
Iron-Speciation Control of Chalcopyrite Dissolution from a Carbonatite Derived Concentrate with Acidic Ferric Sulphate Media


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chalcopyrite Sample Preparation and Characterisation
2.3. Leaching Media and Tests
2.4. Leachate Speciation
2.5. Residue Characterisation
3. Results and Discussion
3.1. Initial Fe Speciation of the Media
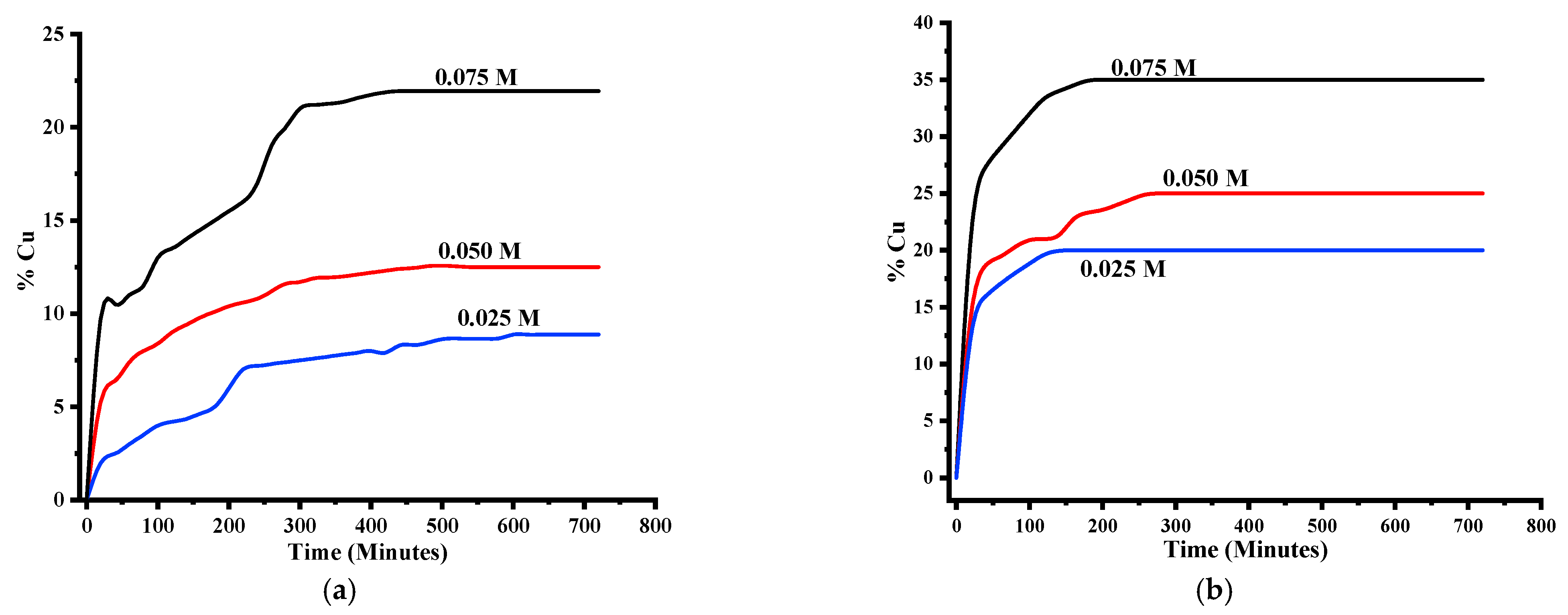
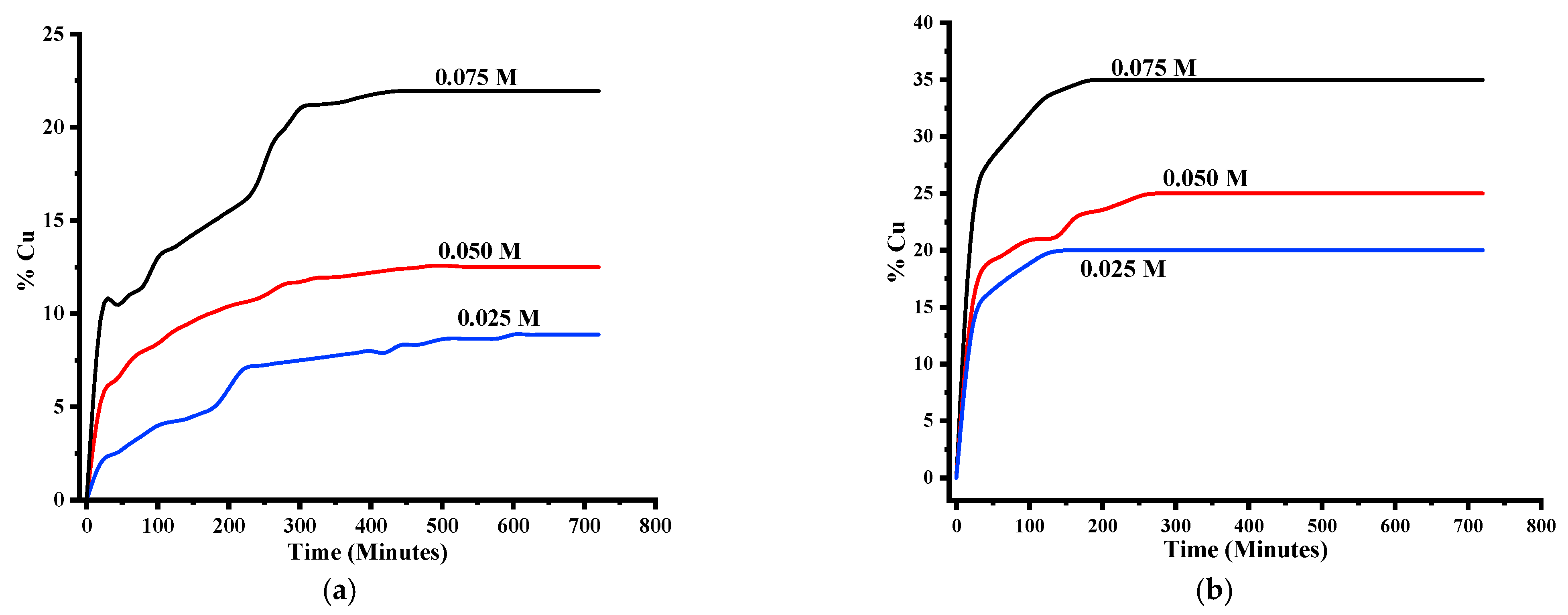
3.1.1. Cu Dissolution Rate and Recovery
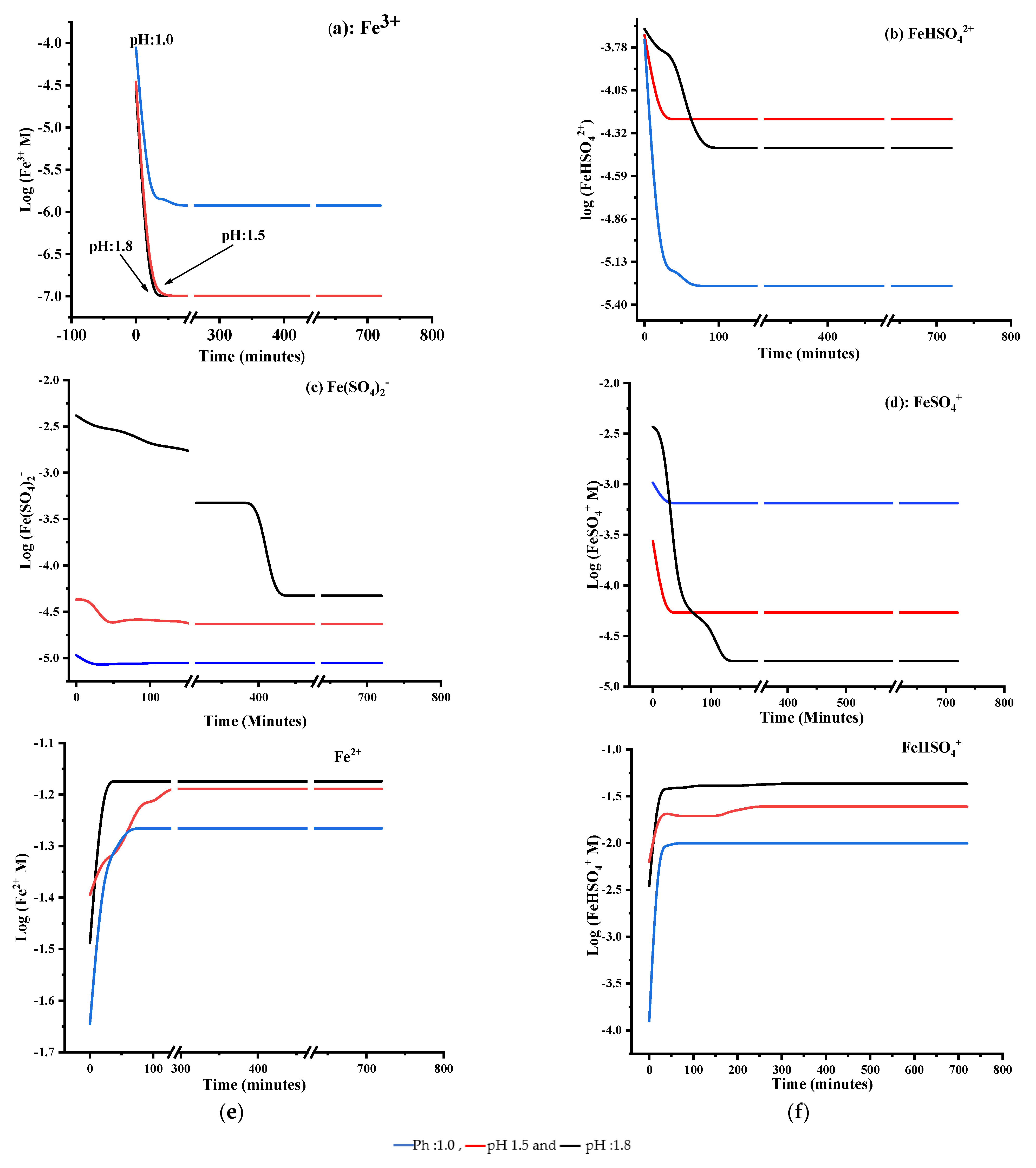
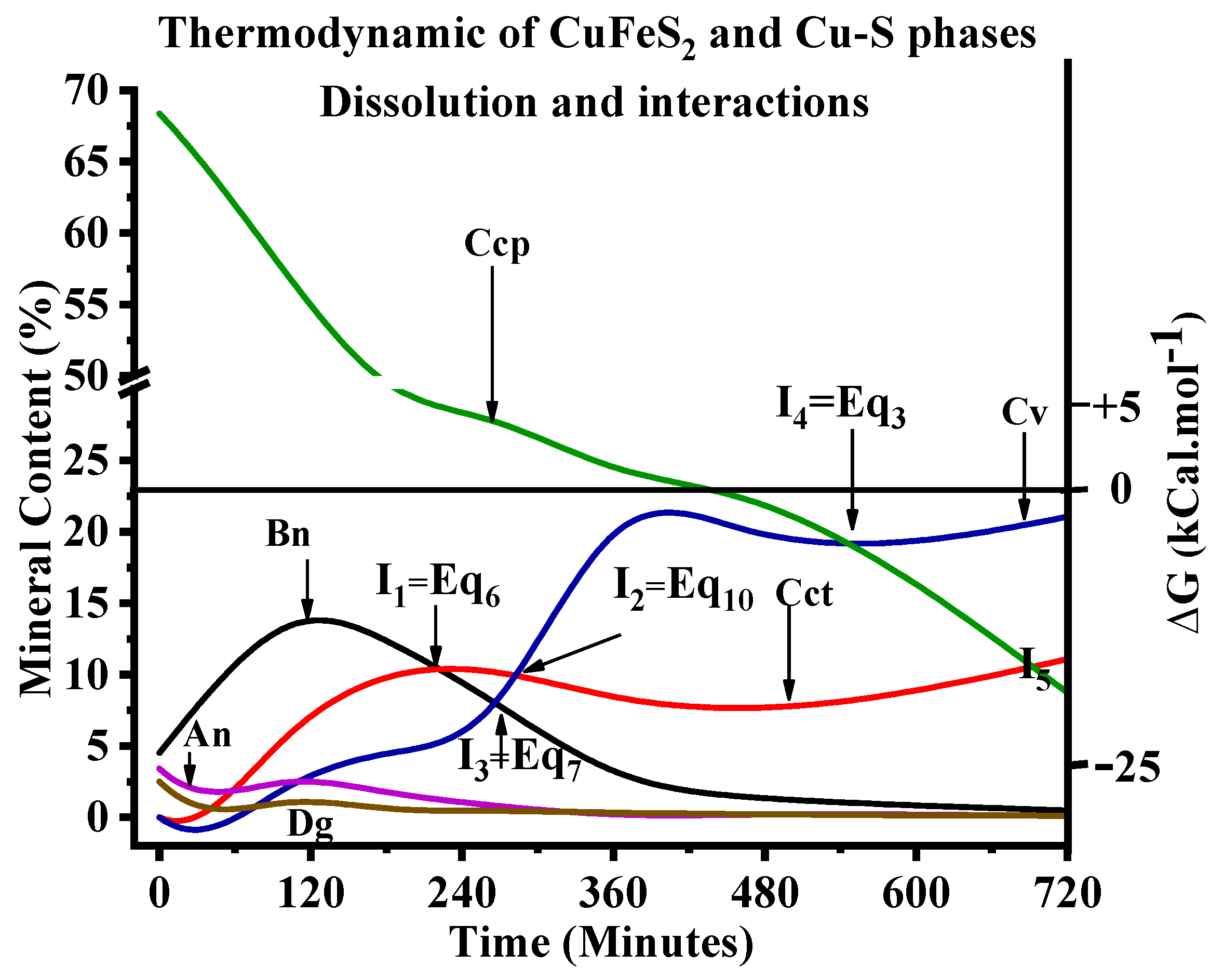
3.1.2. Ferric Speciation during Chalcopyrite Leaching
3.1.3. Effect of Temperature
3.2. Effect of Initial Solution Potential
3.3. Effect of Initial Fe3+
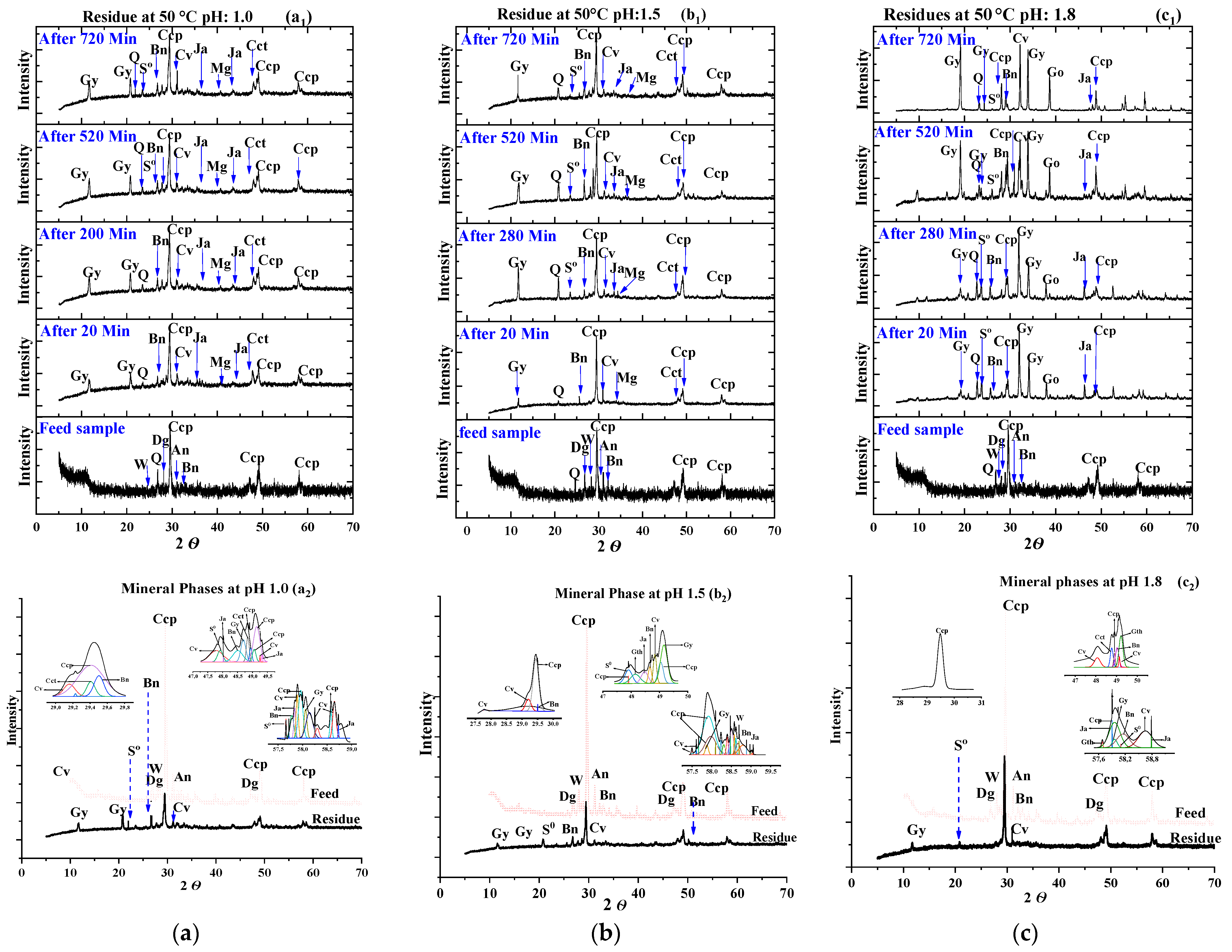
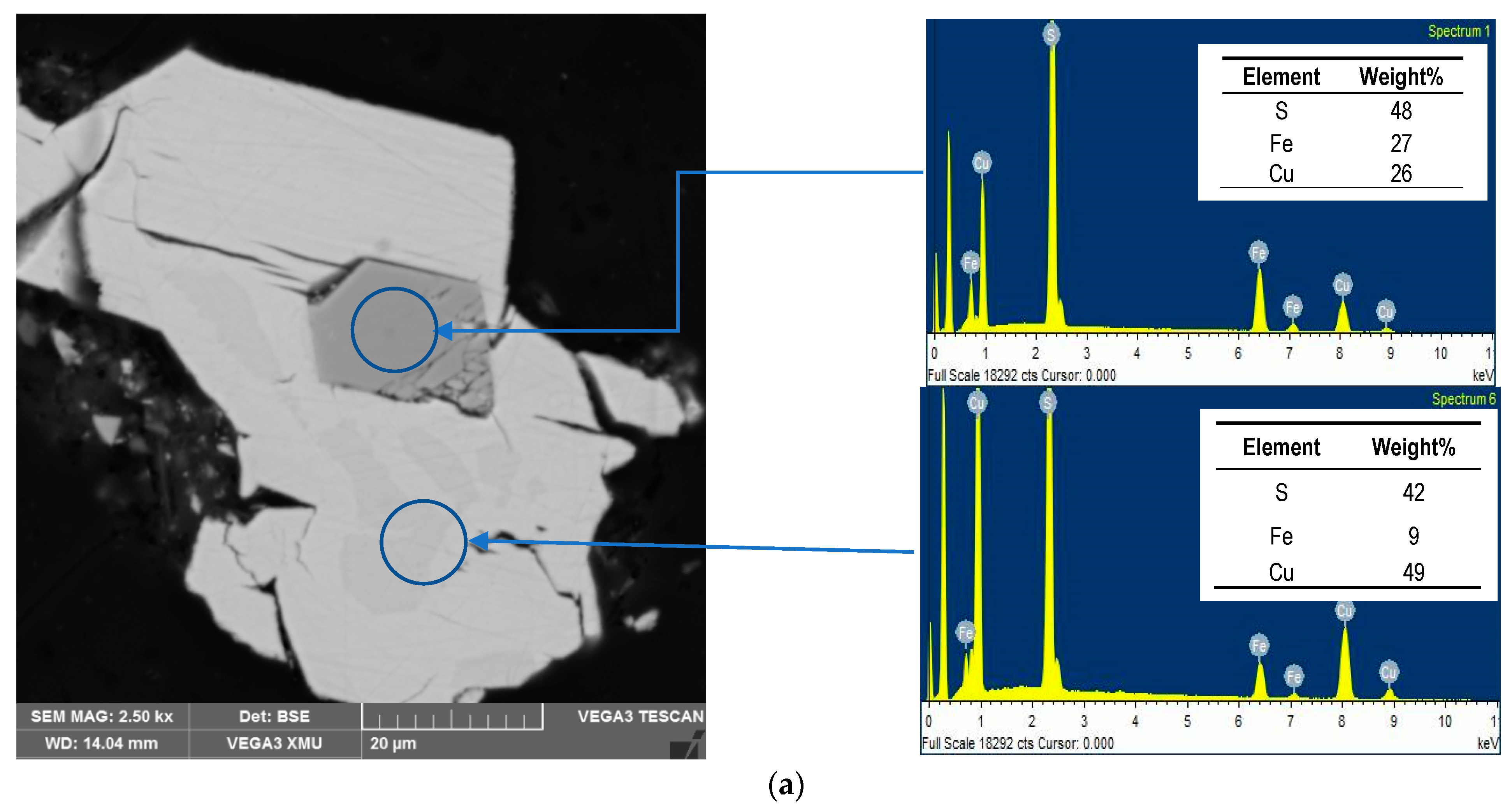
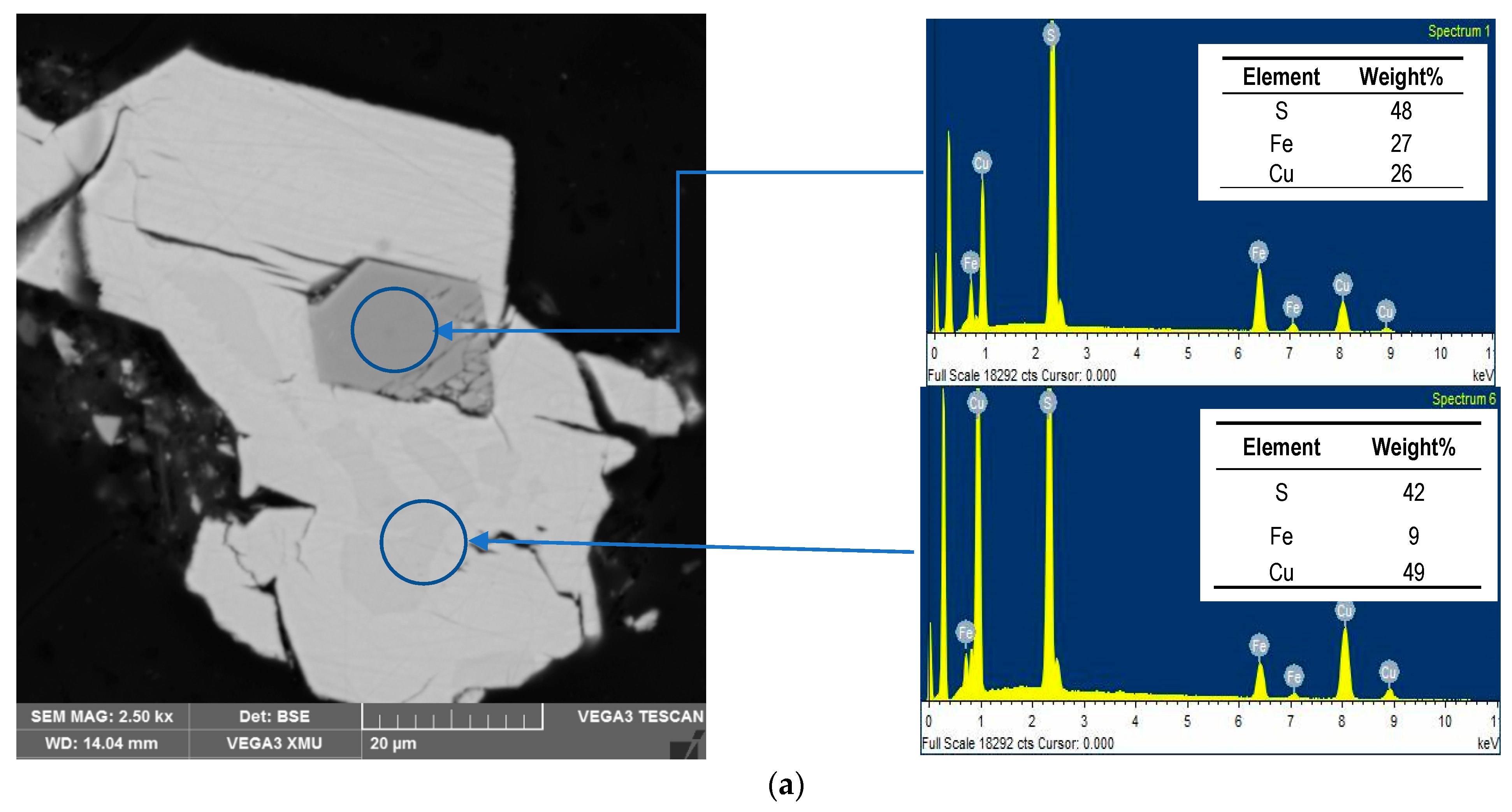
3.4. Residues Characterisation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.; NKawashima, N.; Li, J.; Chandra, A.P.; Gerson, A.R. A review of the structure, and fundamental mechanisms and kinetics of the leaching of chalcopyrite. Adv. Colloid Interface Sci. 