

Revisiting the Raman Spectra of Carbonate Minerals

 , , and
, , and
Abstract
:
1. Introduction
2. Methods
3. Vibrational Analysis
4. Results and Discussion
4.1. Raman Shift
4.2. Bandwidth
4.3. Relative Intensities of Raman Bands
4.4. Size of the Laser Spot
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nesse, W.D. Introduction to Optical Mineralogy, 3rd ed.; Oxford University Press: New York, NY, USA, 2017. [Google Scholar]
- Wenk, H.R.; Bulakh, A. Minerals. Their Constitution and Origin, 2nd ed.; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Sun, J.; Wu, Z.; Cheng, H.; Zhang, Z.; Frost, R.L. A Raman Spectroscopic Comparison of Calcite and Dolomite. Spectrochim. Acta Mol. Biomol. Spectrosc. 2014, 117, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Teoh, C.P.; Laya, J.C.; Rose, K.; Kaczmarek, S. The effects of magnesium concentration in high-magnesium calcite allochems on dolomitization: Insights from high-temperature dolomite synthesis experiments. J. Sediment. Res. 2022, 92, 134–143. [Google Scholar] [CrossRef]
- Allen, B.L.; Hajeck, B.F. Mineral occurrence in soil environments. In Minerals in Soil Environments, 2nd ed.; Dixon, J., Weed, S.B., Eds.; Soil Science Society of America: Madison, WI, USA, 1989. [Google Scholar]
- Yoder, C.H. Ionic Compounds Applications of Chemistry to Mineralogy, 1st ed.; John Wiley & Sons: New Jersey, NY, USA, 2006. [Google Scholar]
- Kaczmarek, S.E.; Thornton, B.P. The effect of temperature on stoichiometry, cation ordering, and reaction rate in high-temperature dolomitization experiments. Chem. Geol. 2017, 468, 32–41. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Anbalagan, G.; Pandi, S. Raman and infrared spectra of carbonates of calcite structure. J. Raman Spectrosc. 2006, 37, 892–899. [Google Scholar] [CrossRef]
- Dufresne, W.J.B.; Rufledt, C.J.; Marshall, C.P. Raman spectroscopy of the eight natural carbonate minerals of calcite structure. J. Raman Spectrosc. 2018, 49, 1999–2007. [Google Scholar] [CrossRef]
- Biellmann, C.; Gillet, P. High-pressure and high-temperature behavior of calcite, aragonite and dolomite: A Raman spectroscopic study. Eur. J. Mineral. 1992, 4, 389–394. [Google Scholar] [CrossRef]
