[Au(CN)2]—Adsorption on a Graphite (0001) Surface: A First Principles Study

Abstract
1. Introduction
2. Model and Calculations
2.1. Computational Method
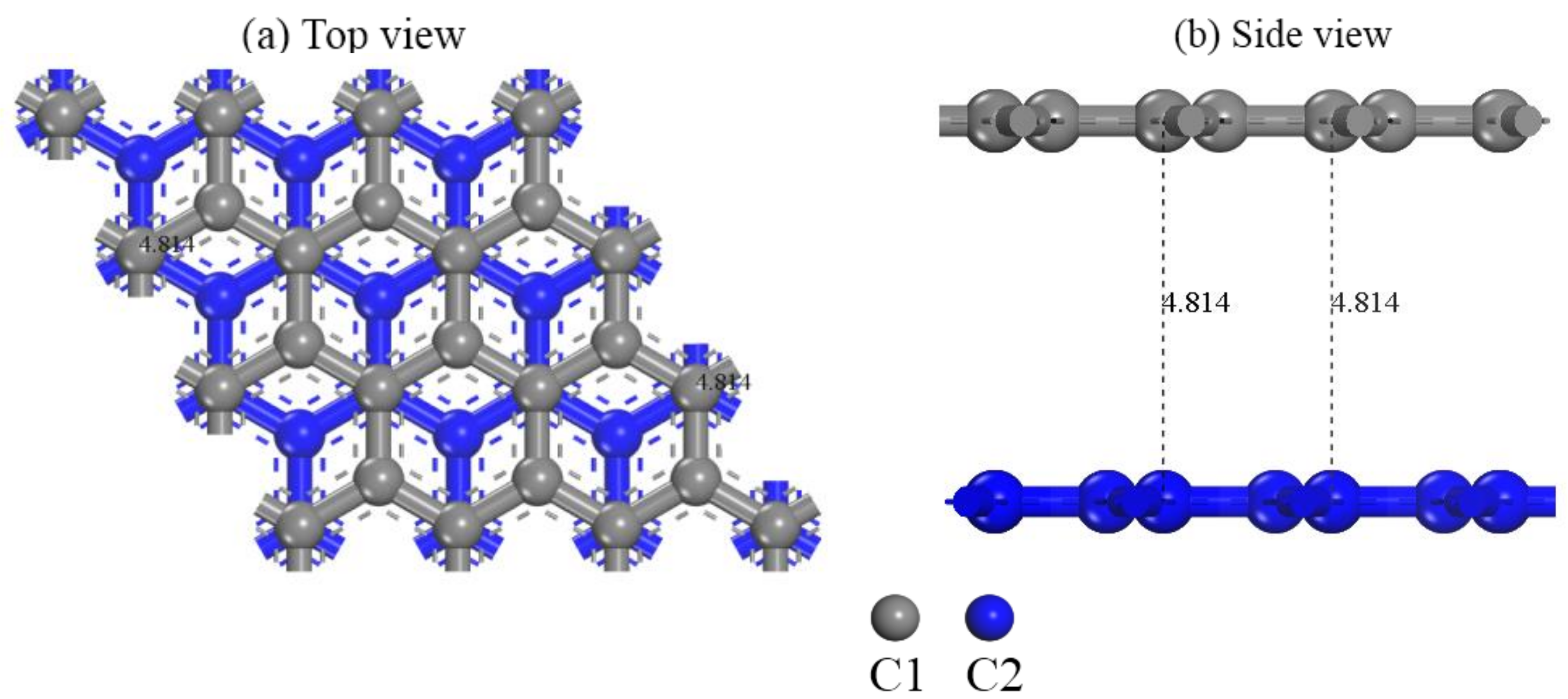
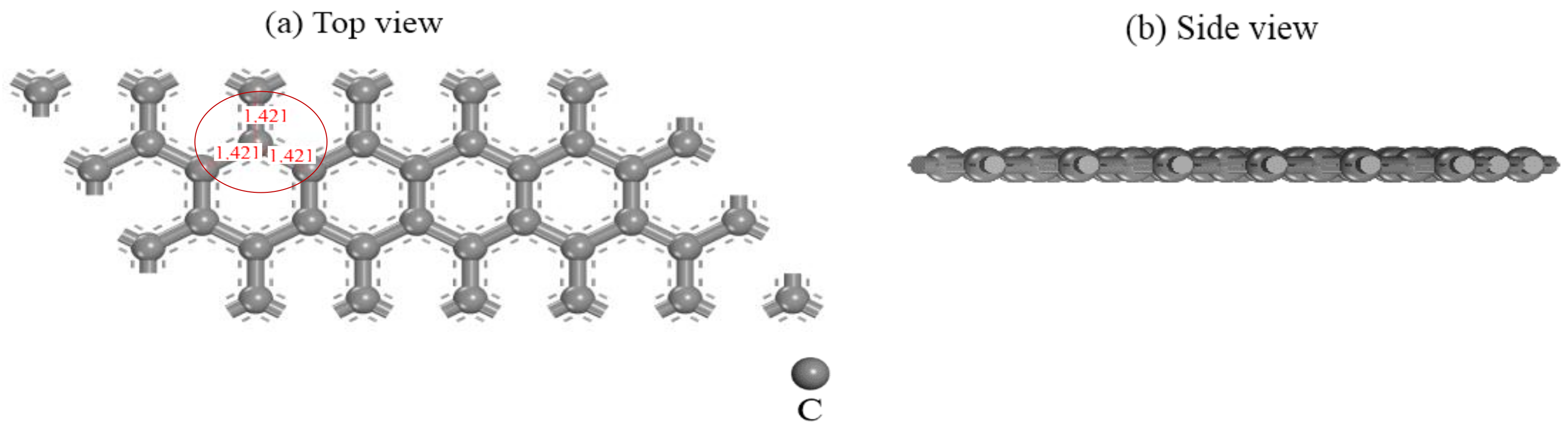
2.2. Computational Models
3. Results and Discussion
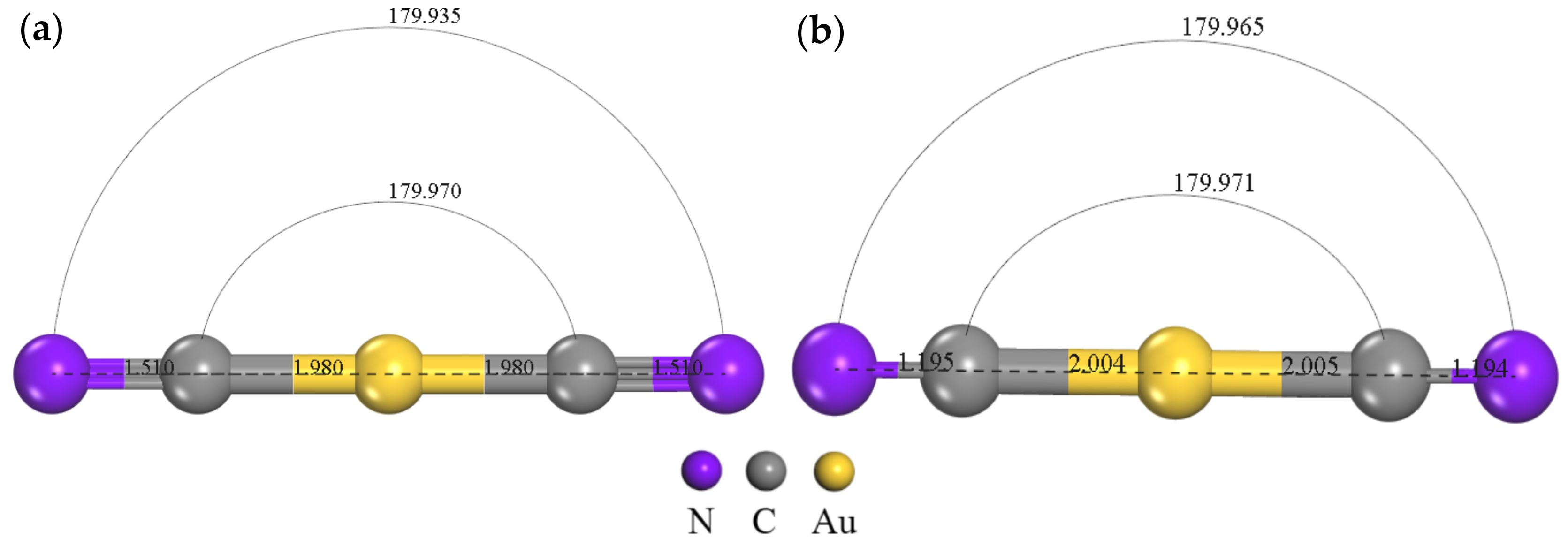
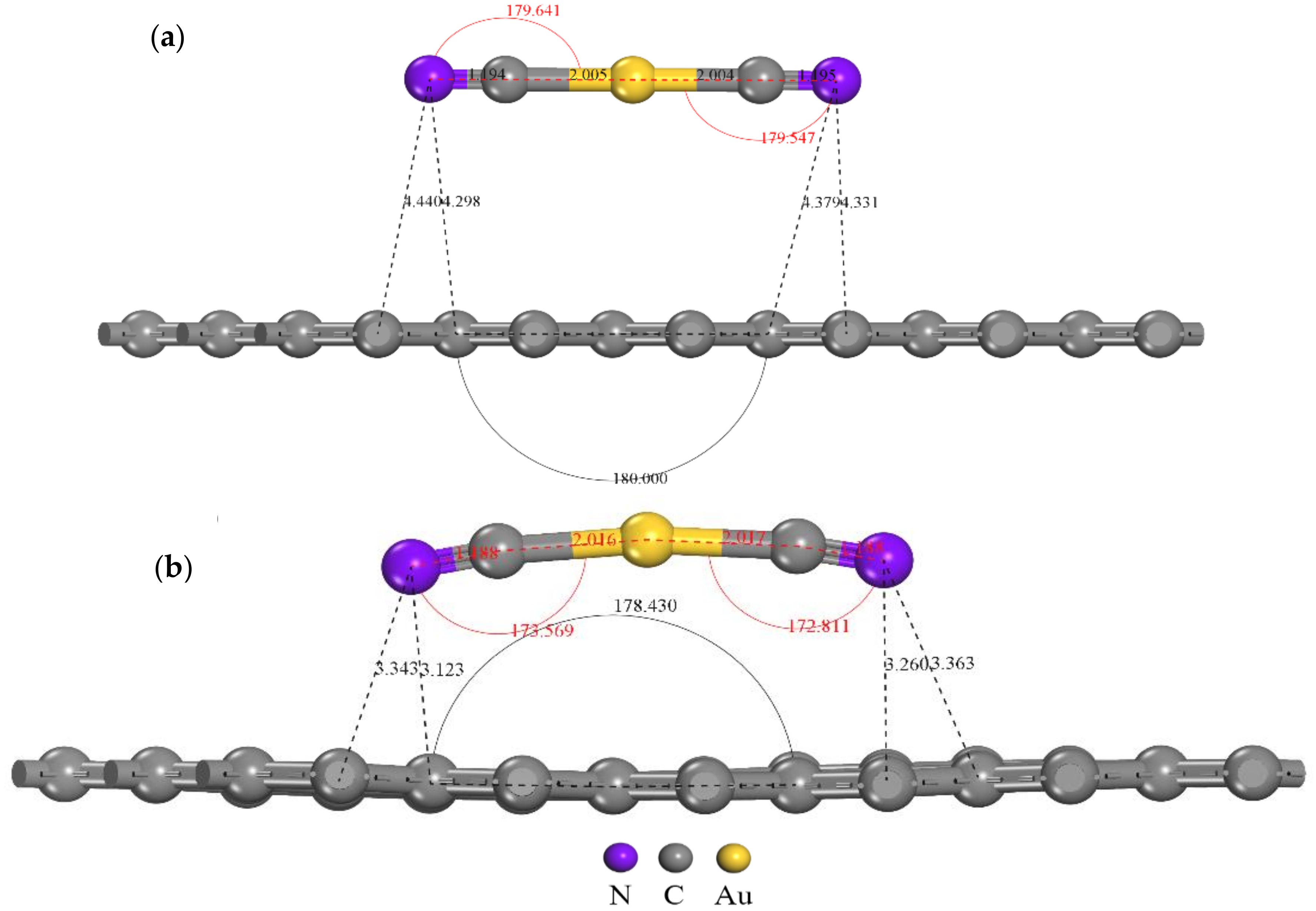
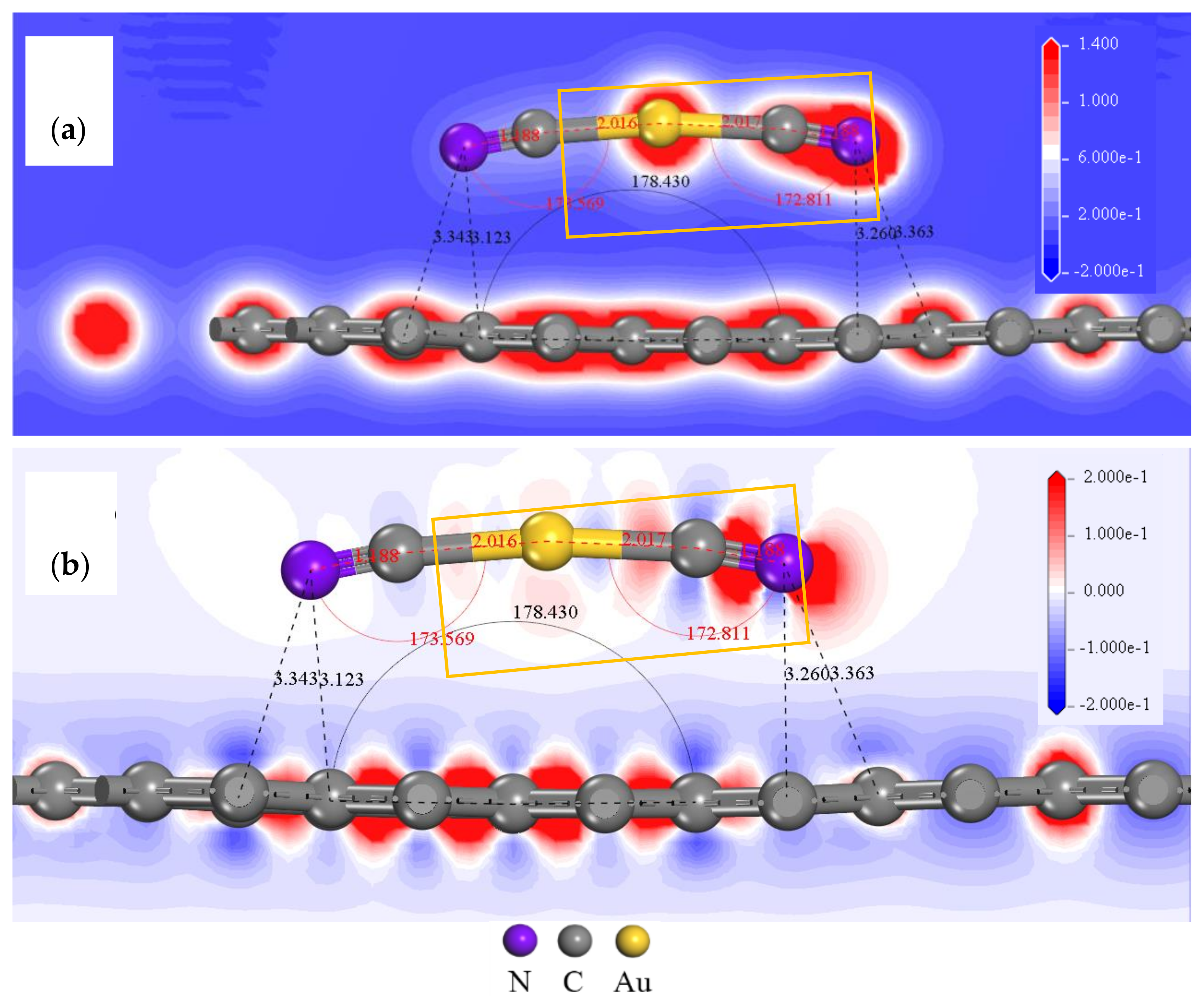
3.1. Adsorption Configuration Analysis of [Au(CN)2]− on Graphite (0001) Surface
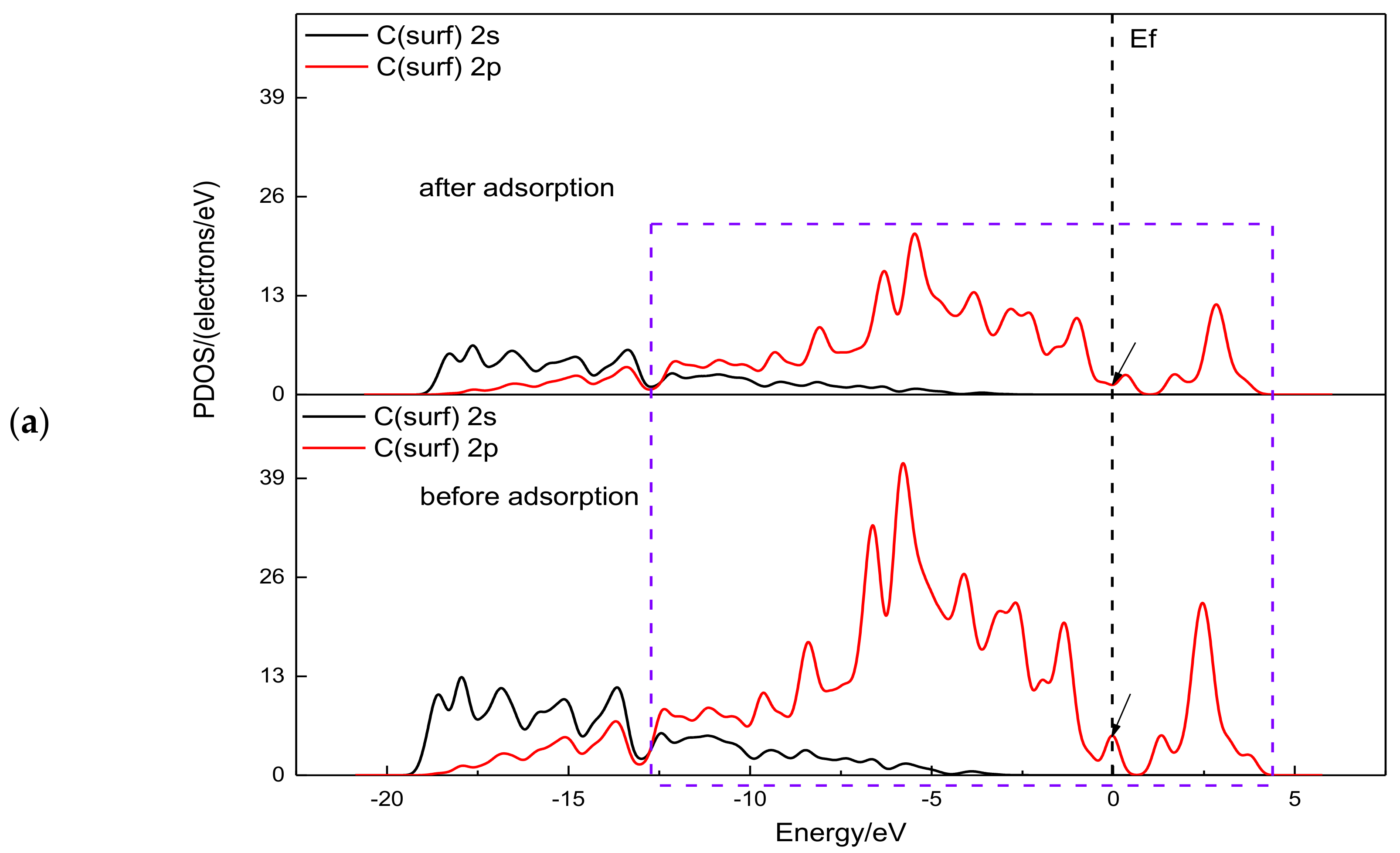
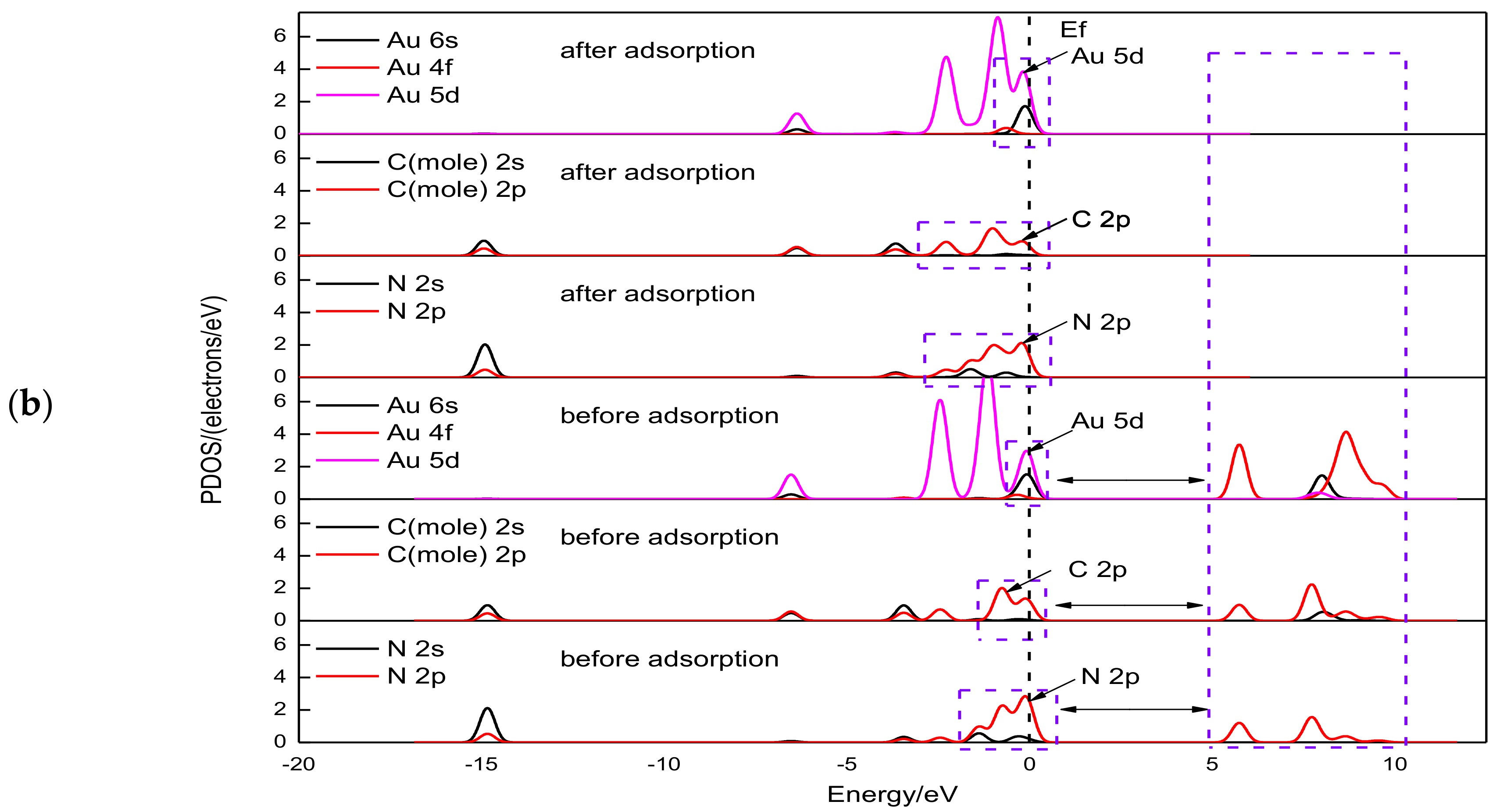
3.2. Electronic Structure Analysis of Gold Cyanide on Graphite (0001) Surface
3.3. Mulliken Population Analysis of [Au(CN)2]− on the Graphite (0001) Surface
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xie, Z.; Xia, Y.; Cline, J.S.; Yan, B.; Wang, Z.; Tan, Q.; Wei, D. Comparison of the native antimony-bearing Paiting gold deposit, Guizhou Province, China, with Carlin-type gold deposits, Nevada, USA. Miner. Depos. 2017, 52, 69–84. [Google Scholar] [CrossRef]
- Muntean, J.L.; Cline, J.S.; Simon, A.C.; Longo, A.A. Magmatic–hydrothermal origin of Nevada’s Carlin-type gold deposits. Nat. Geosci. 2011, 4, 122–127. [Google Scholar] [CrossRef]
- Gómez-Díaz, J.; Honkala, K.; López, N. A Density Functional Theory study on gold cyanide interactions: The fundamentals of ore cleaning. Surf. Sci. 2010, 604, 1552–1557. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Q.; Song, X.; Dong, J. Research status of carbonaceous matter in carbonaceous gold ores and bio-oxidation pretreatment. Trans. Nonferrous Met. Soc. China 2013, 23, 3405–3411. [Google Scholar] [CrossRef]