2013, 197, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Moraga, C.; Castillo, J.; Edelmira, G.; Robles, P.; Toro, N. Submarine Tailings in Chile—A Review. Meatls 2008, 11, 780. [Google Scholar]
- Lundström, M.; Aromaa, J.; Forsén, O.; Barker, M.H. Reaction product layer on chalcopyrite in cupric chloride leaching. Can. Metall. Q 2008, 47, 245–252. [Google Scholar] [CrossRef]
- Olvera, O.G.; Rebolledo, M.; Asselin, E. Atmospheric ferric sulfate leaching of chalcopyrite: Thermodynamics, kinetics and electrochemistry. Hydrometallurgy 2016, 165, 148–158. [Google Scholar] [CrossRef]
- Dutrizac, J.E. Elemental sulphur formation during the ferric chloride leaching of chalcopyrite. Hydrometallurgy 1990, 23, 153–176. [Google Scholar] [CrossRef]
- Veloso, T.C.; Peixoto, J.J.M.; Pereira, M.S.; Leao, V.A. Kinetics of chalcopyrite leaching in either ferric sulphate or cupric sulphate media in the presence of NaCl. Int. J. Miner. Process. 2016, 148, 147–154. [Google Scholar] [CrossRef]
- Nikkhou, F.; Xia, F.; Deditius, A.P. Variable surface passivation during direct leaching of sphalerite by ferric sulfate, ferric chloride, and ferric nitrate in a citrate medium. Hydrometallurgy 2019, 188, 201–215. [Google Scholar] [CrossRef]