- Farsang, S.; Facq, S.; Redfern, S.A.T. Raman modes of carbonate minerals as pressure and temperature gauges up to 6 GPa and 500 °C. Am. Mineral. 2018, 103, 1988–1998. [Google Scholar]
- Tomić, Z.; Makreski, P.; Gajić, B. Identification and spectra-structure determination of soil minerals: Raman study supported by IR spectroscopy and x-ray powder diffraction. J. Raman Spectrosc. 2010, 41, 582–586. [Google Scholar] [CrossRef]
- De La Pierre, M.; Carteret, C.; Maschio, L.; André, E.; Orlando, R.; Dovesi, R. The Raman spectrum of caco3 polymorphs calcite and aragonite: A combined experimental and computational study. J. Chem. Phys. 2014, 140, 164509–164521. [Google Scholar] [CrossRef]
- Kim, Y.; Caumon, M.C.; Barres, O.; Sall, A.; Cauzid, J. Identification and composition of carbonate minerals of the calcite structure by Raman and infrared spectroscopies using portable devices. Spectrochim. Acta Part A 2021, 261. [Google Scholar] [CrossRef]
- Shi, W.; Fleet, M.E.; Shieh, S.R. High-pressure phase transitions in Ca-Mn carbonates (Ca, Mn)CO3 studied by Raman spectroscopy. Am. Mineral. 2012, 97, 999–1001. [Google Scholar] [CrossRef]
- Frost, R.L.; Martens, W.N.; Rintoul, L.; Mahmutagic, E.; Kloprogge, J.T. Raman Spectroscopic Study of Azurite and Malachite at 298 and 77 K. J. Raman Spectrosc. 2002, 33, 252–259. [Google Scholar] [CrossRef]
- Yu, B.S.; Fang, J.N.; Huang, E.P. Characteristics of the Raman spectra of archaeological malachites. J. Raman Spectrosc. 2013, 44, 630–636. [Google Scholar] [CrossRef]
- Jehlička, J.; Culka, A.; Košek, F. Obtaining Raman spectra of minerals and carbonaceous matter using a portable sequentially shifted excitation raman spectrometer—A few examples. J. Raman Spectrosc. 2017, 48, 1583–1589. [Google Scholar] [CrossRef]
- Farfan, G.A.; Boulard, E.; Wang, S.; Mao, W.L. Bonding and electronic changes in rhodochrosite at high pressure. Am. Mineral. 2013, 98, 1817–1823. [Google Scholar] [CrossRef]
- Wang, X.; Ye, Y.; Wu, X.; Smyth, J.R.; Yang, Y.; Zhang, Z.; Wang, Z. High-temperature Raman and FTIR study of aragonite-group carbonates. Phys. Chem. Min. 2019, 46, 51–62. [Google Scholar] [CrossRef]
- Krishnamurti, D. The Raman spectra of aragonite, strontianite and witherite. Proc. Indian Acad. Sci. Sect. 1960, 51, 285–295. [Google Scholar] [CrossRef]
