Abstract
Spinel-structured hausmannite (Mn(II)Mn(III)2O4) is a vital intermediate in Mn mineralogy and a key player in redox chemistry in the environment. Its transformation into other Mn oxides is a critical factor in controlling its environmental occurrence and reactivity. Yet structural impurities and solution pH, as well as the fate of impurities during transformation, which influence hausmannite transformation processes and products, remain largely unknown. In the present work, we address this knowledge gap by investigating pristine and metal-substituted hausmannite, specifically nickel (Ni) or cobalt (Co), equilibrated at two time periods (8 h and 30 days) and three different pH levels (4, 5, and 7). Solution chemistry data revealed that both the equilibration period and pH had a significant impact on hausmannite dissolution rates and the concomitant repartitioning of Ni or Co. Hausmannite with Ni or Co substitution exhibited lower dissolution rates than pristine mineral under acidic conditions. Mineralogy and crystal chemistry data indicated that hausmannite was the major host phase after 30-day equilibration, followed by minor transformed products, including birnessite and manganite. Although minor, birnessite became more abundant than manganite at low pHs. Analytical high-resolution transmission electron microscopy (HRTEM) analyses revealed a poorly crystalline, nano-scaled MnO2 formed from hausmannite and the majority of metal impurities remaining in the host hausmannite. Yet Co was associated with both hausmannite and the newly formed birnessite, whereas Ni was only found with hausmannite, indicating the strong sequestration of Co by Mn(II/III) and Mn(IV) mineral phases. This study highlights the significant impacts of metal impurities and pH on the stability of hausmannite and its transformation into birnessite, as well as the control of Mn-oxide minerals on the solubility and sequestration of transition metals in the environment.
Keywords:
hausmannite; transformation; metal substitution; nickel; cobalt; LCF; XAS; analytical HRTEM 1. Introduction
Widespread occurrences of manganese (Mn) oxides are evident in various environments on the Earth’s surface. These range from Mn-enriched ore deposits to Mn modules found in the oceans and freshwater lakes, as well as Mn nanoparticulates or coatings of minerals, rocks, and soil/sediment particles [1,2,3,4,5,6,7,8,9,10,11,12]. Mn oxides are diverse in both phase and structure and commonly occur as mixed valence states (Mn(II/III/IV)) in layered, tunneled, or spinel structures [10,13]. Such variability in atomic structures and crystal chemistries makes Mn oxides highly reactive, particularly in terms of adsorption, oxidation–reduction, and cation-exchange reactions with heavy metals, metalloids, and trace elements in soils, sediments, and associated aqueous systems [14,15,16,17,18,19,20,21,22,23]. Thus, Mn oxides exert a considerable influence on the distribution, mobility, and environmental fate of inorganic nutrients and contaminants of concern in various geological settings.
There are more than 30 Mn oxide/hydroxide minerals identified on Earth’s surface [10], with hausmannite, Mn(II)Mn(III)2O4, being the fifth most common Mn oxide in soils and sediments [19] and occurring in both nano and bulk phases [24]. Hausmannite has a spinel structure comprising mixed valence states (Mn(II/III)), where Mn(II) occupies the tetrahedral sites and Mn(III) the octahedral sites [10]. Spinel-structured hausmannite can also host a range of transition metals [15,18,25,26]. For instance, hausmannite incorporates divalent nickel (Ni) in the octahedral sites, divalent zinc (Zn) in the tetrahedral sites, and divalent cobalt (Co) in the tetrahedral and trivalent Co in the octahedral sites [25,27,28]. While the extent differs by the type and concentration of metal substituent, changes in hausmannite chemistries profoundly affect the mineral’s oxidation reactivity toward arsenite, As(III) [27,28]. In addition, hausmannite is generally metastable. This intermediate mineral transforms into manganite (γ-Mn(III)OOH) and/or pyrolusite/birnessite (Mn(IV)O2), with the solution pH being crucial in determining the transformation pathways and products [7,29,30,31,32,33]. For instance, under circumneutral conditions, the hausmannite to manganite transformation is preferred [32], whereas under acidic conditions, hausmannite transformation to Mn(IV) oxides is favored [7,34]. Reverse transformations of birnessite to hausmannite may occur in the presence of Mn2+ ions, with these reductive transformation reactions and products also being highly dependent on solution pH [35].
While the abundance and importance of hausmannite in Mn mineralogy and geochemical processes in the environment have been generally recognized, the impacts of structural impurities and solution pH on the hausmannite-to-birnessite transformation are poorly understood. Further, the fate of structural impurities during the transformation process remains largely unknown. Conversely, the effect of structural impurities on birnessite transformation processes and key parameters associated with transformations have been elucidated [36,37,38]. Thus, the present study aims to investigate the effects of structural impurities and solution pH on the transformation of hausmannite to birnessite, as well as the solubility and fate of structural Mn and metal impurities during this process. To this end, we used freshly synthesized, pristine, and trace-level Ni- or Co-substituted hausmannite minerals (at 2 wt%). We equilibrated them at two different equilibration periods (8 h and 30 days) at three pH values (4, 5, and 7). The results of the present work demonstrate the importance and contribution of metal impurities to the temporal stability of hausmannite and its transformation to birnessite, thereby improving our understanding of the vital role of hausmannite in Mn mineralogy and the control of the solubility or sequestration of associated transition metals in the environment.
2. Materials and Methods
2.1. Materials
All chemical agents used in the present study were ACS-grade or better, including manganese acetate tetrahydrate (Mn(CH3COO)2·4H2O, 99 +%, Acros Organics, Waltham MA, USA), nickel acetate tetrahydrate (Ni(CH3COO)2·4H2O, 99 +%, Acros Organics, Waltham, MA, USA), cobalt acetate tetrahydrate (Co(CH3COO)2·4H2O, 99 +%, Acros Organics, Waltham, MA, USA), acetone (C3H6O, ≥99.5%, Thermo Fisher Chemicals, Waltham, MA, USA), ethyl alcohol (C2H5OH, 200 proof, Pharmco-Aaper, Brookfield, CT, USA), sodium chloride (NaCl, ≥99%, crystalline, certified ACS, Fisher Scientific, Pittsburgh, PA, USA), nitric acid (HNO3, 67 to 70% (w/w), trace metal-grade, Fisher Chemical, Fair Lawn, NJ, USA), hydrochloric acid (HCl, 36.5 to 38% (w/w), certified ACS plus, Fisher Chemical, Fair Lawn, NJ, USA), and sodium hydroxide (NaOH, ≥97.0%, pellet, certified ACS, Fisher Chemical, Fair Lawn, NJ, USA). Mn, Ni, and Co standard solutions for the inductively coupled plasma–optical emission spectroscopy (ICP-OES) analysis were obtained from J.T. Baker® (1000 μg/mL (±0.10% w/v) in HNO3 (<3% w/w), reagent-grade for trace metal analysis).
2.2. Mineral Synthesis and Preparation
Pristine and Ni- or Co-substituted hausmannite minerals at 2 wt%–3 wt% were synthesized, and their purity was confirmed using methods described in our earlier studies [27,28,39]. When analyzing the synthetic Ni- or Co-substituted hausmannite minerals using the methods employed in [27,28,39], 2.3 wt% Ni and 2.8 wt% Co were found to be included in these minerals.