- Tan, H.; Lukey, G.C.; van Devebter, J.S.J. The behaviour of carbonaceous matter in cyanide leaching of gold. Hydrometallurgy 2005, 78, 226–235. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Shen, Z. Study of the Separation of Carbonaceous Matter in Micro-grained Gold Ore in Guizhou Province. Nonferrous Met. Miner. Process. Sect. 2016, 3, 33–37. [Google Scholar]
- Razvozzhaeva, E.A.; Nemerov, V.K.; Spiridonov, A.M.; Prokopchuk, S.I. Carbonaceous substance of the Sukhoi Log gold deposit (East Siberia). Russ. Geol. Geophys. 2008, 49, 371–377. [Google Scholar] [CrossRef]
- Su, W.; Hu, R.; Xia, B.; Liu, Y. Calcite Sm-Nd isochron age of the Shuiyindong Carlin-type gold deposit, Guizhou, China. Chem. Geol. 2009, 258, 269–274. [Google Scholar] [CrossRef]
- Konadu, K.T.; Sasaki, K.; Kaneta, T.; Ofori-Sarpong, G.; Osseo-Asare, K. Bio-modification of carbonaceous matter in gold ores: Model experiments using powdered activated carbon and cell-free spent medium of Phanerochaete chrysosporium. Hydrometallurgy 2017, 168, 76–83. [Google Scholar] [CrossRef]
- Helm, M.M.; Vaughan, J.P.; Staunton, W.P. Evaluation of preg-robbing in goldstrike carbonaceous ore using raman spectroscopy. In Proceedings of the 50th Annual Conference of Metallurgists of CIM, Montreal, QC, Canada, 2–5 October 2011; pp. 595–606. [Google Scholar]
- Ofori-Sarpong, G.; Osseo-Asare, K. Preg-robbing of gold from cyanide and non-cyanide complexes: Effect of fungi pretreatment of carbonaceous matter. Int. J. Miner. Process. 2013, 119, 27–33. [Google Scholar] [CrossRef]