- Bersani, D.; Lottici, P.P. Raman spectroscopy of minerals and mineral pigments in archaeometry. J. Raman Spectrosc. 2016, 47, 499–530. [Google Scholar] [CrossRef]
- Frech, R.; Wang, E.C.; Bates, J.B. The i.r. and Raman Spectra of CaCO3 (Aragonite). Spectrochim. Acta Mol. Biomol. Spectrosc. 1980, 36, 915–919. [Google Scholar] [CrossRef]
- Carteret, C.; Dandeu, A.; Moussaoui, S.; Muhr, H.; Humbert, B.; Plasari, E. Polymorphism studied by lattice phonon Raman spectroscopy and statistical mixture analysis method. Application to calcium carbonate polymorphs during batch crystallization. Cryst. Growth Des. 2009, 9, 807–812. [Google Scholar] [CrossRef]
- Fong, M.Y.; Nicol, M. Raman spectrum of calcium carbonate at high pressures. J. Chem. Phys. 1971, 54, 575–578. [Google Scholar] [CrossRef]
- Boulard, E.; Guyot, F.; Fiquet, G. The influence on Fe content on Raman spectra and unit cell parameters of magnesite-siderite solid solutions. Phys. Chem. Min. 2012, 39, 239–246. [Google Scholar] [CrossRef]
- Liu, L.; Lv, C.; Zhuang, C.; Yi, L.; Liu, H.; Du, J. Effects of differential stress on the structure and Raman spectra of calcite from first-principles calculations. Am. Mineral. 2016, 101, 1892–1897. [Google Scholar] [CrossRef]
- Müller, J.; Efthimiopoulos, I.; Jahn, S.; Koch-Müller, M. Effect of temperature on the pressure-induced spin transition in siderite and iron-bearing magnesite: A Raman spectroscopy study. Eur. J. Mineral. 2017, 29, 785–793. [Google Scholar] [CrossRef]
- Aru, M.; Burgio, L.; Rumsey, M.S. Mineral impurities in azurite pigments: Artistic or natural selection? J. Raman Spectrosc. 2014, 45, 1013–1018. [Google Scholar] [CrossRef]
- Borromeo, L.; Zimmermann, U.; Andò, S.; Coletti, G.; Bersani, D.; Basso, D.; Gentile, P.; Schulz, B.; Garzanti, E. Raman spectroscopy as a tool for magnesium estimation in Mg-calcite. J. Raman Spectrosc. 2017, 48, 983–992. [Google Scholar] [CrossRef]
- Farfan, G.; Wang, S.; Ma, H.; Caracas, R.; Mao, W.L. Bonding and structural changes in siderite at high pressure. Am. Mineral. 2012, 97, 1421–1426. [Google Scholar] [CrossRef]
