
A New Constraint on the Physicochemical Condition of Mars Surface during the Amazonian Epoch Based on Chemical Speciation for Secondary Minerals in Martian Nakhlites

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Meteorite Specimen
2.2. Optical Microscopy
2.3. SEM and EPMA
2.4. Focused Ion Beam
2.5. Adsorption Experiment for Cr Adsorption on Mn Oxides in Water
2.6. Synchrotron Radiation X-ray Analysis
3. Results
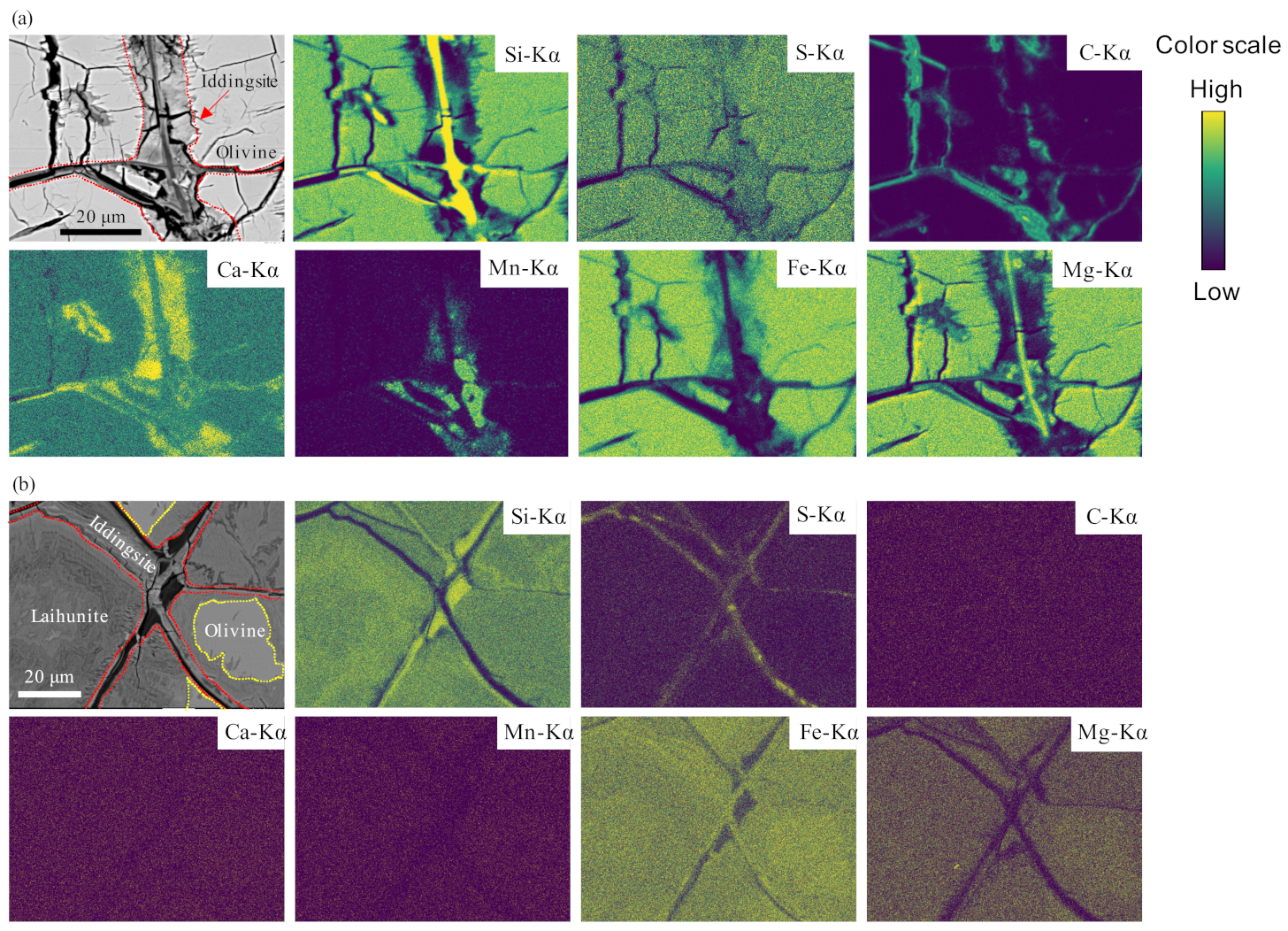
3.1. Identification of the Carbonate Phase in Iddingsite