- Tshilombo, A.F. Mechanism and Kinetics of Chalcopyrite Passivation and Depassivation during Ferric and Microbial Leaching; The University of British Columbia: Vancouver, BC, Canada, 2004. [Google Scholar] [CrossRef]
- Naderi, H.; Abdollahy, M.; Mostoufi, N.; Koleini, M.; Shojaosadati, S.A.; Manafi, Z. Kinetics of chemical leaching of chalcopyrite from low grade copper ore: Behavior of different size fractions. Int. J. Miner. Met. Mater. 2011, 18, 638–645. [Google Scholar] [CrossRef]
- Klauber, C.; Parker, A.; Van Bronswijk, W.; Watling, H. Sulphur speciation of leached chalcopyrite surfaces as determined by x-ray photoelectron spectroscopy. Int. J. Miner. Process. 2001, 62, 65–94. [Google Scholar] [CrossRef]
- Salehi, S.; Noaparast, M.; Ziaeddin, S. The role of catalyst in chalcopyrite passivation during leaching. Int. J. Min. Geo-Eng. 2017, 1, 91–96. [Google Scholar]
- Debernardi, G.; Carlesi, C. Chemical-electrochemical approaches to the study passivation of chalcopyrite. Miner. Process. Extr. Met. Rev. 2013, 34, 10–41. [Google Scholar] [CrossRef]
