Nano Precipitates Formed during the Dissolution of Calcite Incorporated with Cu and Mn †


Abstract
1. Introduction
2. Materials and Methods
2.1. Solid Preparation and Characterization
2.1.1. Coprecipitation Procedures
2.1.2. Characterization
2.2. Dissolution Experiments
3. Results
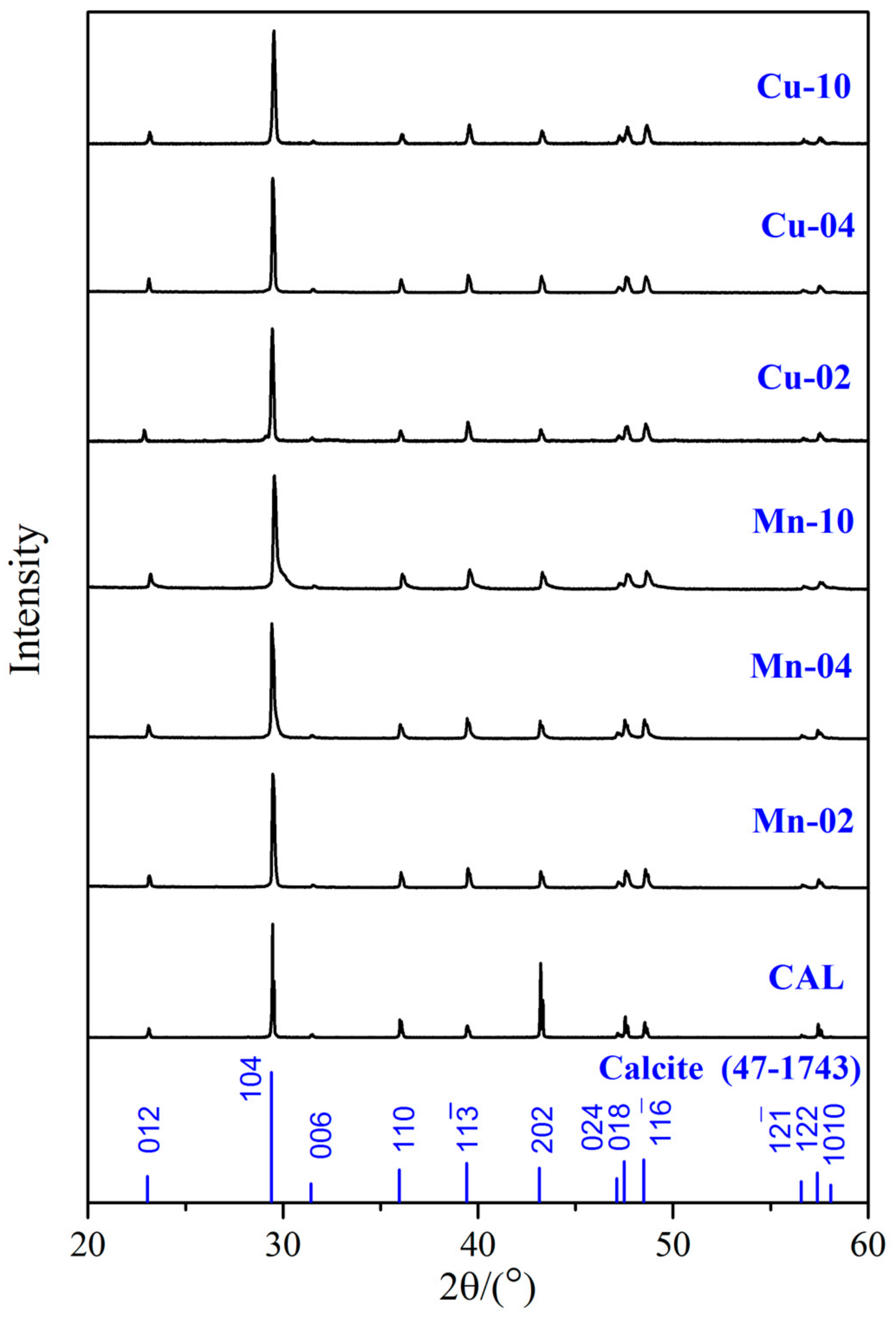
3.1. Structure and Chemical Compositions
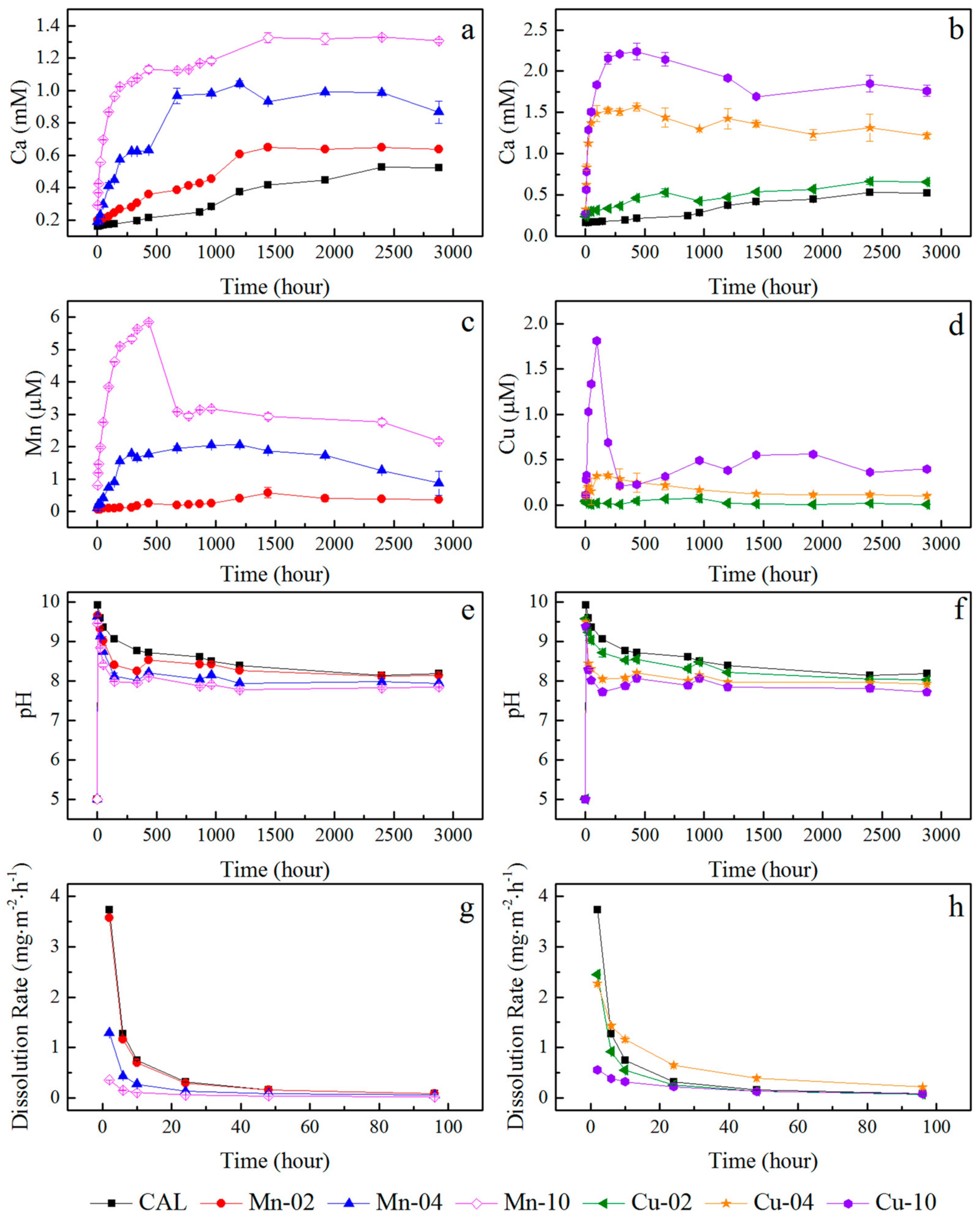
3.2. Release of Metals
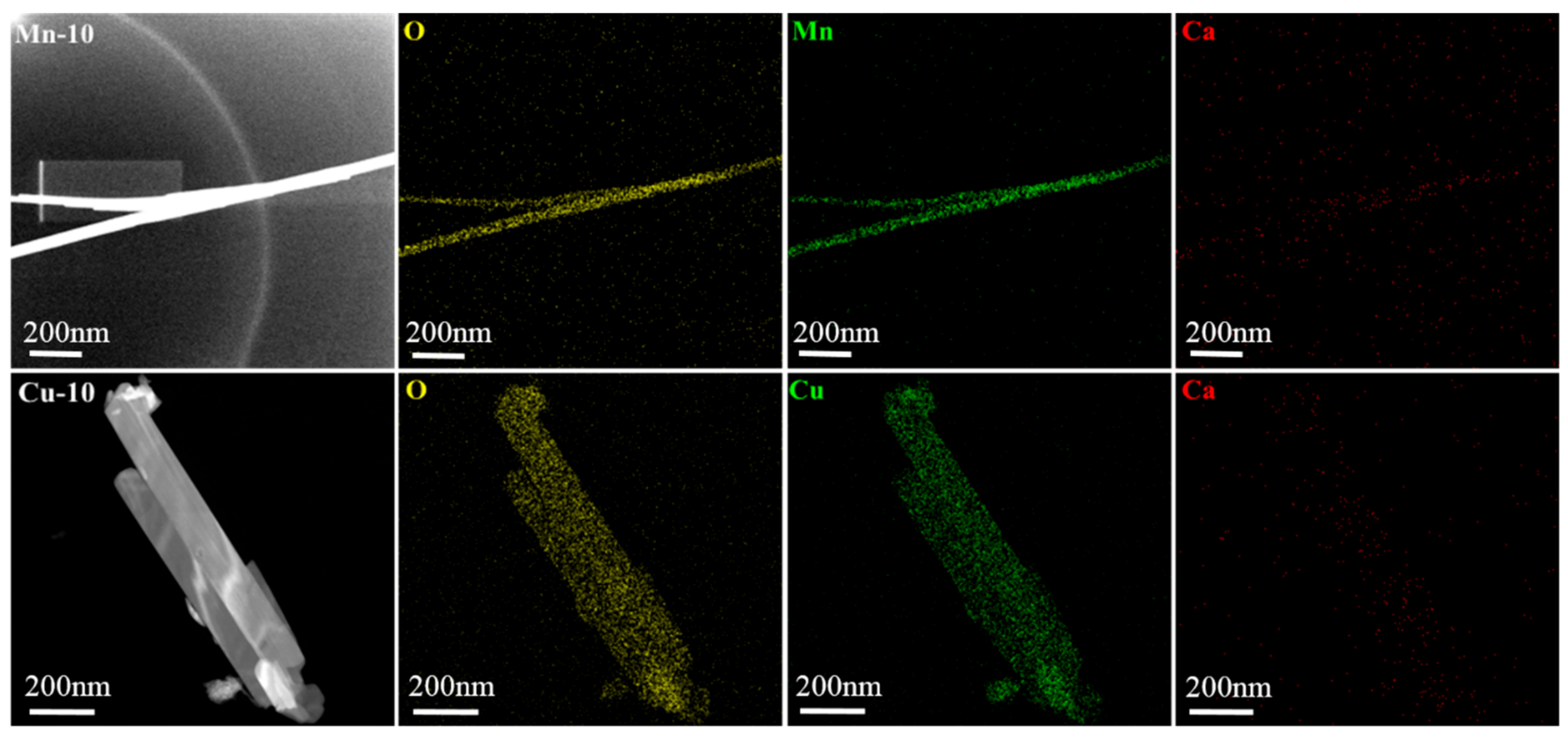
3.3. Morphology
4. Discussion
- (1)
- Water contacts the solid surface and initiates dissolution.
- (2)
- Release of Ca2+, M2+, and CO32- ions from the crystal surface.
- (3)
- Reprecipitation of the chemicals from the solution back onto the solid surface.