A series of mineral suspension samples were prepared by placing 1.0 g L−1 of pristine or Ni- or Co-substituted hausmannite in a centrifuge tube filled with a background electrolyte of 10 mM NaCl solution, followed by titration with dilute HCl or NaOH to reach the target pH of 4, 5, or 7. The resulting suspension samples were allowed to equilibrate for 8 h (t = 8 h) or 30 days (t = 30 days) with agitation under the oxic condition. Batch reactions were run in duplicates. During the equilibration period, the pH of the suspension was regularly checked and manually re-adjusted to maintain the target pH as needed. After equilibration was completed, a small aliquot (~2 mL) of suspension was withdrawn, filtered through a 0.22 μm syringe filter (Millipore, Millex®, Burlington, MA, USA), and the solute acidified with 2% HNO3 for the analysis of total dissolved Mn and Ni (or Co) concentrations by ICP-OES (iCAP 7000, Thermo Scientific, Waltham, MA, USA). The mineral solids were collected by centrifugation, thoroughly rinsed with deionized (DI) water, dried at room temperature, and then stored in a glove bag under N2(g) (Erlab 2200ANM, ErlabTM, Rowley, MA, USA) prior to post-reaction analysis. Hereafter, hausmannite minerals equilibrated for 8 h (t = 8 h) are referred to as reacted hausmannite (R-Haus), reacted Ni-substituted hausmannite (R-NiHaus), and reacted Co-substituted hausmannite (R-CoHaus). Hausmannite minerals equilibrated for 30 days (t = 30 days) are referred to as aged hausmannite (A-Haus), aged Ni-substituted hausmannite (A-NiHaus), and aged Co-substituted hausmannite (A-CoHaus), respectively.
2.3. Mineral Characterization
The mineralogy of all samples was determined using powder X-ray diffraction (PXRD), collected on a Bruker D8 Advance X-ray diffractometer equipped with Ni-filtered Cu Kα radiation and a high-speed energy-dispersive linear detector (LYNEXE). Sample powders were placed on a non-diffracted silicon plate holder and scanned 2θ from 10° to 80° with a step size of 0.01°/s–0.02°/s. Peak identification was achieved using the DIFFRAC.EVA software with the American Mineralogy Crystal Structure Database (AMCSD).
The crystal chemistry of Mn and Ni or Co in the mineral samples was investigated using X-ray absorption spectroscopy (XAS), employing both the X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) energy regions. XAS data were collected at the 6-BM beamline of the National Synchrotron Light Source II (NSLS II), Brookhaven National Lab (BNL (Upton, NY)). Due to the low content of Ni or Co in the hausmannite samples, the Ni and Co K-edge spectra were collected in fluorescence mode at room temperature. In contrast, the Mn K-edge spectra were collected in both transmission and fluorescence modes. A metallic Ni, Co, or Mn foil was run under the same conditions as the mineral samples, serving as a reference for energy calibration during XAS analysis. In addition, a series of reference compounds, including hausmannite (Mn3O4) [27,28,39], manganite [39,40], and birnessite (δ-MnO2) [41], were examined to determine the Mn oxidation state and speciation. These reference compounds were synthesized in the laboratory using the methods described in the cited papers. The XANES spectra of Ni, Co, and Mn were merged, background-corrected, and normalized using the Athena graphical user interface [42]. Linear combination fitting (LCF) of Mn K-edge EXAFS spectra was processed from 3.0 Å−1 to 12.0 Å−1, and the XANES spectra were processed in the range of −20 eV to 30 eV of E° [42].
The morphology, elemental composition, and crystal structure of mineral samples were determined using a JEOL JEM 2100 TEM operating at 200 kV. The microscope was equipped with a Gatan Orius 833 slow-scan CCD camera for recording (high-resolution) images and selected area electron diffraction (SAED) patterns and with an energy-dispersive X-ray (EDX) spectrometer (EDAX r-TEM) for chemical analysis. Where possible, lattice fringes were directly measured by hand using Gatan’s Digital Micrograph and Image J. For TEM sample preparation, approximately 10 mg of powdered sample was added to a centrifuge tube containing 30 mL of DI water (Barnstead, 18.2 MΩ-cm water with 1–5 ppb total organic carbon). The suspension was then sonicated for 10 s using a probe sonicator (Model 505 Sonic Dismembrator, Fisher Scientific, Pittsburgh, PA, USA). A small aliquot of the dispersed suspension was withdrawn, placed onto a 400-mesh copper grid with carbon support films (Electron Microscopy Sciences, Hatfield, PA, USA), and allowed to evaporate at room temperature.
3. Results and Discussion
3.1. Dissolution Reaction at Selected pHs
To detect any changes on pristine and Ni (or Co)-substituted hausmannite minerals in batch reactions of varying equilibration periods and solution pHs, the dissolution rates of these minerals, i.e., the total [Me]aq measured divided by the total equilibration hours, were calculated and compared for two time periods (t = 8 h and t = 30 days) at three pHs, 4, 5, and 7. Here, t = 8 h was chosen based on our earlier investigation with hausmannite minerals [27,28,39], where the release of structural Mn and Ni (or Co) reached a plateau after 8 h of acidic dissolution of hausmannite and Ni (or Co)-substituted hausmannite minerals. t = 30 days was chosen based on the results of previous studies [7,43], where visible color changes of hausmannite suspension were noted after 15 days of equilibration due to the surface oxidation or transformation of hausmannite to a secondary mineral phase. Therefore, in the present work, t = 8 h and t = 30 days serve as an initial and prolonged equilibration period to monitor the mineral transformation processes and are referred to as R (reacted, R-Haus, R-NiHaus, and R-CoHaus) and A (aged, A-Haus, A-NiHaus, and A-CoHaus) systems, respectively.
The [Mn(II)]aq release rates for pristine and Ni (or Co)-substituted hausmannite minerals were compared for different equilibration periods and solution pHs (Figure 1a,b). All R systems produced significantly higher release rates than the A systems, indicating that mineral dissolution rates decreased with increasing equilibration period. Among the R systems, the highest release rates were found at pH 4 (yellow in Figure 1a), followed by pH 5 (blue), and the lowest rate was recorded at pH 7 (pink). This finding is consistent with other studies [7,34,44], where low solution pH accelerated mineral dissolution, and the Mn2+ ion release rate from the hausmannite structure increased with decreasing pH. At lower pHs, where mineral dissolution was favored, pristine hausmannite (R-Haus) released much more [Mn(II)]aq than metal-substituted hausmannite minerals, implying that the presence of structural impurities affects the mineral dissolution rates. By comparison, when mineral dissolution was suppressed by higher pH (e.g., pH 7, pink), all R minerals presented a similar dissolution rate, indicating little to no effect of structural impurities on mineral dissolution. While much lower Mn release rates were observed in the A systems (Figure 1b), the highest rates were found at pH 4 (yellow), followed by pH 5 (blue), similar to the R systems. However, no measurable Mn release was observed at pH 7. Similar to the R systems, pristine hausmannite exhibited a higher Mn release rate than the metal-substituted phases, although the extent was not as substantial as in the R systems.
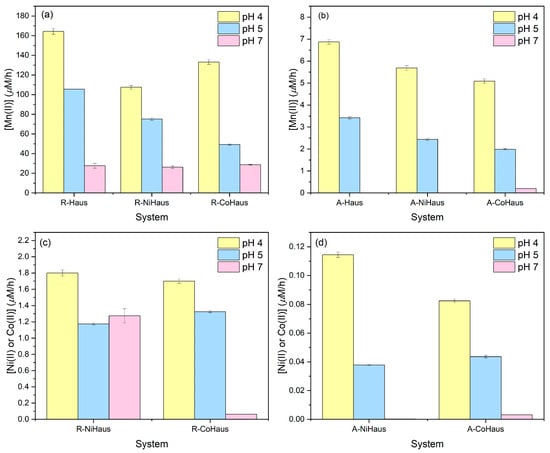
Figure 1.