- Xu, J.; Kuang, Y.; Zhang, B.; Liu, Y.; Fan, D.; Zhou, W.; Xie, H. High-pressure study of azurite Cu3(CO3)2(OH)2 by synchrotron radiation X-Ray diffraction and Raman spectroscopy. Phys. Chem. Min. 2015, 42, 805–816. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, C.; Xu, L.; Liu, J. Effects of hydrostaticity and Mn-substitution on dolomite stability at high pressure. Am. Mineral. 2022, 107, 2234–2241. [Google Scholar] [CrossRef]
- Müller, J.; Speziale, S.; Efthimiopoulos, I.; Jahn, S.; Koch-Müller, M. Raman Spectroscopy of siderite at high pressure: Evidence for a sharp spin transition. Am. Mineral. 2016, 101, 2638–2644. [Google Scholar] [CrossRef]
- Liu, J.; Caracas, R.; Fan, D.; Bobocioiu, E.; Zhang, D.; Mao, W.L. High-pressure compressibility and vibrational properties of (Ca, Mn)CO3. Am. Mineral. 2016, 101, 2723–2730. [Google Scholar] [CrossRef]
- Zhao, C.; Li, H.; Jiang, J.; He, Y.; Liang, W. Phase transition and vibration properties of MnCO3 at high pressure and high-temperature by Raman Spectroscopy. High Press. Res. 2018, 38, 212–223. [Google Scholar] [CrossRef]
- Perrin, J.; Vielzeuf, D.; Laporte, D.; Ricolleau, A.; Rossman, G.R.; Floquet, N. Raman characterization of synthetic magnesian calcites. Am. Mineral. 2016, 101, 2525–2538. [Google Scholar] [CrossRef]
- Cerantola, V.; McCammon, C.; Kupenko, I.; Kantor, I.; Marini, C.; Wilke, M.; Ismailova, L.; Solopova, N.; Chumakov, A.; Pascarelli, S.; et al. High-pressure spectroscopic study of siderite (FeCO3) with a focus on spin crossover. Am. Mineral. 2015, 100, 2670–2681. [Google Scholar] [CrossRef]
- Wang, C.; Ren, L.; Walters, J.B.; Zhang, L.; Tao, R. In situ Raman vibrational spectra of siderite (FeCO3) and rhodochrosite (MnCO3) up to 47 GPa and 1100 K. Am. Mineral. 2023, 108, 312–325. [Google Scholar] [CrossRef]
- Jorge-Villar, S.E.; Edwards, H.G.M. green and blue pigments in roman wall paintings: A challenge for Raman spectroscopy. J. Raman Spectrosc. 2021, 52, 2190–2203. [Google Scholar] [CrossRef]
- Ulian, G.; Valdrè, G. The effect of long-range interactions on the infrared and Raman spectra of aragonite (CaCO3, pmcn) up to 25 GPa. Sci. Rep. 2023, 13, 2725. [Google Scholar] [CrossRef]
- Zhang, J.; Martinez, I.; Guyot, F.; Reeder, R.J. Effects of Mg-Fe+2 Substitution in Calcite-Structure Carbonates: Thermoelastic Properties. Am. Mineral. 1998, 83, 280–287. [Google Scholar] [CrossRef]
