
Structural Distortion and Optoelectronic Signatures in Metal-Substituted Kaolinite: A First-Principles Investigation


Abstract
1. Introduction
2. Calculation
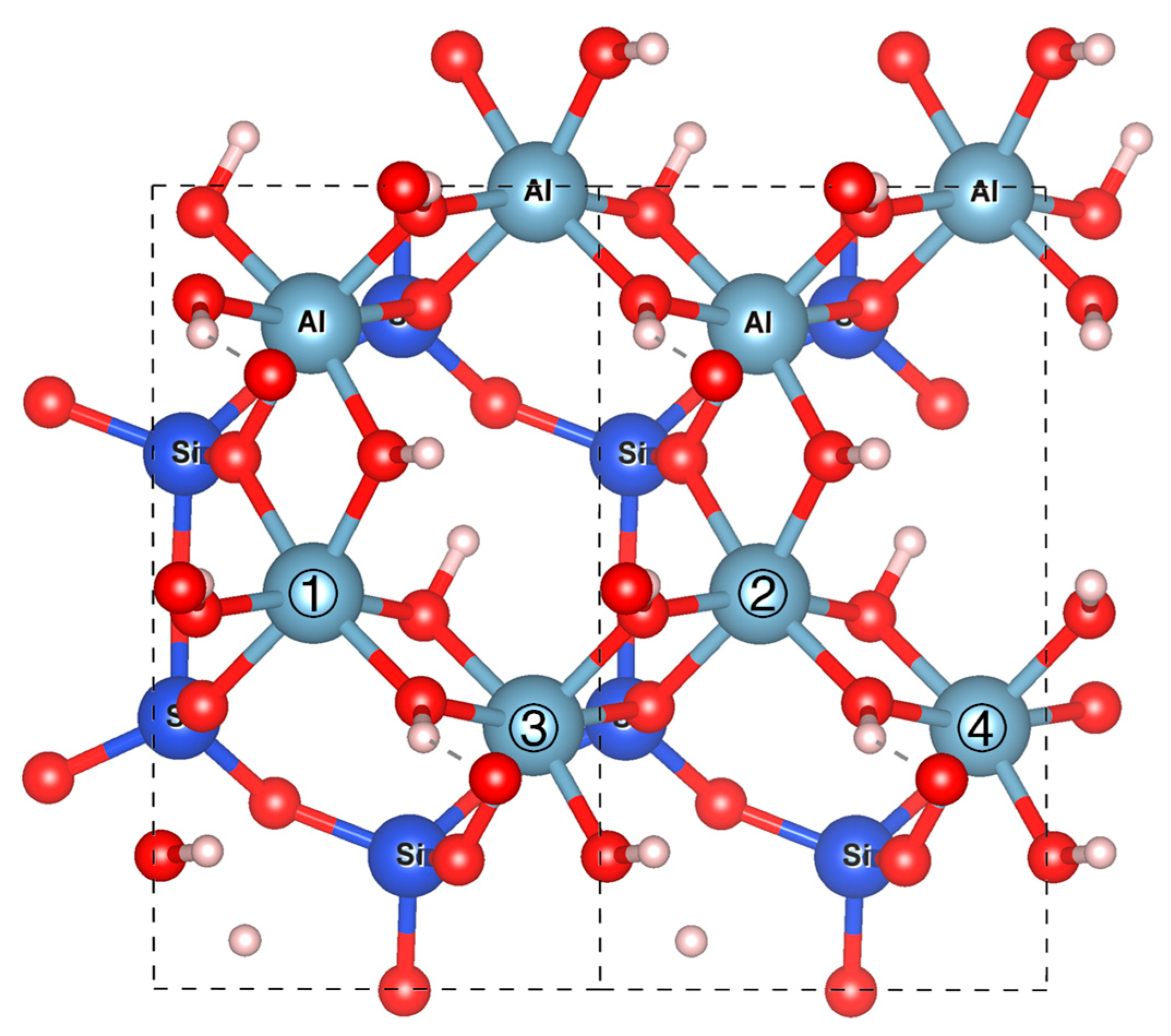
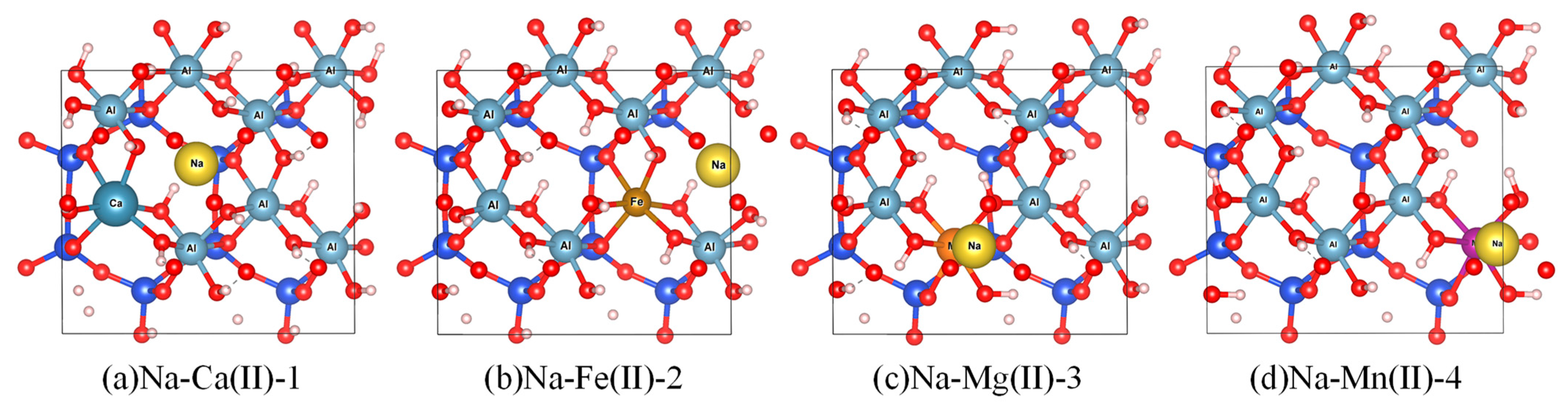
2.1. Model Construction
2.2. Calculation Methods
3. Results and Discussion
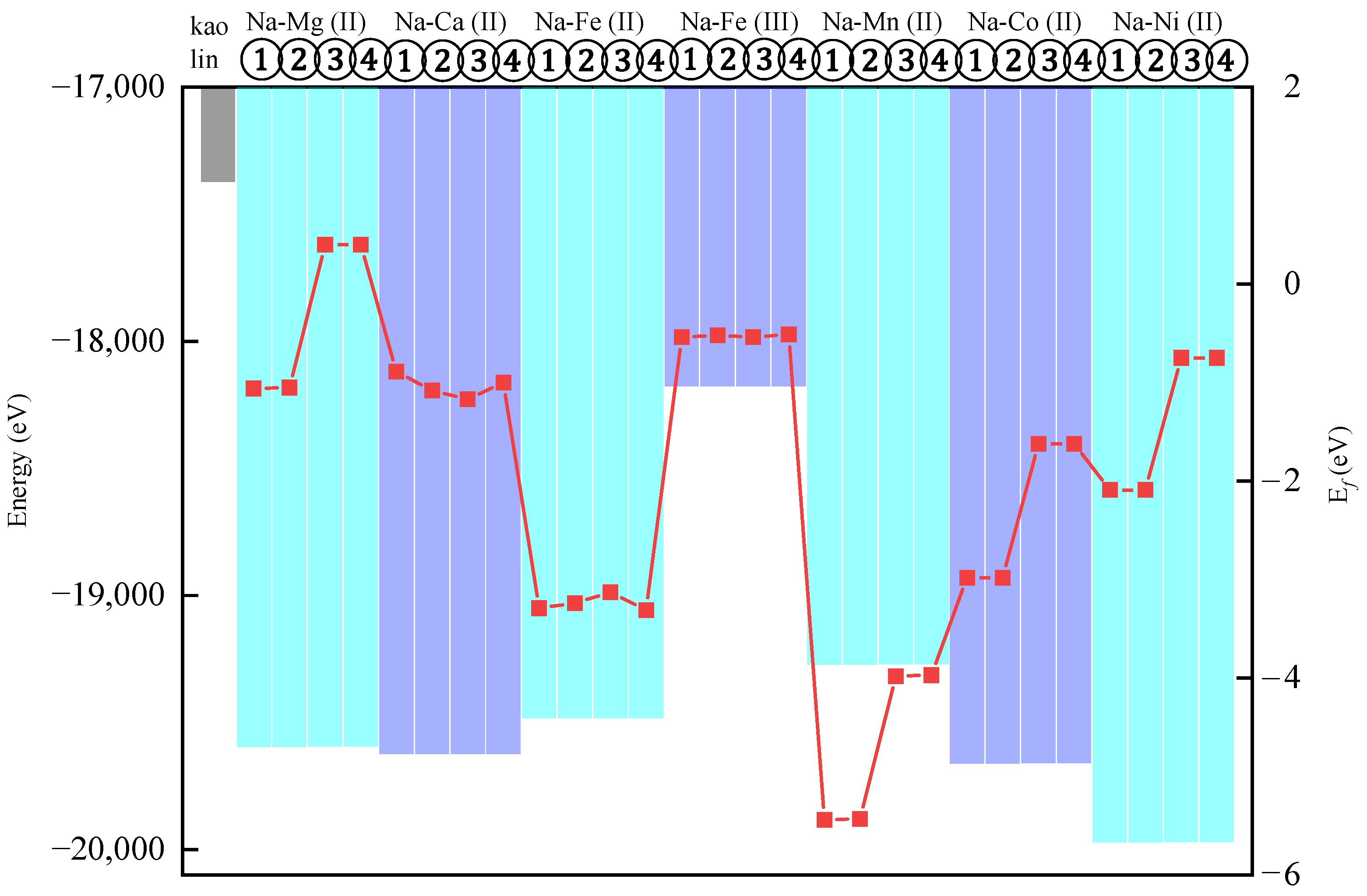
3.1. The Energy of the Substitutional Structure and Its Substitution Energy
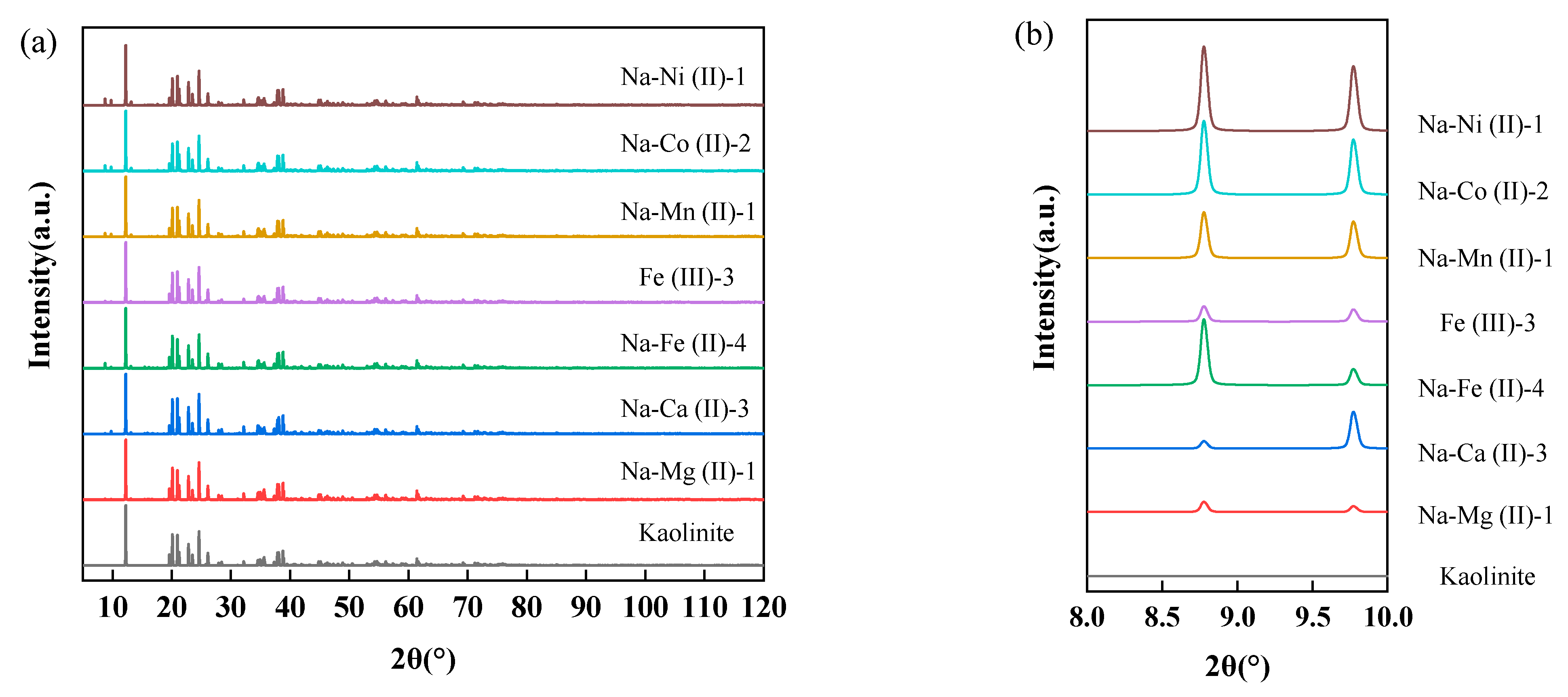
3.2. Substituted Structure Characterization Based on Simulated XRD
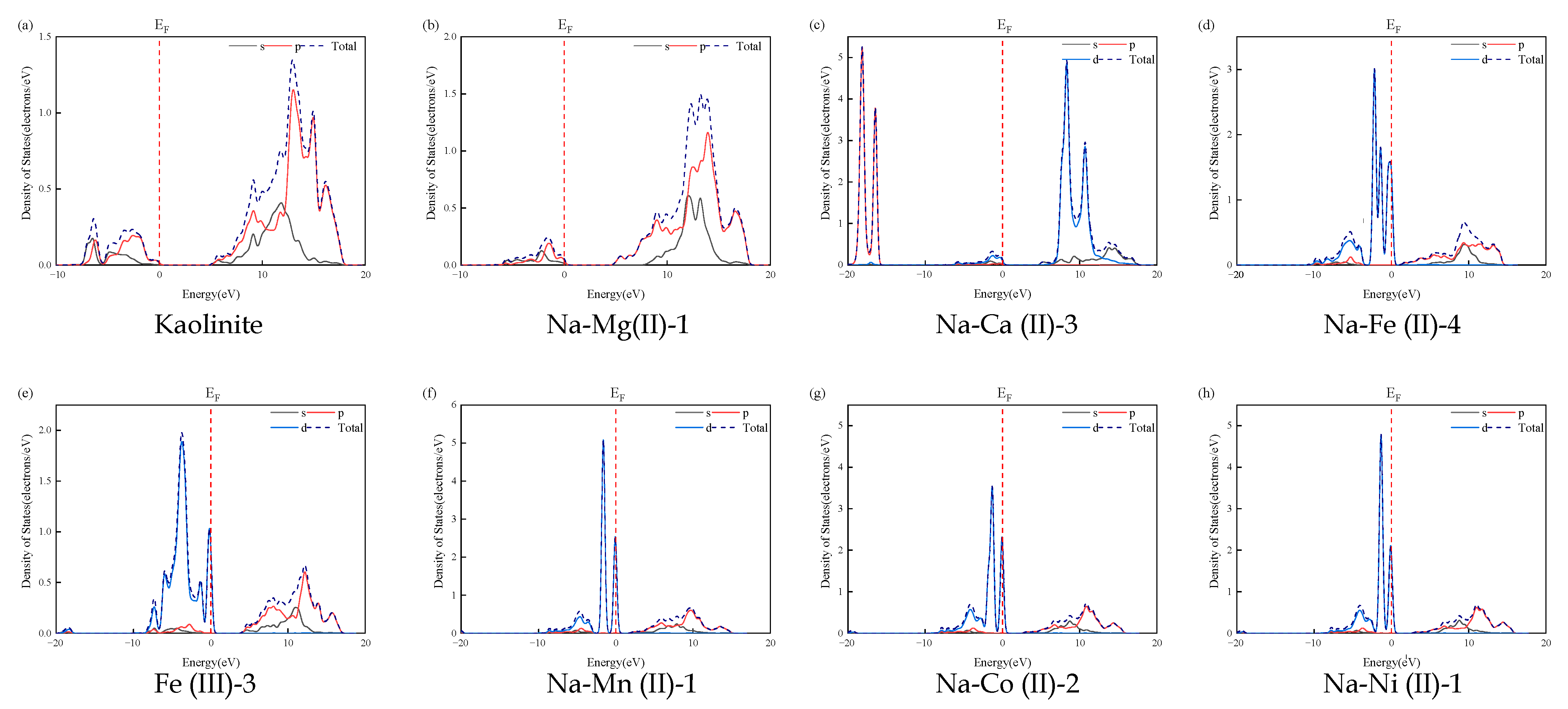
3.3. Band Structure and Density-of-States Analysis
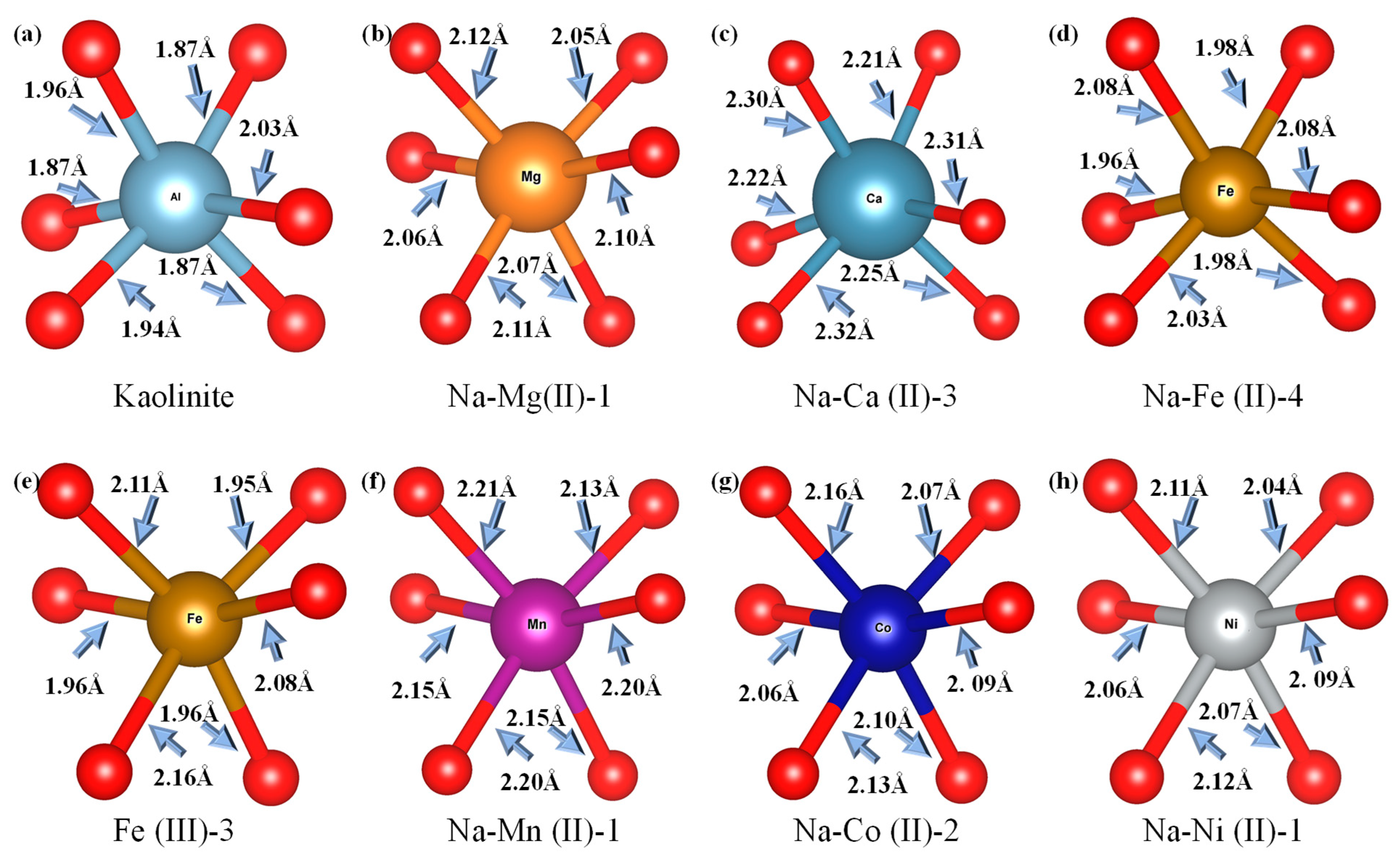
3.4. Localized Structure of Substituted Kalinite on XAFS
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, L.; Sun, Z.; Tao, H.; Wang, J.; Yi, W. Integration of Lightweight Network and Attention Mechanism for the Coal and Gangue Recognition Method. Min. Metall. Explor. 2025, 42, 191–204. [Google Scholar] [CrossRef]