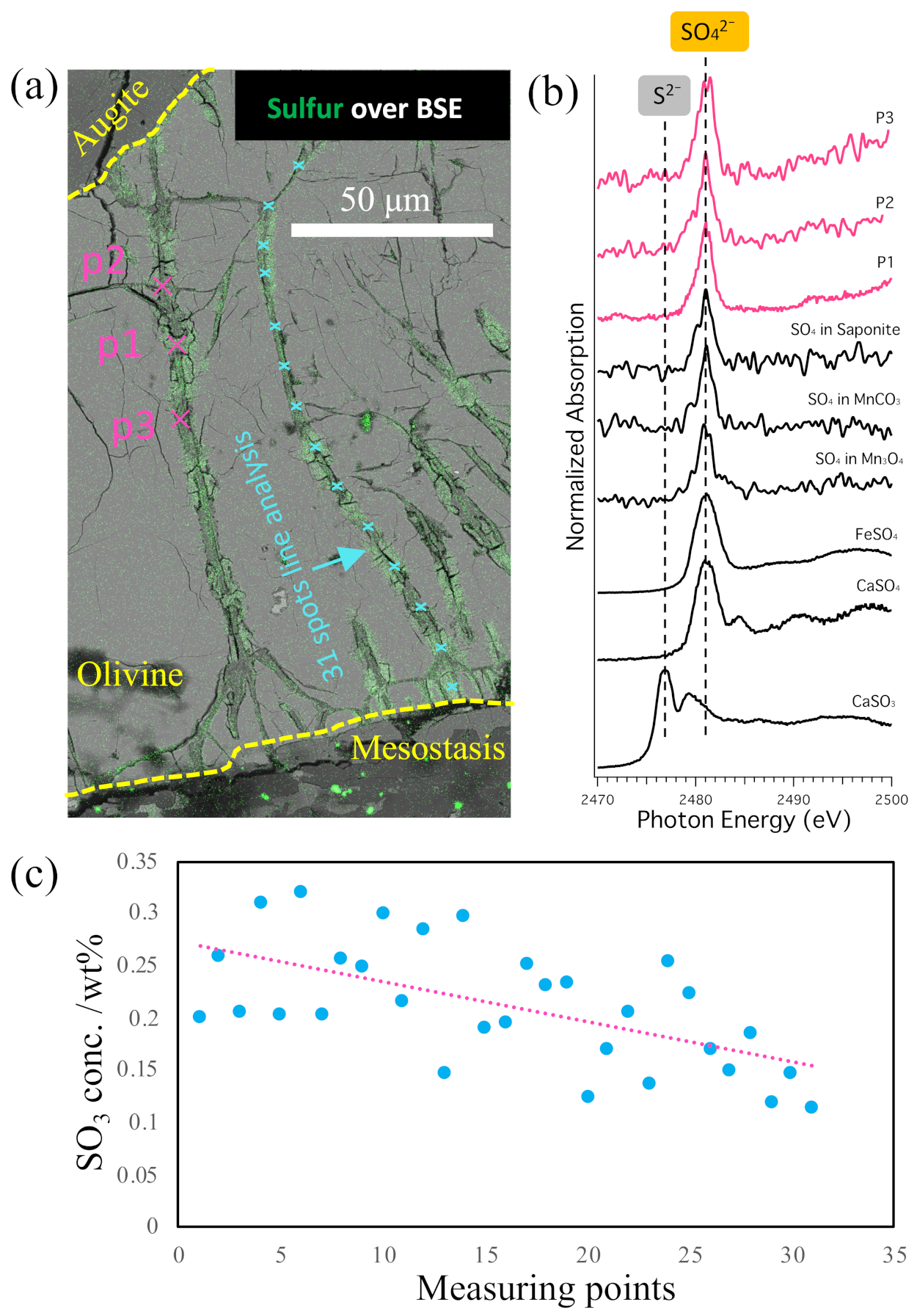
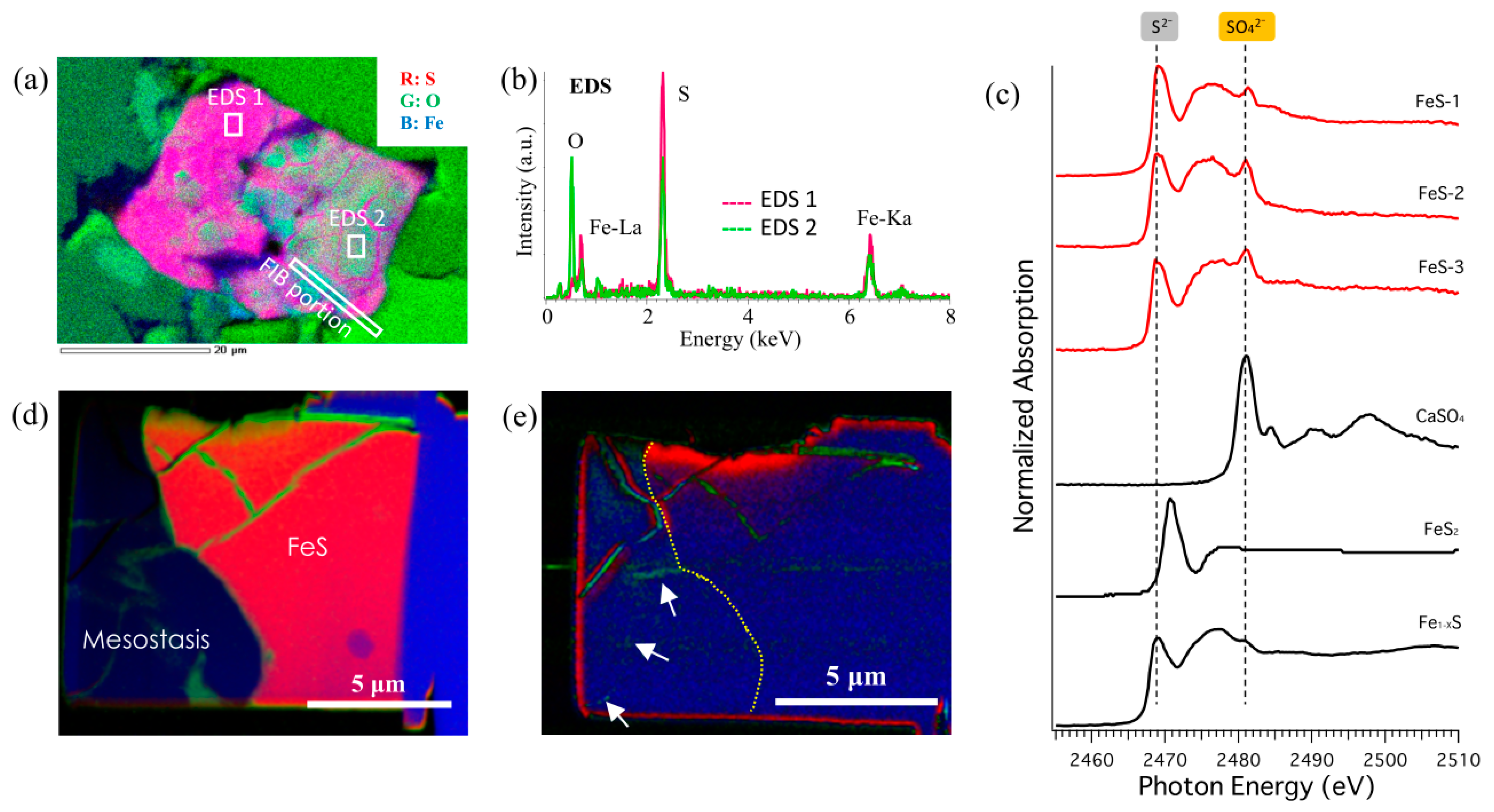
3.2. Speciation and Distribution of S in Iddingsite
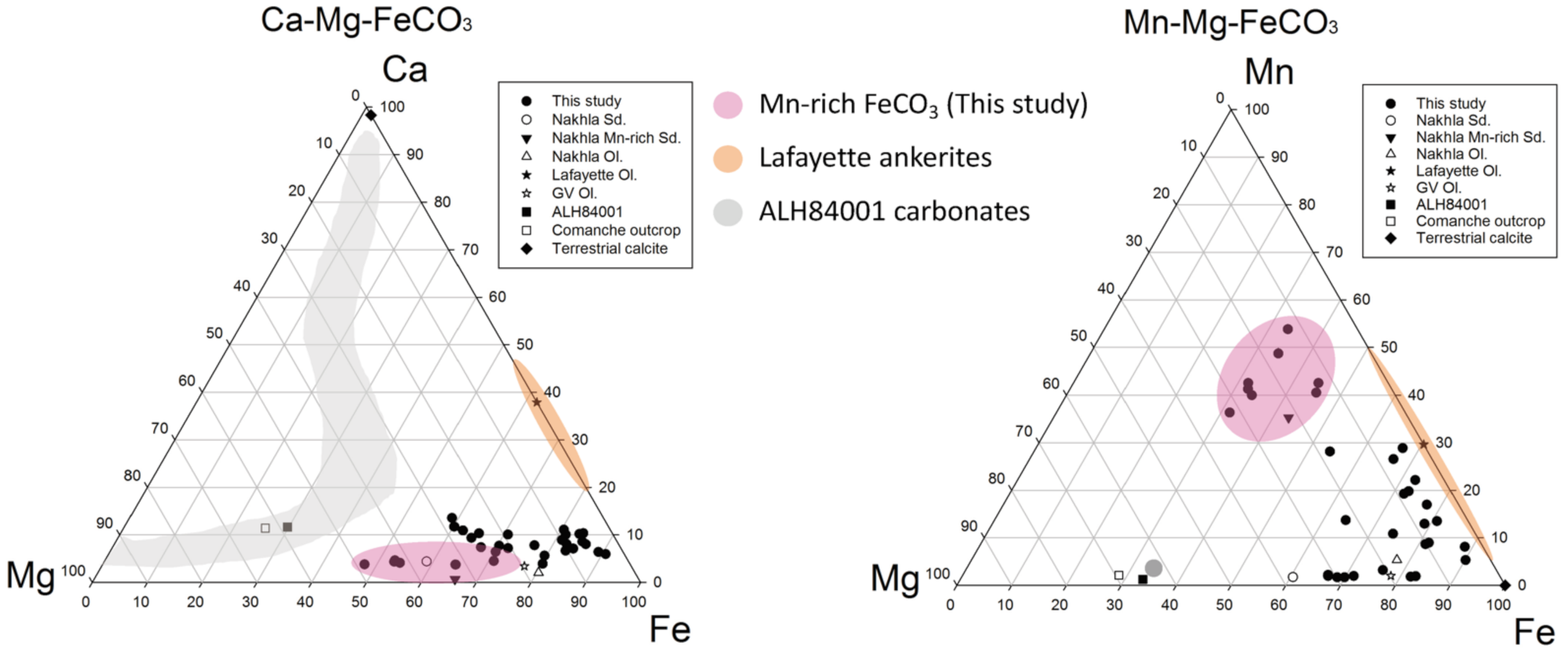
3.3. Characterization of Cr in the Mn-Bearing Siderite
