; Geisler, T.; Putnis, A. Experimental investigations into the silicification of olivine: Implications for the reaction mechanism and acid neutralization. Am. Miner. 2011, 96, 1503–1511. [Google Scholar] [CrossRef]
- Cardew, P.T.; Davey, R.J. The kinetics of solvent-mediated phase transformations. Proc. R. Soc. A Math. Phys. 1985, 398, 415–428. [Google Scholar] [CrossRef]
- Putnis, A. Mineral replacement reactions: From macroscopic observations to microscopic mechanisms. Miner. Mag. 2002, 66, 689–708. [Google Scholar] [CrossRef]
- Putnis, A.; Putnis, C.V. The mechanism of reequilibration of solids in the presence of a fluid phase. J. Solid State Chem. 2007, 180, 1783–1786. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Merino, E. Dynamic weathering model: Constraints required by coupled dissolution and pseudomorphic replacement. Geochim. Cosmochim. Acta 1995, 59, 1559–1570. [Google Scholar] [CrossRef]
- Glikin, A.E. Polymineral-Metasomatic Crystallogenesis; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Xia, F.; Brugger, J.; Chen, G.; Ngothai, Y.; O’Neill, B.; Putnis, A.; Pring, A. Mechanism and kinetics of pseudomorphic mineral replacement reactions: A case study of the replacement of pentlandite by violarite. Geochim. Cosmochim. Acta 2009, 73, 1945–1969. [Google Scholar] [CrossRef]
- Putnis, C.V.; Mezger, K. A mechanism of mineral replacement: Isotope tracing in the model system KCl-KBr-H2O. Geochim. Cosmochim. Acta 2004, 68, 2839–2848. [Google Scholar] [CrossRef]
- Kasioptas, A.; Geisler, T.; Perdikouri, C.; Trepmann, C.; Gussone, N.; Putnis, A. Polycrystalline apatite synthesized by hydrothermal replacement of calcium carbonates. Geochim. Cosmochim. Acta 2011, 75, 3486–3500. [Google Scholar] [CrossRef]
- Perdikouri, C.; Kasioptas, A.; Geisler, T.; Schmidt, B.C.; Putnis, A. Experimental study of the aragonite to calcite transition in aqueous solution. Geochim. Cosmochim. Acta 2011, 75, 6211–6224. [Google Scholar] [CrossRef]
- King, H.E.; Mattner, D.C.; Plümper, O.; Geisler, T.; Putnis, A. Forming cohesive calcium oxalate layers on marble surfaces for stone conservation. Cryst. Growth Des. 2014, 14, 3910–3917. [Google Scholar] [CrossRef]
- Geisler, T.; Janssen, A.; Scheiter, D.; Stephan, T.; Berndt, J.; Putnis, A. Aqueous corrosion of borosilicate glass under acidic conditions: A new corrosion mechanism. J. Non-Cryst. Solids 2010, 356, 1458–1465. [Google Scholar] [CrossRef]
- Janssen, A.; Putnis, A.; Geisler, T. The experimental replacement of ilmenite by rutile in HCl solutions. Miner. Mag. 2010, 74, 633–644. [Google Scholar] [CrossRef]
- Jonas, L.; John, T.; King, H.E.; Geisler, T.; Putnis, A. The role of grain boundaries and transient porosity in rocks as fluid pathways for reaction front propagation. Earth Planet. Sci. Lett. 2014, 386, 64–74. [Google Scholar] [CrossRef]
- Geisler, T.; Pöml, P.; Stephan, T.; Janssen, A.; Putnis, A. Experimental observation of an interface-controlled pseudomorphic replacement reaction in a natural crystalline pyrochlore. Am. Miner. 2005, 90, 1683–1687. [Google Scholar] [CrossRef]
- Pöml, P.; Menneken, M.; Stephan, T.; Niedermeier, D.; Geisler, T.; Putnis, A. Mechanism of hydrothermal alteration of natural self-irradiated and synthetic crystalline titanate-based pyrochlore. Geochim. Cosmochim. Acta 2007, 71, 3311–3322. [Google Scholar] [CrossRef]