- Ye, Z.; Hongwei, M.; Peng, W.; Wenjian, Z.; Xiangang, C.; Mingzhen, Z. Research on Efficient Matching Method of Coal Gangue Recognition Image and Sorting Image. Sci. Rep. 2024, 14, 25536. [Google Scholar] [CrossRef]
- Lv, Z.; Wang, W.; Xu, Z.; Zhang, K.; Lv, H. Cascade Network for Detection of Coal and Gangue in the Production Context. Powder Technol. 2021, 377, 361–371. [Google Scholar] [CrossRef]
- Yin, J.; Zhu, J.; Zhu, H.; Pan, G.; Zhu, W.; Zeng, Q.; Shi, Q. Intelligent Photoelectric Identification of Coal and Gangue—A Review. Measurement 2024, 233, 114723. [Google Scholar] [CrossRef]
- Yang, Y.; Zeng, Q. Impact-Slip Experiments and Systematic Study of Coal Gangue “Category” Recognition Technology Part I: Impact-Slip Experiments between Coal Gangue Mixture and Top Coal Caving Hydraulic Support and the Study of Coal Gangue “Category” Recognition Technology. Powder Technol. 2021, 392, 224–240. [Google Scholar] [CrossRef]
- Li, Z.; Lu, J.; Zhou, S. A Novel Feature Extraction Method for Recognition of Coal and Gangue under Wetting Conditions. Powder Technol. 2023, 428, 118825. [Google Scholar] [CrossRef]