- Senanayake, G. Gold leaching in non-cyanide lixiviant systems: Critical issues on fundamentals and applications. Miner. Eng. 2004, 17, 785–801. [Google Scholar] [CrossRef]
- Zhang, H.; Ritchie, I.M.; La Brooy, S.R. The adsorption of gold thiourea complex onto activated carbon. Hydrometallurgy 2004, 72, 291–301. [Google Scholar] [CrossRef]
- Muir, D.M.; Aylmore, M.G. Thiosulphate as an alternative to cyanide for gold processing—Issues and impediments. Miner. Process. Extr. Metall. 2004, 113, 2–12. [Google Scholar] [CrossRef]
- Raphulu, M.C.; Scurrell, M.S. Cyanide leaching of gold catalysts. Catal. Commun. 2015, 67, 87–89. [Google Scholar] [CrossRef]
- Bas, A.D.; Safizadeh, F.; Zhang, W.; Ghali, E.; Yeonuk, C.H.O.I. Active and passive behaviors of gold in cyanide solutions. Trans. Nonferrous Met. Soc. China 2015, 25, 3442–3453. [Google Scholar] [CrossRef]
- Kianinia, Y.; Reza Khalesi, M.; Abdollahy, M.; Hefter, G.; Senanayake, G.; Hnedkovsky, L.; Darban, A.K.; Shahbazi, M. Predicting Cyanide Consumption in Gold Leaching: A Kinetic and Thermodynamic Modeling Approach. Minerals 2018, 8, 110. [Google Scholar] [CrossRef]
- Sha, X.; Jackson, B. First-principles study of the structural and energetic properties of H atoms on a graphite (0001) surface. Surf. Sci. 2002, 496, 318–330. [Google Scholar] [CrossRef]
- Perea-Ramírez, L.I.; Vargas, R.; Domínguez, Z.; Salas-Reyes, M.; Matus, M.H.; Galván, M. Theoretical study of the adsorption of substituted guaiacol and catechol radicals on a graphite surface. Electrochim. Acta 2017, 242, 66–72. [Google Scholar] [CrossRef]