- Li, J.; Chou, I.; Yuan, S.; Burruss, R.C. Observations on the crystallization of spodumene from aqueous solutions in a hydrothermal diamond-anvil cell. Geofluids 2013, 13, 467–474. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, X.; Chou, I.; Hu, W.; Zhang, Y.; Wang, X. An experimental study of the formation of talc through CaMg(CO3)2–SiO2–H2O interaction at 100–200 °C and vapor-saturation pressures. Geofluids 2017, 2017, 3942826. [Google Scholar] [CrossRef]
- Hänchen, M.; Prigiobbe, V.; Baciocchi, R.; Mazzotti, M. Precipitation in the Mg-carbonate system—Effects of temperature and CO2 pressure. Chem. Eng. Sci. 2008, 63, 1012–1028. [Google Scholar] [CrossRef]
- Geisler, T.; Lenting, C.; Stamm, F.M.; Sulzbach, M. Real-time, in situ hyperspectral Raman imaging of mineral-fluid reactions. Goldschm. Abstr. 2015, 2015, 1016. [Google Scholar]
- Sulzbach, M.; Geisler, T. The replacement of Celestine (SrSO4) by Strontianite (SrCO3) studied in situ, spatially resolved, and real-time by Raman spectroscopy. In Proceedings of the EGU General Assembly Conference, Vienna, Austria, 12–17 April 2015. [Google Scholar]
- Nasdala, L.; Irmer, G.; Wolf, D. The degree of metamictization in zircons: A Raman spectroscopic study. Eur. J. Miner. 1995, 7, 471–478. [Google Scholar] [CrossRef]
- Weissbart, E.J.; Rimstidt, J.D. Wollastonite: Incongruent dissolution and leached layer formation. Geochim. Cosmochim. Acta 2000, 64, 4007–4016. [Google Scholar] [CrossRef]
- De Meer, S.; Spiers, C.J. On mechanisms and kinetics of creep by intergranular pressure solution. In Growth, Dissolution and Pattern Formation in Geosystems; Springer: Dordrecht, The Netherlands, 1999; pp. 345–366. [Google Scholar]
- Felipe, M.A.; Kubicki, J.D.; Rye, D.M. Oxygen isotope exchange kinetics between H2O and H4SiO4 from ab initio calculations. Geochim. Cosmochim. Acta 2004, 68, 949–958. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactant | Product | Temp (°C) | Reference |
|---|---|---|---|
| Calcite | Whewellite | 60 | [76] |
| Wollasonite | Amorphous silica | 90 | [65] |
| Olivine | Amorphous silica | 90 | [66] |
| Borosilicate glass | Amorphous silica | 150 | [77] |
| Ilmenite | Rutile | 150 | [78] |
| Aragonite | Apatite | 150 | [74] |
| Calcite | Apatite | 200 | [79] |
| Aragonite | Calcite | 200 | [75] |
| Leucite | Analcime | 200 | [23] |
| Pyrochlore | Pyrochlore | 200 | [80] |
| Pyrochlore | Rutile | 250 | [81] |
| Oligoclase | Albite | 600 | [59] |
| Labradorite | Albite | 600 | [59] |
| Albite | K-feldspar | 600 | [58] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, H.E.; Geisler, T. Tracing Mineral Reactions Using Confocal Raman Spectroscopy. Minerals 2018, 8, 158. https://doi.org/10.3390/min8040158
King HE, Geisler T. Tracing Mineral Reactions Using Confocal Raman Spectroscopy. Minerals. 2018; 8(4):158. https://doi.org/10.3390/min8040158
Chicago/Turabian StyleKing, Helen E., and Thorsten Geisler. 2018. "Tracing Mineral Reactions Using Confocal Raman Spectroscopy" Minerals 8, no. 4: 158. https://doi.org/10.3390/min8040158
APA StyleKing, H. E., & Geisler, T. (2018). Tracing Mineral Reactions Using Confocal Raman Spectroscopy. Minerals, 8(4), 158. https://doi.org/10.3390/min8040158