- Luan, H.; Xu, H.; Tang, W.; Tian, Y.; Zhang, Q. Coal and Gangue Classification in Actual Environment of Mines Based on Deep Learning. Measurement 2023, 211, 112651. [Google Scholar] [CrossRef]
- Gu, W.; Liu, Z.; Zhao, M.; He, Z.; Ding, Z. Preparation and Characterization of γ-Al2O3 from Coal-Bearing Kaolinite. JOM 2025, 77, 3964–3971. [Google Scholar] [CrossRef]
- Xie, Y.; Han, Z.; Khan, A.; Liang, Y.; Zhong, H.; He, Z. Highly Selective and Green Recovery of Lithium from Coal Gangue Using Recyclable Ammonium Fluoride Leaching System. JOM 2025, 77, 842–850. [Google Scholar] [CrossRef]
- Malden, P.J.; Meads, R.E. Substitution by Iron in Kaolinite. Nature 1967, 215, 844–846. [Google Scholar] [CrossRef]
- Shu, Q.; Min, F.; Chen, J. Influence of Ca2+ Substitution on the Surface Hydration Characteristics of Kaolinite: Combination of Simulations and Experiments. Appl. Surf. Sci. 2024, 678, 161097. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, X.; Gao, W.; Tian, Q. Influences of the Residual Water of Kaolin on the Structure and Properties of Phosphate Acid-Activated Metakaolin-Based Geopolymers. JOM 2023, 75, 4881–4886. [Google Scholar] [CrossRef]