- Man-Chao, H.; Jian, A.Z. Methane adsorption on graphite (0001) films: A first-principles study. Chin. Phys. B 2013, 22, 452–456. [Google Scholar]
- Han, Y.; Liu, W.; Chen, J. DFT simulation of the adsorption of sodium silicate species on kaolinite surfaces. Appl. Surf. Sci. 2016, 370, 403–409. [Google Scholar] [CrossRef]
- Xie, J.; Li, X.; Mao, S.; Li, L.; Ke, B.; Zhang, Q. Effects of structure of fatty acid collectors on the adsorption of fluorapatite (001) surface: A first-principles calculations. Appl. Surf. Sci. 2018, 444, 699–709. [Google Scholar] [CrossRef]
- Han, Y.; Liu, W.; Chen, J.; Han, Y. Adsorption mechanism of hydroxyl calcium on two kaolinite (001) surface. J. China Coal Soc. 2016, 41, 743–750. [Google Scholar]
- Sun, S.P.; Gu, S.; Sun, J.H.; Xia, F.F.; Chen, G.H. First principles investigation of the electronic properties of graphitic carbon nitride with different building block and sheet staggered arrangement. J. Alloys Compd. 2018, 735, 131–139. [Google Scholar] [CrossRef]
- Ancilotto, F.; Toigo, F. First-principles study of potassium adsorption on graphite. Phys. Rev. B Condens. Matter 1993, 47, 13713–13721. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Opara, A.; Du, H.; Miller, J.D. Molecular dynamics simulations of metal–cyanide complexes: Fundamental considerations in gold hydrometallurgy. Hydrometallurgy 2011, 106, 64–70. [Google Scholar] [CrossRef]