- Ghahremaninezhad, A. The Surface Chemistry of Chalcopyrite during Electrochemical Dissolution. Ph.D. Thesis, The University of British Columbia, Vancouver, BC, Canada, 2012. [Google Scholar]
- Zhao, H.; Zhang, Y.; Zhang, X.; Qian, L.; Sun, M.; Yang, Y.; Zhang, Y.; Wang, J.; Kim, H.; Qiu, G. The dissolution and passivation mechanism of chalcopyrite in bioleaching: An overview. Miner. Eng. 2019, 136, 140–154. [Google Scholar] [CrossRef]
- Ruiz-sánchez, A.; Lapidus, G.T. Hydrometallurgy Study of chalcopyrite leaching from a copper concentrate with hydrogen peroxide in aqueous ethylene glycol media. Hydrometallurgy 2017, 169, 192–200. [Google Scholar] [CrossRef]
- Casas, J.M.; Crisóstomo, G.; Cifuentes, L. Speciation of the Fe(II)-Fe(III)-H2SO4-H2O system at 25 and 50 °C. Hydrometallurgy 2005, 80, 254–264. [Google Scholar] [CrossRef]
- Yue, G.; Asselin, E. Kinetics of Ferric Ion Reduction on Chalcopyrite and Its Influence on Leaching up to 150 °C. Electrochim. Acta 2014, 146, 307–321. [Google Scholar] [CrossRef]