- Cui, W.; Chen, J. Insight into Mineral Flotation Fundamentals through the DFT Method. Int. J. Min. Sci. Technol. 2021, 31, 983–994. [Google Scholar] [CrossRef]
- Liu, L.; Kong, C.; Zhao, H.; Lu, F. Elucidating the Enhancement of Kaolinite Flotation by Iron Content through Density Functional Theory: A Study on Sodium Oleate Adsorption Efficiency. Int. J. Min. Sci. Technol. 2024, 34, 855–866. [Google Scholar] [CrossRef]
- Chen, J.; Sun, Y.; Ling, Y.; Chu, X.; Cheng, Y.; Min, F. Effects of Mg(II) Doping Amount on the Hydration Characteristics of Kaolinite Surface: Molecular Dynamics Simulations and Experiments. Surf. Interfaces 2024, 45, 103869. [Google Scholar] [CrossRef]
- Pan, J.-X.; Zhao, L.-S.; Li, Z.; Feng, Z.-Y.; Liu, D.-P.; Chen, J.; Huang, X.-W. Adsorption of Rare Earth Elements and Aluminum on the Surface of Kaolinite: Insights from Sequential Chemical Extractions, XAFS, and DFT. Rare Met. 2025, 44, 4268–4278. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Rehr, J.J.; Kas, J.J.; Vila, F.D.; Prange, M.P.; Jorissen, K. Parameter-Free Calculations of X-Ray Spectra with FEFF9. Phys. Chem. Chem. Phys. 2010, 12, 5503–5513. [Google Scholar] [CrossRef]