Release rate of (a) [Mn(II)]aq for 8 h, (b) [Mn(II)]aq for 30 days, (c) [Ni(II)]aq or [Co(II)]aq for 8 h, and (d) [Ni(II)]aq or [Co(II)]aq for 30 days at pHs 4 (yellow), 5 (blue), and 7 (pink).
The [Ni(II)]aq and [Co(II)]aq release rates from Ni- and Co-substituted hausmannite minerals were also measured and compared by equilibration periods and solution pH values (Figure 1c,d). Similar to [Mn]aq release rates, the R systems (Figure 1c) recorded much higher trace metal release rates than the A systems (Figure 1d). Among R systems, pH 4 produced the highest Ni or Co release rates, followed by pH 5 and 7, which agrees with the increased metal solubility in more acidic conditions. Similar release rates of Ni and Co were observed for pH levels of 4 and 5. However, the Co release rate was significantly reduced at pH 7, but not Ni. The A systems produced much lower Ni or Co release rates (Figure 1d). Similar to the R systems, the metal release was highest at pH 4, followed by pH 5, and was minimal at pH 7.
Our results show that both equilibration period and solution pH control mineral dissolution rates. However, extended equilibration lowers mineral dissolution rates, making it difficult to discern the effect of structural impurities on mineral dissolution. Solution acidity accelerates mineral dissolution, helping to demonstrate the impact of structural impurities on this process. Under acidic conditions, where mineral dissolution is favored, the effects of structural impurities on mineral dissolution are evident both in the early stage (t = 8 h) and long-term (t = 30 days) samples. By contrast, under circumneutral solution conditions, where mineral dissolution is limited, little to no such effect was expected.
3.2. Mineralogy of Aged Hausmannite Minerals
To examine the mineralogical transformations during the equilibration period, A-system samples (t = 30 days) of pristine and metal-substituted hausmannite reacted at pH 5 were analyzed using PXRD (Figure 2). Since the highest mineral dissolution rates were observed at pH 4, this low-pH treatment was not suitable for the PXRD analysis. The pH 7 treatment was also excluded since little to no changes were observed at this circumneutral condition. Indeed, visible color changes of the hausmannite suspension were observed at pHs 4 and 5 during the equilibration period (more evident at pH 4 than at pH 5) but not at pH 7, indicating the persistence and prolonged stability of hausmannite at neutral pH.
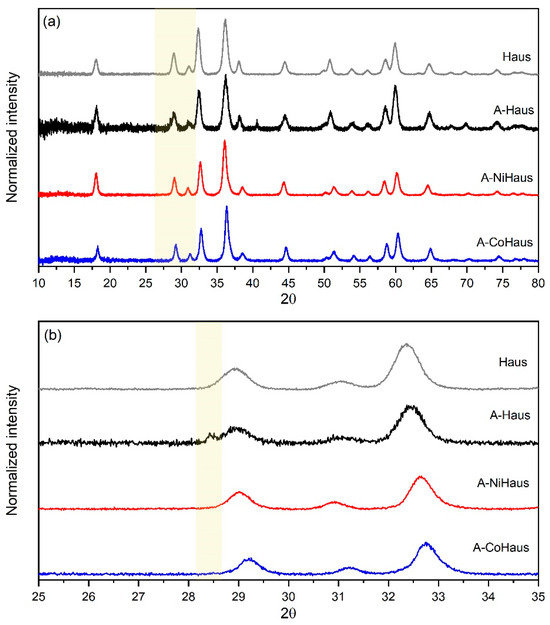
Figure 2.
PXRD analysis on aged hausmannite minerals at pH 5. (a) Diffractograms of A-Haus (black), A-NiHaus (red), and A-CoHaus (blue) with a reference of hausmannite (gray) and (b) enlarged diffractograms in 2θ range from 25° to 35° (orange box) for better presentation of peaks that appeared after 30 days of equilibration.
Diffractograms of A-Haus (black), A-NiHaus (red), and A-CoHaus (blue) matched well with that of freshly synthesized, pristine hausmannite (gray, reference compound), suggesting hausmannite to be the primary mineral phase of aged samples (Figure 2). While hausmannite was dominant, there was a small peak observed at a low angle (2θ = ~ 28.4°) from A-Haus. This diffraction peak is a characteristic feature of the birnessite-type Mn(IV)O2, corresponding to the (002) crystal plane, which supports the formation of a high-valence Mn oxide that was transformed from hausmannite during prolonged equilibration at pH 5. However, no peaks were observed for aged, metal-substituted hausmannite samples at this low angle. This small diffraction peak in the A-Haus sample may indicate that the formation of MnO2 is relatively slow, and the quantity is small.
Furthermore, the broadness of the diffraction peak may also support the small sizes of newly formed MnO2 products. Manganite formation was not observed in any of the aged hausmannite samples, although its formation has been reported in other studies [14,32,35,44]. Thus, the PXRD results support the transformation of hausmannite into birnessite under acidic conditions. Structural impurities seem to play an indirect role in the mineral transformation process by controlling the extent of hausmannite dissolution rates and [Mn]aq availability in the system. However, as shown in the previous section, hausmannite dissolution occurs readily (R systems at t = 8 h), and [Mn]aq availability seems to be a non-limiting factor for both pristine and metal-substituted hausmannite minerals. Thus, the role of structural impurity in the transformation process is challenging to discern using PXRD, especially when the mineral transformation process is slow, the transformation product is present in small quantities and/or is nano-sized and poorly crystalline.
3.3. Crystal Chemistry of Aged Hausmannite Minerals
Synchrotron-based XAS analysis was performed in both the X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) energy regions. The Mn, Ni, and Co K-edge spectra were used to assess any changes in oxidation states and local coordination environments of structural Mn and metal impurities during the experiments.
3.3.1. Mn K-Edge XANES Analysis
The normalized XANES spectra of the aged hausmannite samples, reacted at pH 4, 5, and 7, are shown in Figure 3, along with those of freshly synthesized mineral samples for comparison (green in Figure 3). The overall features of the Mn K-edge XANES spectra are similar. However, the main absorption edge shifted slightly toward higher energy in the reacted samples relative to the starting phase, more noticeably so at pH 4 than at pH 5 and 7 (indicated by an arrow in Figure 3). This shift is consistent with the formation of a Mn phase with a higher oxidation state, such as Mn(IV)O2, in the aged samples.
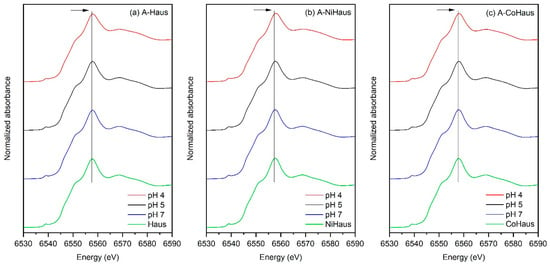
Figure 3.
Mn K-edge XANES regions for (a) A-Haus, (b) A-NiHaus, and (c) A-CoHaus from pH 4 (red), pH 5 (black), and pH 7 (blue). Freshly synthesized, (a) pristine, (b) Ni-substituted, and (c) Co-substituted hausmannite minerals were used as a reference (green) for comparison. The black solid line guides the shift in the location of the absorption edge towards higher energy.