- Hirato, T.; Majima, H.; Awakura, Y. The leaching of chalcopyrite with ferric sulfate. Met. Mater. Trans. A 1987, 18, 489–496. [Google Scholar] [CrossRef]
- Sapieszko, R.S.; Patel, R.C.; Matijevic, E. Ferric hydrous oxide sols. 2. Thermodynamics of aqueous hydroxo and sulfato ferric complexes. J. Phys. Chem. 1977, 81, 1061–1068. [Google Scholar] [CrossRef]
- Simons, K. Soil Sampling; Gilson, Inc.: Geogia, GA, USA, 2014. [Google Scholar]
- Nyembwe, K.J.; Fosso-Kankeu, E.; Waanders, F.; Nyembwe, K.D. Structural, compositional and mineralogical characterization of carbonatitic copper sulfide: Run of mine, concentrate and tailings. Int. J. Miner. Metall. Mater. 2019, 26, 143–151. [Google Scholar] [CrossRef]
- Antonijevic, G.D.; Bogdanovic, M.M. Investigation of the leaching of chalcopyritic ore in acidic solutions. Hydrometallurgy 2004, 73, 245–256. [Google Scholar] [CrossRef]
- Córdoba, E.M.; Muñoz, J.A.; Blázquez, M.L.; González, F.; Ballester, A. Leaching of chalcopyrite with ferric ion. Part I: General aspects. Hydrometallurgy 2008, 93, 81–87. [Google Scholar] [CrossRef]