- Bissengaliyeva, M.R. Calculations of the structural and thermodynamic characteristics of copper carbonates by quantum-chemical methods. Russ. J. Phys. Chem. 2009, 83, 238–244. [Google Scholar] [CrossRef]
- Porto, S.P.S.; Giordmaine, J.A.; Damen, T.C. Depolarization of Raman Scattering in Calcite. Phys. Rev. 1966, 147, 608–611. [Google Scholar] [CrossRef]
- Warr, L.N. a new collection of clay mineral ‘crystallinity’ index standards and revised guidelines for the calibration of Kübler and Árkai indices. Clay Min. 2018, 53, 339–350. [Google Scholar] [CrossRef]
- Kirov, G.K.; Vesselinov, I.; Cherneva, Z. Conditions of formation of calcite crystals of tabular and acute rhombohedral habits. Krist. Und Tech. 1972, 7, 497–509. [Google Scholar]
- Clark, R.J.H.; Dines, T.J. Resonance Raman spectroscopy, and its application to inorganic chemistry. new analytical methods (27). Angew. Chem. Int. Ed. Engl. 1986, 25, 131–158. [Google Scholar] [CrossRef]
- Langille, D.B.; O’Shea, D.C. Raman spectroscopy studies of antiferromagnetic FeCO3 and related carbonates. J. Phys. Chem. Solids 1977, 38, 1161–1171. [Google Scholar] [CrossRef]






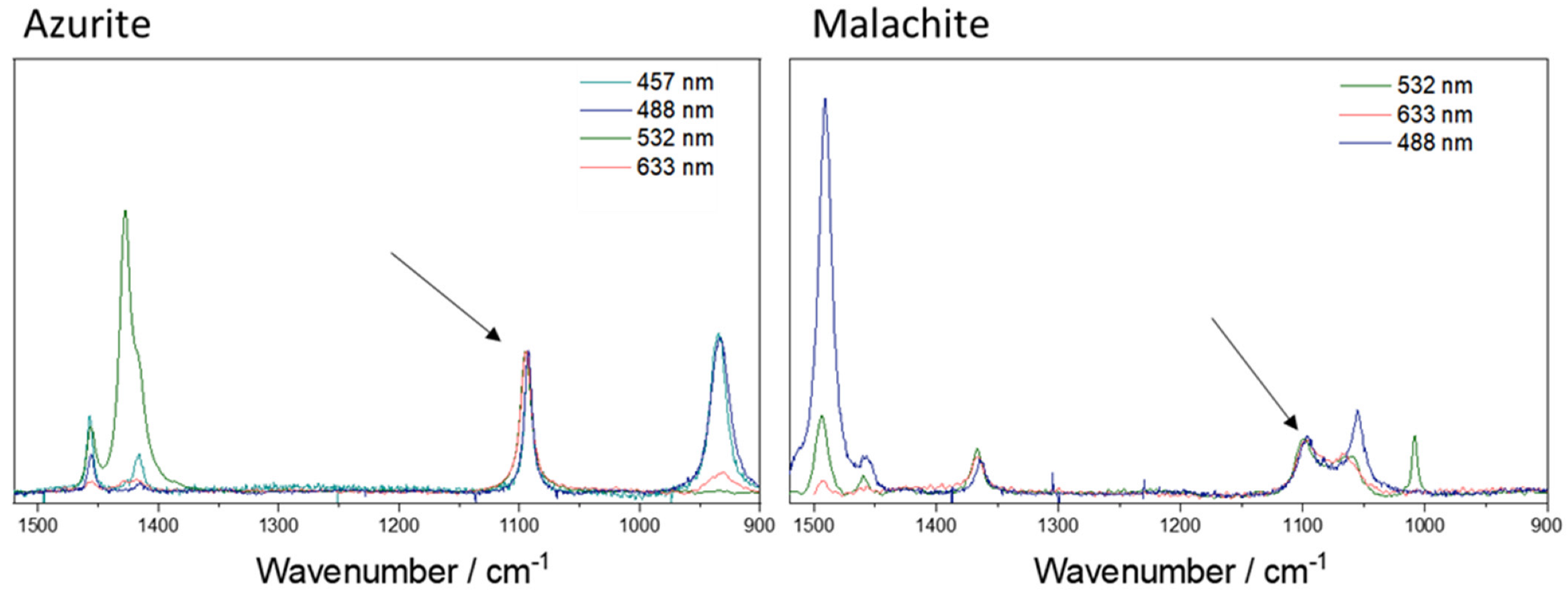


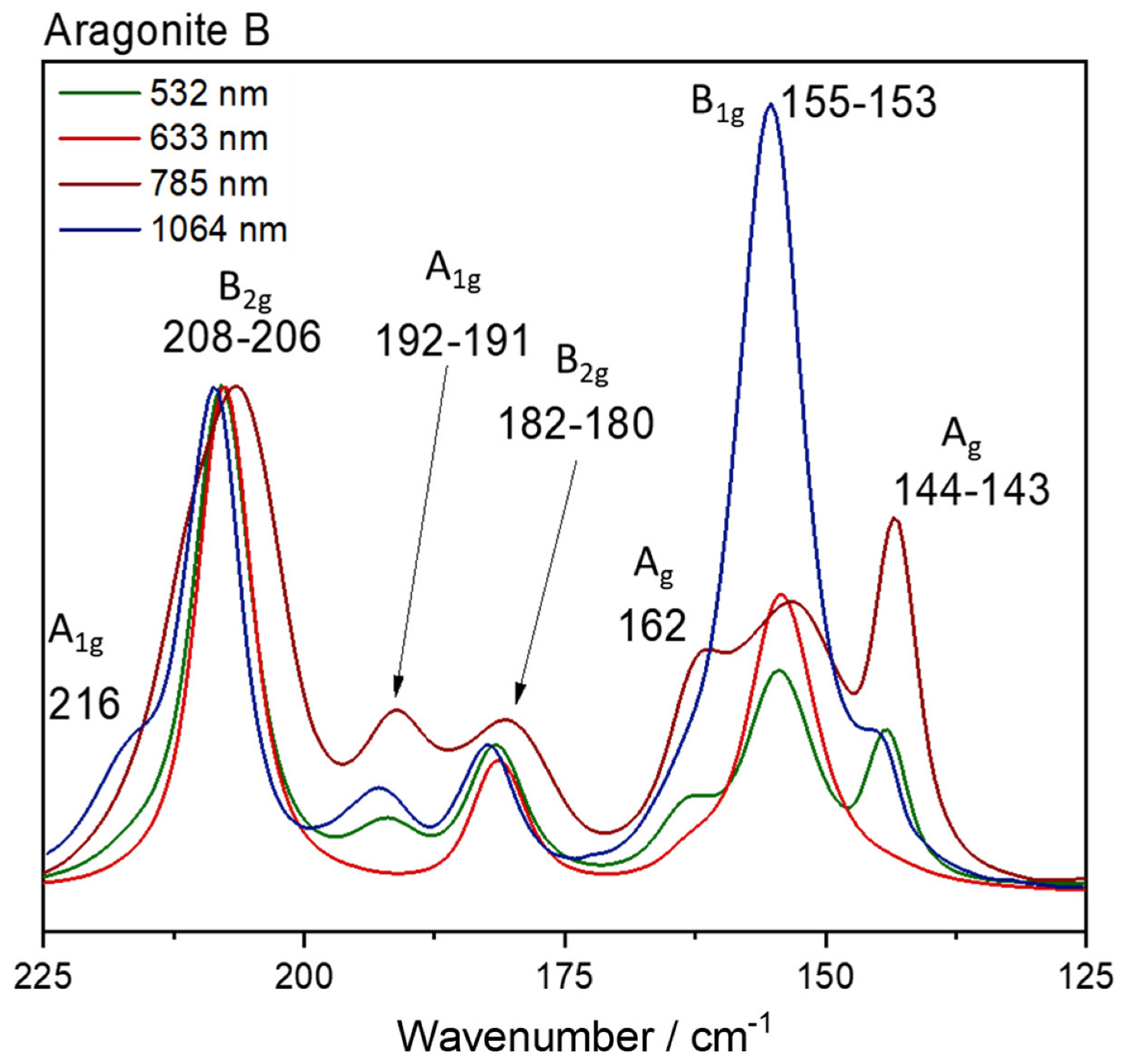


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Bandwidth/cm−1 |
|---|---|
| Calcite | 4 |
| Magnesite | 12 |
| Dolomite | 6 |
| Rhodochrosite | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, J.F.; Edwards, H.G.M.; Korsakov, A.; de Oliveira, L.F.C. Revisiting the Raman Spectra of Carbonate Minerals. Minerals 2023, 13, 1358. https://doi.org/10.3390/min13111358
Alves JF, Edwards HGM, Korsakov A, de Oliveira LFC. Revisiting the Raman Spectra of Carbonate Minerals. Minerals. 2023; 13(11):1358. https://doi.org/10.3390/min13111358
Chicago/Turabian StyleAlves, Julliana F., Howell G. M. Edwards, Andrey Korsakov, and Luiz Fernando C. de Oliveira. 2023. "Revisiting the Raman Spectra of Carbonate Minerals" Minerals 13, no. 11: 1358. https://doi.org/10.3390/min13111358
APA StyleAlves, J. F., Edwards, H. G. M., Korsakov, A., & de Oliveira, L. F. C. (2023). Revisiting the Raman Spectra of Carbonate Minerals. Minerals, 13(11), 1358. https://doi.org/10.3390/min13111358