- Jorissen, K.; Rehr, J.J. New Developments in FEFF: FEFF9 and JFEFF. J. Phys. Conf. Ser. 2013, 430, 012001. [Google Scholar] [CrossRef]
- Wang, J.; Fu, L.; Yang, H. Structural Modulation of Kaolinite Nanoclay via DFT and Molecular Dynamics Simulations: A Review. Appl. Clay Sci. 2024, 258, 107473. [Google Scholar] [CrossRef]
- Jige, M.; Takagi, T.; Takahashi, Y.; Kurisu, M.; Tsunazawa, Y.; Morimoto, K.; Hoshino, M.; Tsukimura, K. Fe-Kaolinite in Granite Saprolite beneath Sedimentary Kaolin Deposits: A Mode of Fe Substitution for Al in Kaolinite. Am. Mineral. 2018, 103, 1126–1135. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Für Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, Z.; Wu, G.; Jiang, D.; Lan, Y. Investigating the Influence of Impurity Defects on the Adsorption Behavior of Hydrated Sc3+ on the Kaolinite (001) Surface Using Density Functional Theory. Materials 2024, 17, 610. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Chen, J.; Min, F.; Ling, Y.; Shang, H.; Wang, T. Exploration of the Interaction between Fe-Doped Kaolinite Surface and H2O Based on DFT Simulation and Experiment. J. Mol. Liq. 2024, 395, 123901. [Google Scholar] [CrossRef]
- Liu, L.; Min, F.; Chen, J.; Zhang, M.; Lu, F. DFT Research on Lattice Substitution of Fe with Different Valence States in Coal Measures Kaolinite. J. China Univ. Min. Technol. 2019, 48, 903–910. [Google Scholar] [CrossRef]
- Chen, Y.; Wen, H.; Tao, N.; Xu, F.; Ye, Q. Mineralogical and Geochemical Investigations of the Li-Rich Clay Strata from Central Yunnan, Southwest China. Ore Geol. Rev. 2025, 181, 106614. [Google Scholar] [CrossRef]
- Ható, Z.; Kristóf, T. On the Role of the Interlayer Interactions in Atomistic Simulations of Kaolinite Clay. Molecules 2024, 29, 4731. [Google Scholar] [CrossRef]
- Kwon, Y.-M.; Chang, I.; Cho, G.-C. Xanthan Biopolymer-Based Soil Treatment Effect on Kaolinite Clay Fabric and Structure Using XRD Analysis. Sci. Rep. 2023, 13, 11666. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, Y.; Chen, K.; Yi, C.; Nong, Y.; Chen, Z.; Chen, J. Adsorption of Polycarboxylate Ether Superplasticizer Incorporating Various Functional Groups on the Kaolin (001) Surface: Theoretical Study and Experimental Testing. MetaResource 2024, 1, 61–73. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, W.; Luan, Z.; He, M. First-Principles Analysis on Phase Transition, Atomic, Electronic, and Mechanical Properties of Kaolinite under Pressures. Phys. B Condens. Matter 2024, 674, 415554. [Google Scholar] [CrossRef]
- Yuan, L.-D.; Zhang, X.; Acosta, C.M.; Zunger, A. Uncovering Spin-Orbit Coupling-Independent Hidden Spin Polarization of Energy Bands in Antiferromagnets. Nat. Commun. 2023, 14, 5301. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Luo, W.; Ji, J.; Barone, P.; Picozzi, S.; Xiang, H. Band Splitting with Vanishing Spin Polarizations in Noncentrosymmetric Crystals. Nat. Commun. 2019, 10, 5144. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bandgap (eV) | |
|---|---|
| Kaolinite | 4.975 |
| Na-Mg (II)-1 | 4.662 |
| Na-Ca (II)-3 | 4.680 |
| Na-Fe (II)-4 | 1.014 |
| Fe (III)-3 | 3.931 |