- Salinas, K.E.; Herreros, O.; Torres, C.M. Leaching of Primary Copper Sulfide Ore. Mineral 2018, 8, 312. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.L.; Peters, E. Extractive Metallurgy of Copper; AIME: New York, NY, USA, 1976. [Google Scholar]
- Hirato, T.; Majima, H.; Awakura, Y. The leaching of chalcopyrite with cupric chloride. Met. Mater. Trans. A 1987, 18, 31–39. [Google Scholar] [CrossRef]
- Striggow, B. Field Measurement of Oxidation-Reduction Potential (ORP); United States Environmental Protection Agency: Georgia, GA, USA, 2013. [Google Scholar]
- Zhao, H.; JWang, J.; Qin, W.; Hu, M.; Qiu, G. Electrochemical dissolution of chalcopyrite concentrates in stirred reactor in the presence of Acidithiobacillus ferrooxidans. Int. J. Electrochem. Sci. 2015, 10, 848–858. [Google Scholar]
- Sokić, M.D.; Marković, B.; Živković, D. Kinetics of chalcopyrite leaching by sodium nitrate in sulphuric acid. Hydrometallurgy 2009, 95, 273–279. [Google Scholar] [CrossRef]
- Acero, P.; Cama, P.; Ayora, J.; Asta, C.M. Chalcopyrite dissolution rate law from pH 1 to 3. Geol. Acta 2009, 7, 389–397. [Google Scholar]