The formation of higher-valence MnO2 may proceed via two pathways: (i) the disproportionation of metastable Mn(III) in hausmannite to Mn(IV) and Mn2+ (Equation (1)) [43] and (ii) the direct oxidation of Mn2+ by dissolved O2 (Equation (2)) [14,17,34].
Equation (1) requires the consumption of protons and the release of divalent Mn ions as the reaction proceeds, while Equation (2) requires divalent Mn ions and dissolved molecular oxygen, producing protons as the reaction progresses. Both equations are sensitive to solution pH conditions but differ in how they affect pH and dissolved Mn2+ levels. While the two pathways may occur simultaneously, we believe that Equation (1) represents the dominant pathway for Mn(IV)O2 formation in our batch reactions. This is supported by the observed increase in solution pH and total dissolved Mn ions as the dissolution reaction progresses (see Section 3.1), as well as by its thermodynamically favorable equilibrium constant (K) (Table S1). In addition, studies have suggested that Equation (2), the oxidation of dissolved Mn2+ by O2, is kinetically slow [45,46].
3.3.2. Ni or Co K-Edge XANES Analysis
Ni and Co K-edge XANES data of A-NiHaus and A-CoHaus were collected at three pH levels and then compared to those of freshly synthesized NiHaus and CoHaus (green in Figure 4). They aligned well (i.e., no edge shifts were observed), suggesting that neither the equilibration period nor solution pH affected the oxidation states of Ni or Co. As expected, Ni is divalent and remains so in the mineral structure over the 30-day equilibration period at these pH levels. In addition, Ni is found to occupy octahedral sites in hausmannite [28]. On the other hand, Co is multivalent, occurring as Co(II) and Co(III) in Mn minerals. Our earlier investigation of freshly synthesized Co-substituted hausmannite revealed that the majority of substituted Co is divalent and occupies tetrahedral sites. In contrast, only a minor fraction of trivalent Co is observed, occupying octahedral sites [27]. The similarities of the Co K-edge XANES spectra (Figure 4b) and the first derivatives of the pre-edge region (Figure 4c) between freshly prepared and aged Co-substituted hausmannite minerals at varying pHs indicate that the ratios of Co(II) to Co(III) remain mostly unchanged. The lack of observable formation of additional Co(III) suggests slow, if any, oxidation of Co(II) to Co(III) in the mineral structure. A detailed analysis of the Co K-edge XANES data for freshly synthesized Co-substituted hausmannite can be found in Song et al. (2021) [27].
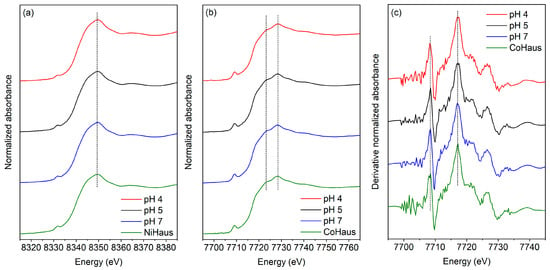
Figure 4.
XANES regions of (a) Ni K-edge, (b) Co K-edge, and (c) Co K-edge in first derivative from freshly synthesized metal-substituted hausmannite minerals (Haus (green)) and A-NiHaus or A-CoHaus equilibrated for 30 days at pH 4 (red), 5 (black), and 7 (blue). The dashed lines indicate that no change was observed at the absorption edge location after aging.
3.3.3. Mn K-Edge EXAFS Analysis
To examine pH-dependent changes in the Mn local environments during the equilibration period, the Mn K-edge EXAFS regions from the aged pristine and metal-substituted hausmannite samples equilibrated at pHs 4, 5, and 7 were analyzed and compared to those of freshly synthesized pristine and metal-substituted minerals (Figure 5). The Mn K-edge EXAFS regions of the Mn reference compounds, including hausmannite, birnessite, and manganite, were used for the analysis (Figure S1). While the amplitudes and shapes of the Mn K-edge EXAFS spectra are generally similar for all samples, notable differences were observed in the k range from 3.5 Å−1 to 9.8 Å−1 in the Mn K-edge EXAFS regions (Figure 5a–c, marked with colored boxes). For instance, all aged, pristine, or metal-substituted hausmannite samples exhibit changes in the amplitude of peaks at 3.5 Å−1 (decrease in peak amplitude, pink arrow) and 4.0 Å−1 (increase in peak amplitude, black arrow), particularly at low pH. The greater changes at pH 4 in this region can be related to the formation of a layered Mn oxide, such as birnessite [37], and confirm more mineral transformation at low pH. Furthermore, spectral changes were also observed in the 7.0 Å−1 to 9.0 Å−1 range (orange-colored box), which is known as an “indicator” region for the identification of Mn oxides [37]. Specifically, there is a decrease in the amplitude of peaks located at ~7.5 Å−1 and 8.5 Å−1, which suggests a decrease in Mn(III) contents and the formation of birnessite-type MnO2. As shown in Equation (1), the Mn(III) in the hausmannite structure can disproportionate to Mn(IV), accompanied by the consumption of H+ and the release of aqueous Mn2+. Thus, low-pH conditions (i.e., pH 4) help accelerate the disproportionation of hausmannite to MnO2 [7,37,43].
Structural changes were also evident in the radial structural functions (RSFs) (Figure 5d–f). Notably, peaks at ~1.5 Å and ~2.5 Å (R+∆R), corresponding to the first Mn-O and second Mn-Mn shell distances, respectively, are the common features of layered Mn oxides [37]. The growth of the Mn-Mn peak at 2.5 Å is attributed to the transformation from spinel-structured hausmannite to layered MnO2, as observed in Mn K-edge EXAFS (Figure S1). This peak growth has also been reported elsewhere [37,47,48,49,50,51]. In addition, a concurrent decrease in the Mn-Mn peaks between 2.8 Å and 3.5 Å (R+∆R), marked with a pink arrow, is consistent with decreasing scattering from hausmannite, further supporting this transformation (Figure S1) [37]. These changes in RSF features exhibit a strong pH dependency, with the most pronounced changes observed at pH 4. While the importance of solution pH on the transformation of hausmannite to birnessite was identified, the impact of structural impurities on the transformation process was difficult to discern due to close similarity in spectral features of A-NiHaus (Figure 5b,e) and A-CoHaus (Figure 5c,f).
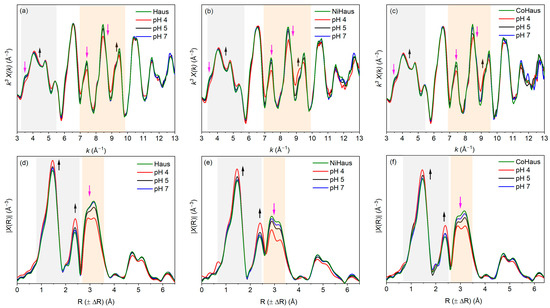
Figure 5.
Mn K-edge EXAFS regions in k3-weighted magnitude for (a) A-Haus, (b) A-NiHaus, and (c) A-CoHaus and in the radial distance (R) for (d) A-Haus, (e) A-NiHaus, and (f) A-CoHaus at pH 4 (red), 5 (black), and 7 (blue). Freshly synthesized, pristine, Ni-substituted, and Co-substituted hausmannite minerals were used as a reference (green) for comparison. The gray and orange boxes indicate the regions where changes in the amplitude and/or shape of the spectra are observed. The black and pink arrows indicate the areas where increases and decreases in peaks are observed, respectively.