- Mohammadnejad, S.; Provis, J.L.; van Deventer, J.S.J. Computational modelling of gold complexes using density functional theory. Comput. Theor. Chem. 2015, 1073, 45–54. [Google Scholar] [CrossRef]
- Mohammadnejad, S.; Provis, J.L.; van Deventer, J.S.J. Computational modelling of interactions between gold complexes and silicates. Comput. Theor. Chem. 2017, 1101, 113–121. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, Y.; Zhou, R.; Feng, J. Stability and elastic properties of Y–C binary compounds investigated by first principles calculations. J. Alloys Compd. 2014, 587, 819–826. [Google Scholar] [CrossRef]
- Liu, Y.; Xing, J.; Li, Y.; Sun, L.; Wang, Y. A first principles study of adhesion and electronic structure at Fe (110)/graphite (0001) interface. Appl. Surf. Sci. 2017, 405, 497–502. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-Principles Simulation: Ideas, Illustrations and the CASTEP Code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Schultz, P.A.; Desjarlais, M.P.; Mattsson, T.R.; Leung, K. Designing meaningful density functional theory calculations in materials science—A primer. Model. Simul. Mater. Sci. Eng. 2005, 13, R1–R31. [Google Scholar] [CrossRef]
- Wu, T.; Cao, D.; Wang, X.; Jiao, Z.; Chen, M.; Luo, H.; Zhu, P. First principle calculations of hexyl thiolate monolayer on Au (1 1 1). Appl. Surf. Sci. 2015, 330, 158–163. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Q.; Li, X.; Ye, J.; Li, L. Structural and Electronic Properties of Different Terminations for Quartz (001) Surfaces as Well as Water Molecule Adsorption on It: A First-Principles Study. Minerals 2018, 8, 58. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Rafailov, P.M.; Maultzsch, J.; Thomsen, C.; Dettlaff-Weglikowska, U.; Roth, S. Kohn Anomaly and Electron–Phonon Interaction at the K-Derived Point of the Brillouin Zone of Metallic Nanotubes. Nano Lett. 2009, 9, 3343–3348. [Google Scholar] [CrossRef] [PubMed]
- Ireta, J.; Neugebauer, J.; Scheffler, M. On the Accuracy of DFT for Describing Hydrogen Bonds: Dependence on the Bond Directionality. J. Phys. Chem. A 2004, 108, 5692–5698. [Google Scholar] [CrossRef]
- Perdew, J.P. Generalized gradient approximations for exchange and correlation: A look backward and forward. Phys. B Condens. Matter 1991, 172, 1–6. [Google Scholar] [CrossRef]
- Ortmann, F.; Bechstedt, F.; Schmidt, W.G. Semiempirical van der Waals correction to the density functional description of solids and molecular structures. Phys. Rev. B 2006, 73, 205101. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Luo, X.; Fang, C.; Li, X.; Lai, W.; Liang, T. Adsorption behaviors of Cs and I atoms on the graphite surface by the first-principles. J. Nucl. Mater. 2013, 441, 113–118. [Google Scholar] [CrossRef]
- Khosravi, R.; Azizi, A.; Ghaedrahmati, R.; Gupta, V.K.; Agarwal, S. Adsorption of gold from cyanide leaching solution onto activated carbon originating from coconut shell—Optimization, kinetics and equilibrium studies. J. Ind. Eng. Chem. 2017, 54, 464–471. [Google Scholar] [CrossRef]
- Kondos, P.D.; Deschênes, G.; Morrison, R.M. Process optimization studies in gold cyanidation. Hydrometallurgy 1995, 39, 235–250. [Google Scholar] [CrossRef]