- Harmer-bassell, S.L.; Thomas, J.E.; Harmer, S.L.; Thomas, J.E. The evolution of surface layers formed during chalcopyrite leaching The evolution of surface layers formed during chalcopyrite leaching. Geochim. Cosmochim. Acta 2006, 70, 4392–4402. [Google Scholar] [CrossRef]
- Sokic, M.D.; Markovic, B.R.; Matkovic, V.L.; Strbac, N.D.; Zivkovic, D.T. Mechanism of Chalcopyrite Leaching in Oxidative Sulphuric Acid Solution. J. Chem. Chem. Eng. 2011, 5, 37–41. [Google Scholar]
- Dutrizac, J.E.; Chen, T.T.; Jambor, J.L. Mineralogical changes occurring during the ferric lon leaching of bornite. Met. Mater. Trans. A 1985, 16, 679–693. [Google Scholar] [CrossRef]
- Nyembwe, K.J.; EFosso-kankeu, E.; Waanders, F.; Mkandawire, M. pH-dependent leaching mechanism of carbonatitic chalcopyrite in ferric sulfate solution. Trans. Nonferrous Met. Soc. China 2021, 31, 2139–2152. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | Fe2(SO4)3 (M) | Rate Cu2+ | K M−1S−1 | Rxt0 Order Fe3+ | Ea (kJ/mol) |
|---|---|---|---|---|---|
| 298.15 | 0.025 | 8.31 × 10−5 | 3.3 × 10−3 | 0.979 | 79.04 |
| 0.050 | 1.61 × 10−4 | 3.2 × 10−3 | |||
| 0.075 | 2.45 × 10−4 | 3.3 × 10−3 | |||
| 323.5 | 0.025 | 9.70 × 10−4 | 3.9 × 10−2 | - | |
| 0.050 | 1.91 × 10−3 | 3.8 × 10−2 | |||