| Na-Mn (II)-1 | 1.734 |
| Na-Co (II)-2 | 2.492 |
| Na-Ni (II)-1 | 2.580 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Q.; Xie, J.; Zhu, J.; Yin, J.; Zhu, W. Structural Distortion and Optoelectronic Signatures in Metal-Substituted Kaolinite: A First-Principles Investigation. Minerals 2025, 15, 541. https://doi.org/10.3390/min15050541
Zeng Q, Xie J, Zhu J, Yin J, Zhu W. Structural Distortion and Optoelectronic Signatures in Metal-Substituted Kaolinite: A First-Principles Investigation. Minerals. 2025; 15(5):541. https://doi.org/10.3390/min15050541
Chicago/Turabian StyleZeng, Qiuyu, Jun Xie, Jinbo Zhu, Jianqiang Yin, and Wenliang Zhu. 2025. "Structural Distortion and Optoelectronic Signatures in Metal-Substituted Kaolinite: A First-Principles Investigation" Minerals 15, no. 5: 541. https://doi.org/10.3390/min15050541
APA StyleZeng, Q., Xie, J., Zhu, J., Yin, J., & Zhu, W. (2025). Structural Distortion and Optoelectronic Signatures in Metal-Substituted Kaolinite: A First-Principles Investigation. Minerals, 15(5), 541. https://doi.org/10.3390/min15050541