- Miller, D.J.; Hong, H.L. An experimental method for determining heat of physical adsorption. J. Catal. 1983, 81, 281–290. [Google Scholar] [CrossRef]
- Levita, G.; Kajita, S.; Righi, M.C. Water adsorption on diamond (111) surfaces: An ab initio study. Carbon 2018, 127, 533–540. [Google Scholar] [CrossRef]
- Sibrell, P.L.; Miller, J.D. Significance of Graphitic Structural Features in Gold Adsorption by Carbon. Miner. Metall. Process. 1992, 9, 189–195. [Google Scholar]
- Yalcin, M.; Arol, A.I. Gold cyanide adsorption characteristics of activated carbon of non-coconut shell origin. Hydrometallurgy 2002, 63, 201–206. [Google Scholar] [CrossRef]
- Wei, H. Studies on the Gold Adsorption of the Coal Based Activated Carbon in the Contained CN Solution. Master’s Thesis, Beijing University of Technology, Beijing, China, June 2010; pp. 1–65. [Google Scholar]
- Li, S.; Yao, J.; Lin, D. Graphite; Chemical Industry Press: Beijing, China, 1991. [Google Scholar]
- Xiang, H.U.; Duan, P.; Song, J.; Li, W.; Chen, L.; Bian, X. Study on the influences of ionization region material arrangement on Hall thruster channel discharge characteristics. Plasma Sci. Technol. 2018, 20, 024008. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements | Atomic Orbitals |
|---|---|
| H | 1s1 |
| C | 2s22p2 |
| N | 2s22p3 |
| Au | 5d106s1 |
| Adsorption Model | Bond | Adsorption Status | Population | Length (Å) |
|---|---|---|---|---|
| Gra–[Au(CN)2]− | C–N | Before | 1.61 | 1.194 |
| After | 1.71 | 1.187 | ||
| C–Au | Before | 0.46 | 2.004 | |