3.4. Behavior and XANES Features of Cr Absorbed Onto MnOOH
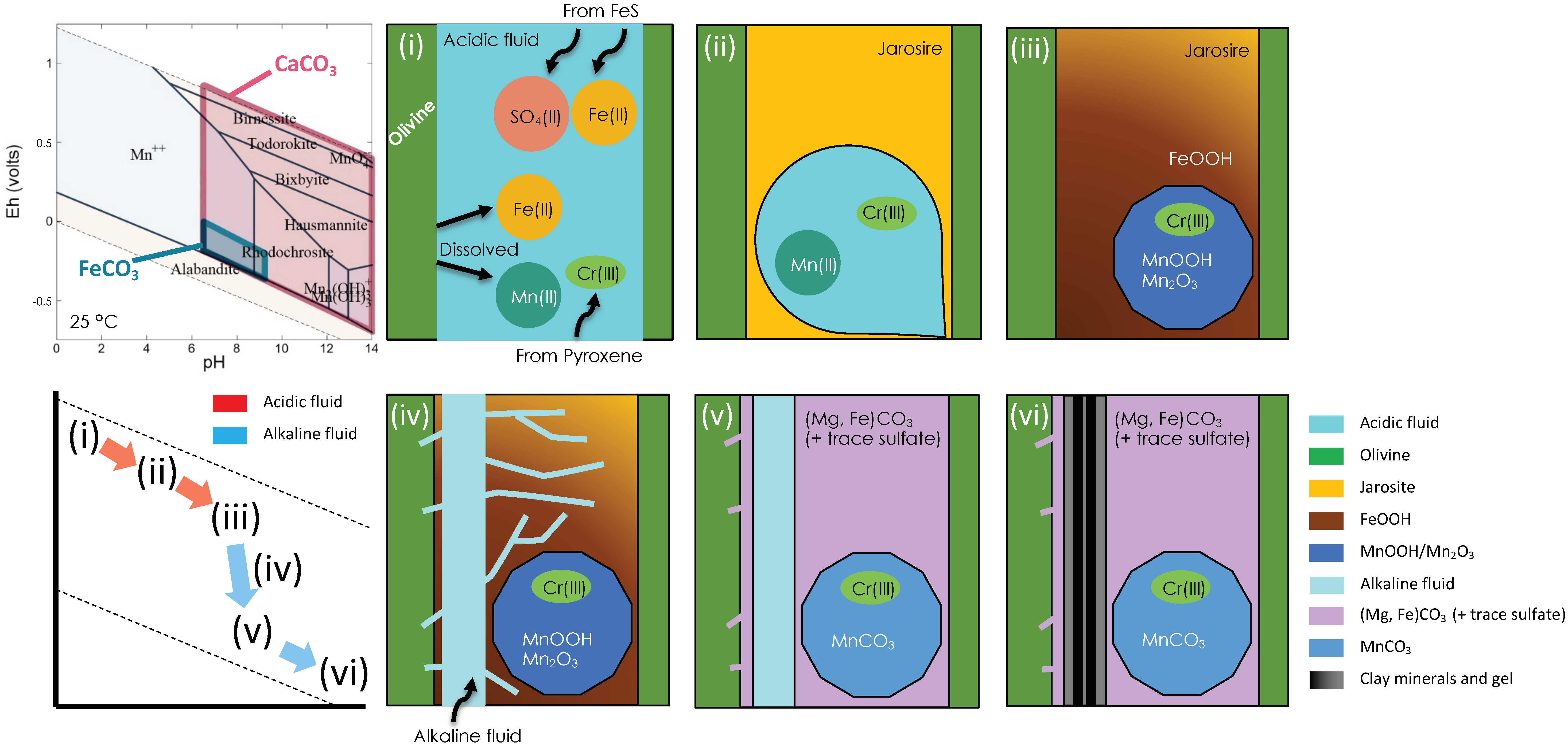
4. Discussion
4.1. Characterization of Siderite and Mn-Rich Siderite in Y 000593
4.2. S Chemistry
4.3. Mn and Cr Chemistries
4.4. Reaction Pathway during Iddingsite Formation