3.3.4. Linear Combination Fitting (LCF) of the Mn K-Edge XAS Data
Linear combination fitting (LCF) of the Mn K-edge XANES and EXAFS data was performed for aged pristine and metal-substituted hausmannite samples to determine the fractional contribution of the compositions in the transformed Mn oxide phases and also to detect the effects of structural impurities on the fractional contribution [52]. Three reference Mn minerals, including pristine hausmannite (Haus), manganite (Mang), and birnessite (Birn), were used as standards during the fitting process. Fits of the Mn XANES and EXAFS data yielded similar results; the LCF fits based on the XANES data are presented in Figure 6 and Table 1, while those using the EXAFS data are shown in SI Figure S2 and Table S2.

Table 1.
The LCF results of Mn K-edge XANES regions for A-Haus, A-NiHaus, and A-CoHaus at pH 4, 5, and 7. The fitting results are from Figure 6.
Similar to the PXRD results, the fitting results indicate that hausmannite is the dominant mineral in all aged samples (Figure 6). In general, the relative contribution of hausmannite increases with the solution pH, indicating that the mineral is stable at circumneutral pH. Both birnessite and manganite are transformation products from hausmannite. While both were minor, the relative contribution of birnessite was significantly greater than that of manganite for virtually all the samples, which supports the PXRD and XAS results. At pH 4, all samples produced 15% birnessite; however, a trivial contribution was found from birnessite at pHs 5 and 7 and manganite at all pH levels. This trend is reflected in the increase in the average oxidation states (AOS) of Mn, where the AOS value increases as the pH decreases, reflecting the formation of higher Mn AOS minerals, such as birnessite (Table 1). The following pathways may be involved in the formation and possible transformation of manganite (γ-MnOOH): the transformation of hausmannite to manganite through Mn(II)(s) loss (Equation (3)) and the direct oxidation of Mn(II) by dissolved O2 to form manganite (Equation (4)). These reactions are all thermodynamically viable (K values for the Equations provided in Table S1) and highly solution pH- and Mn(II)-dependent.
To determine the impact of structural impurities on the transformation of hausmannite into birnessite, the relative contributions of hausmannite in A-NiHaus and A-CoHaus were compared to those in A-Haus. The results indicate that A-NiHaus underwent the lowest extent of transformation to birnessite and manganite, whereas A-CoHaus transformation was the most extensive under all pH conditions (Table 1). Although the low quantity of transformation products prevents a detailed understanding of the prevailing transformation processes, these findings do suggest that the hausmannite transformation process and the resulting products are affected by the type of impurity in the mineral structure, similar to the impact of structural impurities on the mineral dissolution rates noted in Section 3.1.
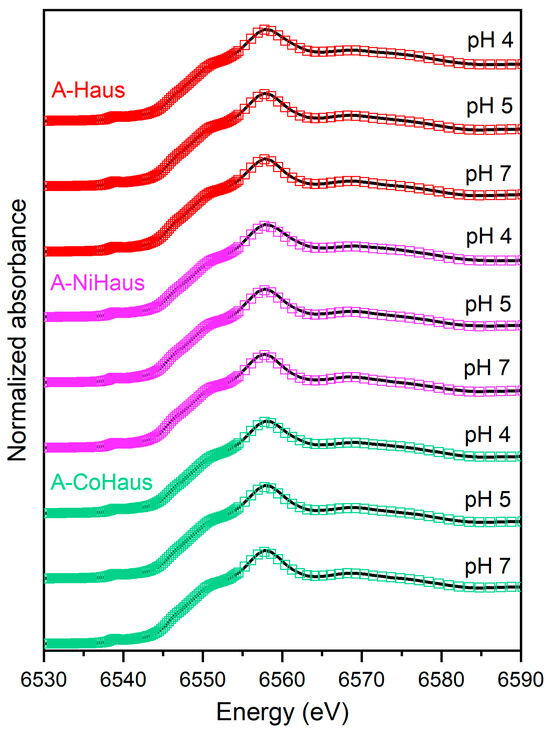
Figure 6.
LCF analysis of the Mn K-edge XANES data for A-Haus (red), A-NiHaus (purple), and A-CoHaus (green) at pH 4, 5, and 7. Black solid lines represent the original spectra, and colored blank squares refer to the fitting results. The corresponding fitting results are also summarized in Table 1 with reference compounds.
3.4. Morphology and Crystal Chemistry of Aged Hausmannite Minerals
To visually confirm the formation of transformation products and examine their morphology and crystallinity, analytical high-resolution HRTEM analyses were performed on pristine hausmannite and aged hausmannite minerals (the A system equilibrated at pH 4) (Figure 7). The latter is the focus of the present work, as pristine hausmannite serves as a reference for mineral size and morphology prior to equilibration. Detailed analyses of pristine and metal-substituted hausmannite minerals are presented in our earlier work [27]. In brief, pristine hausmannite has an average particle size of 21.1 (±4.5) × 16.7 (±3.7) nm with a pseudo-octahedral morphology (Figure 7a). Trace levels of Ni or Co substitution in the hausmannite structure do not affect the mineral morphology. However, Ni substitution seems to cause a slight increase in the average mineral particle size [28], whereas Co does not. A study by Yoshinaga et al. (2018) [53] also supports the finding that Co substitution up to 40 mol% in Mn3O4 does not induce a change in the mineral’s size and morphology.
TEM images revealed that, regardless of whether they possess structural impurities or not, hausmannite particles are dominant at all pH levels, presenting in large quantities after the 30-day equilibration period (Figure 7b–d). This finding is in good agreement with the PXRD and XAS results. While dominant, edge sites of hausmannite with pseudo-octahedral shape (Figure 7a) appeared to be rounded by solution acidity (in pHs 4 and 5). These are shown in Figure 7b[1] with white arrows and dotted lines, indicating the absence of edge sites of hausmannite particles. This phenomenon was also noted in our earlier study with hausmannite and arsenite As(III) [39], where edge sites were consumed first during the reductive dissolution of hausmannite by As(III). These results suggest that the hausmannite edge sites are the active surface sites in both acidic and reductive dissolution processes.
In addition to rounded edges, the formation of nanorods/wires was also observed in aged pristine and metal-substituted hausmannite minerals at pH levels of 4 and 5 (not at pH 7). Since nanorods/wires look distinctly different from hausmannite, it was easier to detect them from the mineral mixtures. Studies have suggested that MnO2 with a nanorod/wire morphology serves as a precursor to the layered δ-MnO2 (birnessite) [7,54,55]. In this work, the spacing of the lattice fringes (Figure 7b[1,2] and Figure 7c,d with red arrows) is 5.1 Å, which is reasonably close to the reported d-spacing of birnessite [7,54,55,56]. It is also worthwhile to note that no lattice fringes extend throughout the rod/wire (Figure 7b[1,2] and Figure 7c,d, enlarged, red arrows); instead, in the places where they can be recognized, the fringes are localized within and surrounded by amorphous areas. This suggests an assemblage of short-range ordered (at least one dimension, <5 nm) MnO2 nanocrystals, which may explain the lack of discernible peaks in the PXRD patterns and the low contribution in the XAS LCF results. HRTEM imaging analyses thus help detect the initial stage of hausmannite transformation to birnessite. All known natural birnessite minerals are fine-grained and relatively poorly crystalline [10].
EDX analysis for the chemical composition of aged hausmannite minerals showed that a majority of Ni and Co in hausmannite remains in the mineral structure after the 30-day equilibration period (Figure 7e,g). EDX results on the hausmannite aggregates in the aged samples presented 1.75 wt% of Ni and 4.01 wt% of Co, which seemed reasonably close to the target wt% of Ni (2.3 wt%) or Co (2.8 wt%) substitution in the hausmannite structure (see Section 2.2.). This finding also agrees with the dissolution data in Figure 1c,d, where lower Ni or Co release rates were noted.