| After | 0.52 | 2.016 | ||
| C(surf)–C(surf) | Before | 1.08 | 1.420 | |
| After | (1.06~1.11) | (1.413~1.424) |
| Adsorption Model | Atom Name | Adsorption Status | Valence Electrons Number | Charge (e) | ||||
|---|---|---|---|---|---|---|---|---|
| s | p | d | f | Total | ||||
| Gra–[Au(CN)2]− | Au | Before | 0.87 | 0.00 | 9.42 | 0.08 | 10.37 | 0.63 |
| After | 1.04 | 0.00 | 9.56 | 0.03 | 10.63 | 0.37 | ||
| N | Before | 1.74 | 3.53 | 0.00 | 0.00 | 5.27 | –0.26 | |
| After | 1.72 | 3.73 | 0.00 | 0.00 | 5.45 | –0.46 | ||
| C | Before | 1.29 | 2.77 | 0.00 | 0.00 | 4.06 | −0.05 | |
| After | 1.28 | 2.81 | 0.00 | 0.00 | 4.09 | –0.09 | ||
| C(surf) | Before | 1.05 | 2.95 | 0.00 | 0.00 | 4.00 | 0.00 | |
| After | 1.05 | 2.92 | 0.00 | 0.00 | 3.97 | 0.03 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhang, Q.; Xie, J.; Shen, Z. [Au(CN)2]—Adsorption on a Graphite (0001) Surface: A First Principles Study. Minerals 2018, 8, 425. https://doi.org/10.3390/min8100425
Li X, Zhang Q, Xie J, Shen Z. [Au(CN)2]—Adsorption on a Graphite (0001) Surface: A First Principles Study. Minerals. 2018; 8(10):425. https://doi.org/10.3390/min8100425
Chicago/Turabian StyleLi, Xianhai, Qin Zhang, Jun Xie, and Zhihui Shen. 2018. "[Au(CN)2]—Adsorption on a Graphite (0001) Surface: A First Principles Study" Minerals 8, no. 10: 425. https://doi.org/10.3390/min8100425
APA StyleLi, X., Zhang, Q., Xie, J., & Shen, Z. (2018). [Au(CN)2]—Adsorption on a Graphite (0001) Surface: A First Principles Study. Minerals, 8(10), 425. https://doi.org/10.3390/min8100425