| 0.075 | 2.91 × 10−3 | 3.8 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nyembwe, K.J.; Fosso-Kankeu, E.; Waanders, F.; Mkandawire, M. Iron-Speciation Control of Chalcopyrite Dissolution from a Carbonatite Derived Concentrate with Acidic Ferric Sulphate Media. Minerals 2021, 11, 963. https://doi.org/10.3390/min11090963
Nyembwe KJ, Fosso-Kankeu E, Waanders F, Mkandawire M. Iron-Speciation Control of Chalcopyrite Dissolution from a Carbonatite Derived Concentrate with Acidic Ferric Sulphate Media. Minerals. 2021; 11(9):963. https://doi.org/10.3390/min11090963
Chicago/Turabian StyleNyembwe, Kolela J., Elvis Fosso-Kankeu, Frans Waanders, and Martin Mkandawire. 2021. "Iron-Speciation Control of Chalcopyrite Dissolution from a Carbonatite Derived Concentrate with Acidic Ferric Sulphate Media" Minerals 11, no. 9: 963. https://doi.org/10.3390/min11090963
APA StyleNyembwe, K. J., Fosso-Kankeu, E., Waanders, F., & Mkandawire, M. (2021). Iron-Speciation Control of Chalcopyrite Dissolution from a Carbonatite Derived Concentrate with Acidic Ferric Sulphate Media. Minerals, 11(9), 963. https://doi.org/10.3390/min11090963