EDX analysis was also performed on a newly formed, nanorod/wire-shaped birnessite to detect the presence of Ni or Co. Due to the aggregation of hausmannite and birnessite shown in Figure 7f,h, spot analysis was applied on the surface of birnessite by focusing the beam to a ~ 3 nm spot. Therefore, the same spot (marked + on the image) was used for imaging, as well as structural and chemical analyses. While the formation of birnessite was observed in both aged Ni- and Co-substituted hausmannite minerals, Ni was not detected from birnessite (Figure 7f) while Co was (Figure 7h). The presence of Co was detected at 0.81 wt% from the nanorod/wire-shaped birnessite phase (Figure 7h). To ensure Co is associated with birnessite and not with hausmannite or secondary Co-oxide, SAED measurements were taken at the same + spot (Figure 7h). The SAED patterns confirmed the sole presence of birnessite [56] at the location where the EDX was taken. Therefore, Ni is strongly associated with the primary mineral, hausmannite, but tends to be released into solution during transformation rather than be adsorbed on or incorporated into a newly formed birnessite. In contrast, once Co is released from hausmannite during the transformation process, it appears to be adsorbed onto or incorporated into the newly formed birnessite, distributed over two mineral phases. A strong sequestration of Co by birnessite [57] and todorokite [37] has already been shown. The results of analytical HRTEM analyses in this work provide, for the first time, nanoparticle-level information on the hausmannite transformation process to birnessite and subsequent Co sequestration by both Mn minerals during the transformation process.

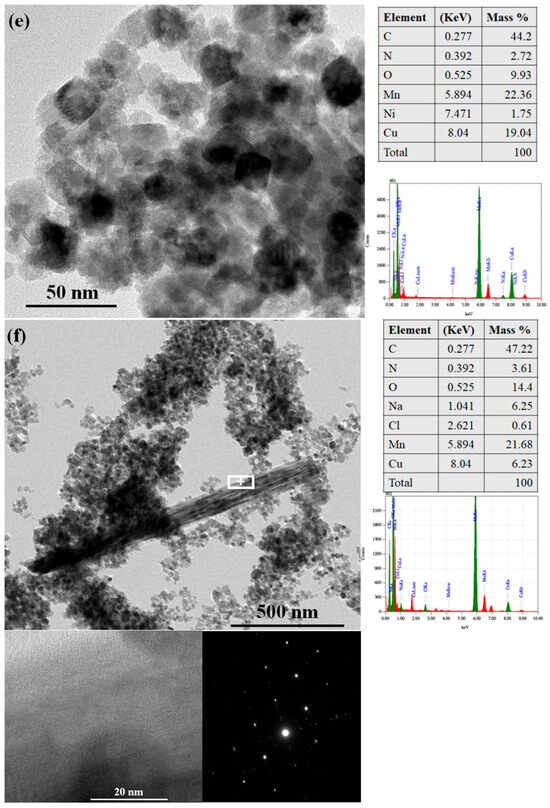
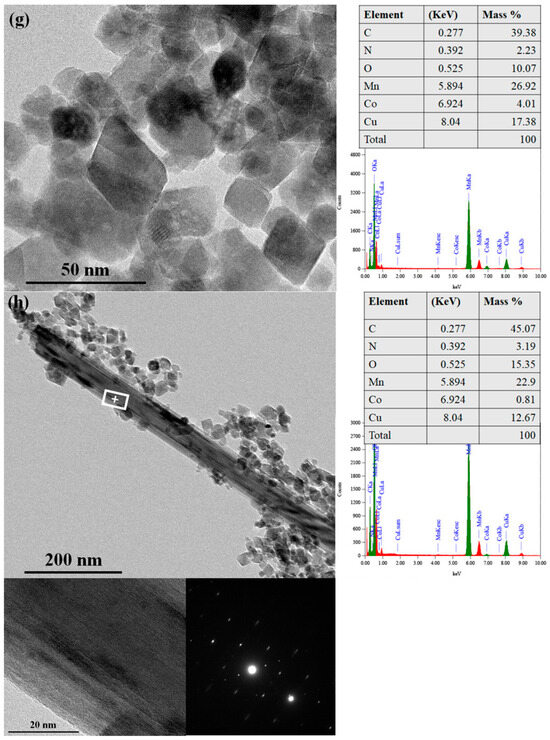
Figure 7.
HRTEM images of (a) freshly made pristine hausmannite; (b) 30-day-aged pristine hausmannite sample with 1 and 2 boxed areas; [1] and [2] enlarged boxed areas; (c) 30-day-aged pristine hausmannite sample with enlarged boxed area; (d) 30-day-aged pristine hausmannite sample with enlarged boxed area; (e) 30-day-aged Ni-substituted hausmannite sample with the EDX results; (f) 30-day-aged Ni-substituted hausmannite sample with the EDX and SAED results; (g) 30-day-aged Co-substituted hausmannite sample with the EDX results; and (h) 30-day-aged Co-substituted hausmannite sample with the EDX and SAED results.
4. Conclusions
The present study provides a detailed investigation into the effects of solution pH, equilibration time, and structural impurities on the stability and transformation of hausmannite. Both solution acidity and equilibration time significantly influence the dissolution rate and the transformation of hausmannite into birnessite. Structural impurities also play a critical role by reducing mineral dissolution rates and altering the relative proportions of transformation products compared to the pristine mineral. For the first time, this work presents nanoparticle-level insights into the transformation of hausmannite to birnessite, along with subsequent Co sequestration by both Mn(II/III) and Mn(IV) oxide phases. HRTEM images reveal that the newly formed birnessite consists of short-range ordered (at least one dimension, <5 nm) MnO2 nanocrystals within an amorphous matrix. The results of this study highlight the importance of considering structural impurities in geochemical processes, such as dissolution and transformation, and contribute to a better understanding of Mn mineralogy and the solubility, mobility, and sequestration of trace elements in the environment.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/min15070697/s1, Table S1: The equilibrium constant (K) values for Equations (1)–(4); Figure S1: The Mn K-edge XANES and EXAFS regions in normalized absorbance (left), k3-weighed magnitude (middle), and radial distance (R, right) for comparison among (a) hausmannite, (b) birnessite, and (c) manganite; Figure S2: LCF analysis of the Mn K-edge EXAFS data for A-Haus (red), A-NiHaus (purple), and A-CoHaus (green) at pH 4, 5, and 7. Black solid lines represent the original spectra, and colored blank squares refer to the fitting results. The corresponding fitting results are also summarized in Table S2 with reference compounds; Table S2: LCF results of Mn K-edge EXAFS regions for A-Haus, A-NiHaus, and A-CoHaus at pH 4, 5, and 7 (k-fitting range from 3 Å−1 to 12 Å−1).
Author Contributions
Data curation, B.S., M.M.R., E.J.E. and B.K.; formal analysis, B.S., M.M.R. and B.K.; funding acquisition, E.J.E. and B.K.; investigation, B.S., M.M.R. and B.K.; methodology, B.S., M.M.R., E.J.E. and B.K.; supervision, E.J.E. and B.K.; validation, B.S., M.M.R., E.J.E. and B.K.; writing—original draft, B.S.; writing—review and editing, M.M.R., E.J.E. and B.K. All authors have read and agreed to the published version of the manuscript.