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reeder, R.J. Crystal chemistry of the rhombohedral carbonates. In Carbonates: Mineralogy and Chemistry: Reviews in Mineralogy; Mineralogical Society of America: Chantilly, VA, USA, 1990; Volume 11, pp. 1–48. [Google Scholar]
- Berner, R.A. Kinetics of weathering and diagenesis. In Kinetics of Geochemical Processes: Reviews in Mineralogy and Geochemistry; Mineralogical Society of America: Chantilly, VA, USA, 1981; Volume 8, pp. 111–134. [Google Scholar]
- Plummer, L.N.; Wigley, T.M.L.; Parkhurst, D.L. The kinetics of calcite dissolution in CO2-water systems at 5 °C to 60 °C and 0.0 to 1.0 atm CO2. Am. J. Sci. 1978, 278, 179–216. [Google Scholar] [CrossRef]
- Plummer, L.N.; Busenberg, E. The solubilities of calcite, aragonite and vaterite in CO2-H2O solutions between 0 and 90 °C, and an evaluation of the aqueous model for the system CaCO3-CO2-H2O. Geochim. Cosmochim. Acta 1982, 46, 1011–1040. [Google Scholar] [CrossRef]
- Gong, Q.; Deng, J.; Wang, Q.; Yang, L.; She, M. Calcite dissolution in deionized water from 50 °C to 250 °C at 10 MPa: Rate Equation and Reaction Order. Acta Geol. Sin. 2008, 82, 994–1001. [Google Scholar] [CrossRef]
- Alkattan, M.; Oelkers, E.H.; Dandurand, J.-L.; Schott, J. An experimental study of calcite and limestone dissolution rates as a function of pH from −1 to 3 and temperature from 25 °C to 80 °C. Chem. Geol. 1998, 151, 199–214. [Google Scholar] [CrossRef]
- Arvidson, R.S.; Collier, M.; Davis, K.J.; Vinson, M.D.; Amonette, J.E.; Luttge, A. Magnesium inhibition of calcite dissolution kinetics. Geochim. Cosmochim. Acta 2006, 70, 583–594. [Google Scholar] [CrossRef]
- Lea, A.S.; Amonette, J.E.; Baer, D.R.; Liang, Y.; Colton, N.G. Microscopic effects of carbonate, manganese, and strontium ions on calcite dissolution. Geochim. Cosmochim. Acta 2001, 65, 369–379. [Google Scholar] [CrossRef]
- Cubillas, P.; Köhler, S.; Prieto, M.; Causserand, C.; Oelkers, E.H. How do mineral coatings affect dissolution rates? An experimental study of coupled CaCO3 dissolution-CdCO3 precipitation. Geochim. Cosmochim. Acta 2005, 69, 5459–5476. [Google Scholar] [CrossRef]
- Hazen, R.M.; Downs, R.T.; Jones, A.P.; Kah, L. Carbon Mineralogy and Crystal Chemistry. In Carbon in Earth: Reviews in Mineralogy and Geochemistry; Mineralogical Society of America: Chantilly, VA, USA, 2013; Volume 75, pp. 7–46. [Google Scholar]
- Elzinga, E.J.; Rouff, A.A.; Reeder, R.J. The long-term fate of Cu2+, Zn2+, and Pb2+ adsorption complexes at the calcite surface: An X-ray absorption spectroscopy study. Geochim. Cosmochim. Acta 2006, 70, 2715–2725. [Google Scholar] [CrossRef]
- Tang, Y.; Elzinga, E.J.; Jae Lee, Y.; Reeder, R.J. Coprecipitation of chromate with calcite: Batch experiments and X-ray absorption spectroscopy. Geochim. Cosmochim. Acta 2007, 71, 1480–1493. [Google Scholar] [CrossRef]
- Lakshtanov, L.Z.; Stipp, S.L.S. Experimental study of nickel (II) interaction with calcite: Adsorption and coprecipitation. Geochim. Cosmochim. Acta 2007, 71, 3686–3697. [Google Scholar] [CrossRef]
- Curti, E. Coprecipitation of radionuclides with calcite: Estimation of partition coefficients based on a review of laboratory investigations and geochemical data. Appl. Geochem. 1999, 14, 433–445. [Google Scholar] [CrossRef]
- Prieto, M. Thermodynamics of Solid Solution-Aqueous Solution Systems. Rev. Mineral. Geochem. 2009, 70, 47–85. [Google Scholar] [CrossRef]
- Böttcher, M. Experimental dissolution of CaCO3-MnCO3 solid solutions in CO2-H2O solutions at 20°C: I. Synthetic low-temperature carbonates. Solid State Ionics 1997, 101–103, 1263–1266. [Google Scholar] [CrossRef]