Funding
The National Science Foundation funded the present study under Grant No. 2003866 and 2003364.
Data Availability Statement
The data are contained within the article and its Supplementary Materials.
Acknowledgments
We thank the 6 BM beamline scientist, Bruce Ravel, for his support on XAS data acquisition and analysis. The present study utilized the 6BM beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated by Brookhaven National Laboratory under Contract No. DE-SC0012704. In addition, the analytical HRTEM analysis in the present study was supported by the Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure (NanoEarth), a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the NSF (ECCS 1542100).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Bauman, A.J. Desert varnish and marine ferromanganese oxide nodules: Congeneric phenomena. Nature 1976, 259, 387–388. [Google Scholar] [CrossRef]
- Chapnick, S.D.; Moore, W.S.; Nealson, K.H. Microbially mediated manganese oxidation in a freshwater lake. Limnol. Oceanogr. 1982, 27, 1004–1014. [Google Scholar] [CrossRef]
- Chukhrov, F.V.; Gorshkov, A.I.; Rudnitskaya, E.S.; Beresovskaya, V.V.; Sivtsov, A.V. Manganese minerals in clays: A review. Clays Clay Miner. 1980, 28, 346–354. [Google Scholar] [CrossRef]
- Haack, E.A.; Warren, L.A. Biofilm hydrous manganese oxyhydroxides and metal dynamics in acid rock drainage. Environ. Sci. Technol. 2003, 37, 4138–4147. [Google Scholar] [CrossRef]
- Lee, S.; Xu, H. XRD and TEM studies on nanophase manganese oxides in freshwater ferromanganese nodules from Green Bay, Lake Michigan. Clays Clay Miner. 2016, 64, 523–536. [Google Scholar] [CrossRef]
- Lu, A.; Li, Y.; Ding, H.; Xu, X.; Li, Y.; Ren, G.; Liang, J.; Liu, Y.; Hong, H.; Chen, N.; et al. Photoelectric conversion on Earth’s surface via widespread Fe-and Mn-mineral coatings. Proc. Natl. Acad. Sci. USA 2019, 116, 9741–9746. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Tan, W.; Suib, S.L.; Qiu, G.; Liu, F. Dissolution and phase transformation processes of hausmannite in acidic aqueous systems under anoxic conditions. Chem. Geol. 2018, 487, 54–62. [Google Scholar] [CrossRef]
- Maynard, J.B. The chemistry of manganese ores through time: A signal of increasing diversity of earth-surface environments. Econ. Geol. 2010, 105, 535–552. [Google Scholar] [CrossRef]
- Murray, J.W.; Balistrieri, L.S.; Paul, B. The oxidation state of manganese in marine sediments and ferromanganese nodules. Geochim. Cosmochim. Acta 1984, 48, 1237–1247. [Google Scholar] [CrossRef]
- Post, J.E. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 1999, 96, 3447–3454. [Google Scholar] [CrossRef]
- Post, J.E. Crystal Structures of Manganese Oxide Minerals. In Biomineralization: Processes of Iron and Manganese: Modern and Ancient Environments; Skinner, H.C.W., Fitzpatrick, R.W., Eds.; Catena Verlag: Cremlingen-Destedt, Germany, 1992; pp. 51–73. [Google Scholar]
- Schindler, M.; Dorn, R.I. Coatings on rocks and minerals: The interface between the lithosphere and the biosphere, hydrosphere, and atmosphere. Elem. Int. Mag. Mineral. Geochem. Petrol. 2017, 13, 155–158. [Google Scholar] [CrossRef]
- Johnson, J.E.; Webb, S.M.; Ma, C.; Fischer, W.W. Manganese mineralogy and diagenesis in the sedimentary rock record. Geochim. Cosmochim. Acta 2016, 173, 210–231. [Google Scholar] [CrossRef]
- Elzinga, E.J. Reductive transformation of birnessite by aqueous Mn (II). Environ. Sci. Technol. 2011, 45, 6366–6372. [Google Scholar] [CrossRef] [PubMed]
- Green, W.J.; Stage, B.R.; Bratina, B.J.; Wagers, S.; Preston, A.; O’bryan, K.; Shacat, J.; Newell, S. Nickel, copper, zinc and cadmium cycling with manganese in Lake Vanda (Wright Valley, Antarctica). Aquat. Geochem. 2004, 10, 303–323. [Google Scholar] [CrossRef]
- Greene, A.C.; Madgwick, J.C. Microbial formation of manganese oxides. Appl. Environ. Microbiol. 1991, 57, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Hem, J.D. Redox processes at surfaces of manganese oxide and their effects on aqueous metal ions. Chem. Geol. 1978, 21, 199–218. [Google Scholar] [CrossRef]
- Lefkowitz, J.P.; Elzinga, E.J. Impacts of aqueous Mn(II) on the sorption of Zn (II) by hexagonal birnessite. Environ. Sci. Technol. 2015, 49, 4886–4893. [Google Scholar] [CrossRef]
- McKenzie, R.M. The sorption of some heavy metals by the lower oxides of manganese. Geoderma 1972, 8, 29–35. [Google Scholar] [CrossRef]
- Pardee, J.T. Manganese-Bearing Deposits near Lake Crescent and Humptulips; US Geological Survey: Washington, DC, USA, 1927; No. 795-A; pp. 1–24.
- Tebo, B.M.; Bargar, J.R.; Clement, B.G.; Dick, G.J.; Murray, K.J.; Parker, D.; Verity, R.; Webb, S.M. Biogenic manganese oxides: Properties and mechanisms of formation. Annu. Rev. Earth Planet. Sci. 2004, 32, 287–328. [Google Scholar] [CrossRef]
- Vodyanitskii, Y.N. Mineralogy and geochemistry of manganese: A review of publications. Eurasian Soil Sci. 2009, 42, 1170–1178. [Google Scholar] [CrossRef]
- Wang, H.; Adeleye, A.S.; Huang, Y.; Li, F.; Keller, A.A. Heteroaggregation of nanoparticles with biocolloids and geocolloids. Adv. Colloid Interface Sci. 2015, 226, 24–36. [Google Scholar] [CrossRef]
- Birkner, N.; Navrotsky, A. Rapidly reversible redox transformation in nanophase manganese oxides at room temperature triggered by changes in hydration. Proc. Natl. Acad. Sci. USA 2014, 111, 6209–6214. [Google Scholar] [CrossRef] [PubMed]
- Antao, S.M.; Cruickshank, L.A.; Hazrah, K.S. Structural trends and solid-solutions based on the crystal chemistry of two hausmannite (Mn3O4) samples from the kalahari manganese field. Minerals 2019, 9, 343. [Google Scholar] [CrossRef]
- Shacat, J.A.; Green, W.J.; Decarlo, E.H.; Newell, S. The geochemistry of Lake Joyce, McMurdo Dry Valleys, Antarctica. Aquat. Geochem. 2004, 10, 325–352. [Google Scholar] [CrossRef]
- Song, B.; Cerkez, E.B.; Elzinga, E.J.; Kim, B. Effects of structural cobalt on the stability and reactivity of hausmannite and manganite: Cobalt coordination chemistry and arsenite oxidation. Chem. Geol. 2021, 583, 120453. [Google Scholar] [CrossRef]
- Song, B.; Cerkez, E.B.; Elzinga, E.J.; Kim, B. Effects of Ni incorporation on the reactivity and stability of hausmannite (Mn3O4): Environmental implications for Mn, Ni, and As solubility and cycling. Chem. Geol. 2020, 558, 119862. [Google Scholar] [CrossRef]
- Hem, J.D. Rates of manganese oxidation in aqueous systems. Geochim. Cosmochim. Acta 1981, 45, 1369–1374. [Google Scholar] [CrossRef]
- Hem, J.D.; Roberson, C.E.; Fournier, R.B. Stability of β-MnOOH and manganese oxide deposition from springwater. Water Resour. Res. 1982, 18, 563–570. [Google Scholar] [CrossRef]
- Junta, J.L.; Hochella, M.F., Jr. Manganese (II) oxidation at mineral surfaces: A microscopic and spectroscopic study. Geochim. Cosmochim. Acta 1994, 58, 4985–4999. [Google Scholar] [CrossRef]
- Lind, C.J. Hausmannite (Mn3O4) conversion to manganite (γ-MnOOH) in dilute oxalate solution. Environ. Sci. Technol. 1988, 22, 62–70. [Google Scholar] [CrossRef]
- Murray, J.W.; Dillard, J.G.; Giovanoli, R.; Moers, H.; Stumm, W. Oxidation of Mn (II): Initial mineralogy, oxidation state and ageing. Geochim. Cosmochim. Acta 1985, 49, 463–470. [Google Scholar] [CrossRef]
- Peña, J.; Duckworth, O.W.; Bargar, J.R.; Sposito, G. Dissolution of hausmannite (Mn3O4) in the presence of the trihydroxamate siderophore desferrioxamine B. Geochim. Cosmochim. Acta 2007, 71, 5661–5671. [Google Scholar] [CrossRef]
- Lefkowitz, J.P.; Rouff, A.A.; Elzinga, E.J. Influence of pH on the reductive transformation of birnessite by aqueous Mn(II). Environ. Sci. Technol. 2013, 47, 10364–10371. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, P.; Zhu, M. Effects of metal cations on coupled birnessite structural transformation and natural organic matter adsorption and oxidation. Geochim. Cosmochim. Acta 2019, 250, 292–310. [Google Scholar] [CrossRef]
- Wu, Z.; Peacock, C.L.; Lanson, B.; Yin, H.; Zheng, L.; Chen, Z.; Tan, W.; Qiu, G.; Liu, F.; Feng, X. Transformation of Co-containing birnessite to todorokite: Effect of Co on the transformation and implications for Co mobility. Geochim. Cosmochim. Acta 2019, 246, 21–40. [Google Scholar] [CrossRef]
- Zhao, S.; González-Valle, Y.A.; Elzinga, E.J.; Saad, E.M.; Tang, Y. Effect of Zn (II) coprecipitation on Mn (II)-induced reductive transformation of birnessite. Chem. Geol. 2018, 492, 12–19. [Google Scholar] [CrossRef]
- Song, B.; Cerkez, E.B.; Grandstaff, D.E.; Goodwin, C.M.; Beebe, T.P., Jr.; Kim, B. Reactivity of binary manganese oxide mixtures towards arsenite removal: Evidence of synergistic effects. Appl. Geochem. 2021, 130, 104939. [Google Scholar] [CrossRef]
- Hu, C.C.; Wu, Y.T.; Chang, K.H. Low-temperature hydrothermal synthesis of Mn3O4 and MnOOH single crystals: Determinant influence of oxidants. Chem. Mater. 2008, 20, 2890–2894. [Google Scholar] [CrossRef]
- McKenzie, R.M. The synthesis of birnessite, cryptomelane, and some other oxides and hydroxides of manganese. Mineral. Mag. 1971, 38, 493–502. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Bricker, O. Some stability relations in the system Mn-O2-H2O at 25 °C and one atmosphere total pressure. Am. Mineral. J. Earth Planet. Mater. 1965, 50, 1296–1354. [Google Scholar]
- Hem, J.D.; Lind, C.J. Nonequilibrium models for predicting forms of precipitated manganese oxides. Geochim. Cosmochim. Acta 1983, 47, 2037–2046. [Google Scholar] [CrossRef]
- Nealson, K.H.; Tebo, B.M.; Rosson, R.A. Occurrence and mechanisms of microbial oxidation of manganese. In Advances in Applied Microbiology; Laskin, A.I., Ed.; Academic Press: Cambridge, MA, USA, 1988; Volume 33, pp. 279–318. [Google Scholar]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Manceau, A.; Lanson, M.; Geoffroy, N. Natural speciation of Ni, Zn, Ba, and As in ferromanganese coatings on quartz using X-ray fluorescence, absorption, and diffraction. Geochim. Cosmochim. Acta 2007, 71, 95–128. [Google Scholar] [CrossRef]
- Manceau, A.; Marcus, M.A.; Tamura, N.; Proux, O.; Geoffroy, N.; Lanson, B. Natural speciation of Zn at the micrometer scale in a clayey soil using X-ray fluorescence, absorption, and diffraction. Geochim. Cosmochim. Acta 2004, 68, 2467–2483. [Google Scholar] [CrossRef]
- Manceau, A.; Marcus, M.A.; Tamura, N. Quantitative speciation of heavy metals in soils and sediments by synchrotron X-ray techniques. Rev. Mineral. Geochem. 2002, 49, 341–428. [Google Scholar] [CrossRef]
- Manceau, A.; Combes, J.M. Structure of Mn and Fe oxides and oxyhydroxides: A topological approach by EXAFS. Phys. Chem. Miner. 1988, 15, 283–295. [Google Scholar] [CrossRef]
- Peña, J.; Kwon, K.D.; Refson, K.; Bargar, J.R.; Sposito, G. Mechanisms of nickel sorption by a bacteriogenic birnessite. Geochim. Cosmochim. Acta 2010, 74, 3076–3089. [Google Scholar] [CrossRef]
- Manceau, A.; Marcus, M.A.; Grangeon, S. Determination of Mn valence states in mixed-valent manganates by XANES spectroscopy. Am. Mineral. 2012, 97, 816–827. [Google Scholar] [CrossRef]
- Yoshinaga, T.; Saruyama, M.; Xiong, A.; Ham, Y.; Kuang, Y.; Niishiro, R.; Akiyama, S.; Sakamoto, M.; Hisatomi, T.; Domen, K.; et al. Boosting photocatalytic overall water splitting by Co doping into Mn3O4 nanoparticles as oxygen evolution cocatalysts. Nanoscale 2018, 10, 10420–10427. [Google Scholar] [CrossRef]
- Ching, S.; Petrovay, D.J.; Jorgensen, M.L.; Suib, S.L. Sol-gel synthesis of layered birnessite-type manganese oxides. Inorg. Chem. 1997, 36, 883–890. [Google Scholar] [CrossRef]
- Portehault, D.; Cassaignon, S.; Baudrin, E.; Jolivet, J.P. Structural and morphological control of manganese oxide nanoparticles upon soft aqueous precipitation through MnO4−/Mn2+ reaction. J. Mater. Chem. 2009, 19, 2407–2416. [Google Scholar] [CrossRef]
- Drits, V.A.; Silvester, E.; Gorshkov, A.I.; Manceau, A. Structure of synthetic monoclinic Na-rich birnessite and hexagonal birnessite: I. Results from X-ray diffraction and selected-area electron diffraction. Am. Mineral. 1997, 82, 946–961. [Google Scholar] [CrossRef]
- Simanova, A.A.; Peña, J. Time-resolved investigation of cobalt oxidation by Mn(III)-rich δ-MnO2 using quick X-ray absorption spectroscopy. Environ. Sci. Technol. 2015, 49, 10867–10876. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).