- Harstad, A.O.; Stipp, S.L.S. Calcite dissolution: Effects of trace cations naturally present in Iceland spar calcites. Geochim. Cosmochim. Acta 2007, 71, 56–70. [Google Scholar] [CrossRef]
- Katsikopoulos, D.; Fernández-González, Á.; Prieto, M. Precipitation and mixing properties of the “disordered” (Mn,Ca)CO3 solid solution. Geochim. Cosmochim. Acta 2009, 73, 6147–6161. [Google Scholar] [CrossRef]
- Salem, M.R.; Mangood, A.H.; Hamdona, S.K. Dissolution of calcite crystals in the presence of some metal ions. J. Mater. Sci. 1994, 29, 6463–6467. [Google Scholar] [CrossRef]
- Gutjahr, A.; Dabringhaus, H.; Lacmann, R. Studies of the growth and dissolution kinetics of the CaCO3 polymorphs calcite and aragonite II. The influence of divalent cation additives on the growth and dissolution rates. J. Cryst. Growth 1996, 158, 310–315. [Google Scholar] [CrossRef]
- Arvidson, R.S.; Ertan, I.E.; Amonette, J.E.; Luttge, A. Variation in calcite dissolution rates: A fundamental problem? Geochim. Cosmochim. Acta 2003, 67, 1623–1634. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Astilleros, J.M.; Pina, C.M. The morphology of calcite crystals grown in a porous medium doped with divalent cations. Chem. Geol. 2006, 225, 314–321. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Pina, C.M.; Fernndez-Daz, L.; Putnis, A. Molecular-scale surface processes during the growth of calcite in the presence of manganese. Geochim. Cosmochim. Acta 2002, 66, 3177–3189. [Google Scholar] [CrossRef]
- Davis, K.J.; Dove, P.M.; Wasylenki, L.E.; De Yoreo, J.J. Morphological consequences of differential Mg2+ incorporation at structurally distinct steps on calcite. Am. Mineral. 2004, 89, 714–720. [Google Scholar] [CrossRef]
- Putnis, C.V.; Ruiz-Agudo, E.; Hövelmann, J. Coupled fluctuations in element release during dolomite dissolution. Mineral. Mag. 2014, 78, 1355–1362. [Google Scholar] [CrossRef]
- Walton, A.G. The formation and properties of precipitates. In Chemical Analysis; Interscience Publishers: New York, NY, USA, 1967; Volume 23, pp. 1–232. [Google Scholar]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; USGS, Water Resource Investigations Report 99-4259; U.S. Geological Survey, Earth Science Information Center, Open-File Reports Section: Reston, VA, USA, 1999. [Google Scholar]
- McBride, M.B.; Bouldin, D.R. Long-term reactions of copper (II) in a contaminated calcareous soil. Soil Sci. Soc. Am. J. 1984, 48, 56–59. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | M2+/Ca2+ Ratio (in Mole) | BET Specific Surface Area (m2/g) | First Time of New Precipitation Discovery (h) | |
|---|---|---|---|---|
| Added | Measured | |||
| CAL | / | / | 0.173 | / |
| Mn-02 | 0.02 | 0.02 | 0.221 | 1920 |
| Mn-04 | 0.04 | 0.04 | 0.583 | 288 |
| Mn-10 | 0.10 | 0.12 | 3.269 | 96 |
| Cu-02 | 0.02 | 0.02 | 0.389 | 672 |
| Cu-04 | 0.04 | 0.04 | 0.577 | / |
| Cu-10 | 0.10 | 0.12 | 1.953 | 48 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wu, S.; Chen, F. Nano Precipitates Formed during the Dissolution of Calcite Incorporated with Cu and Mn. Minerals 2018, 8, 484. https://doi.org/10.3390/min8110484
Zhang X, Wu S, Chen F. Nano Precipitates Formed during the Dissolution of Calcite Incorporated with Cu and Mn. Minerals. 2018; 8(11):484. https://doi.org/10.3390/min8110484
Chicago/Turabian StyleZhang, Xiaohang, Shijun Wu, and Fanrong Chen. 2018. "Nano Precipitates Formed during the Dissolution of Calcite Incorporated with Cu and Mn" Minerals 8, no. 11: 484. https://doi.org/10.3390/min8110484
APA StyleZhang, X., Wu, S., & Chen, F. (2018). Nano Precipitates Formed during the Dissolution of Calcite Incorporated with Cu and Mn. Minerals, 8(11), 484. https://doi.org/10.3390/min8110484