4.5. Estimation of the Alteration Process in Nakhlite
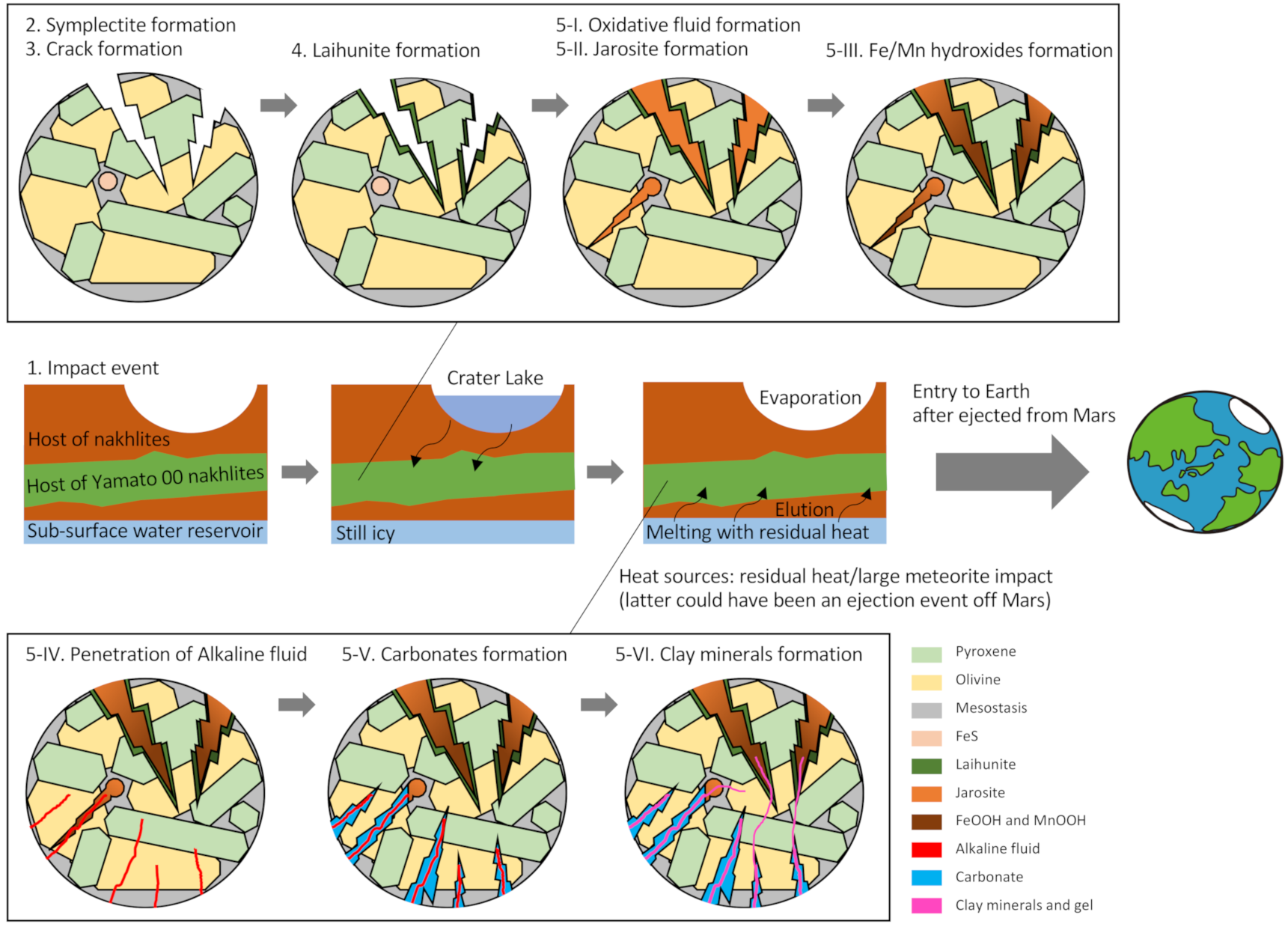
- Impact event: An impact event occurred near the host rock of the nakhlite. Explaining the formation of symplectite and laihunite with the involvement of temporary warm water is difficult. This event was the source of all subsequent reactions as follows. Some of the other small craters are also distributed around the crater of the nakhlite ejection [8]. The timescale of alteration is unknown. However, the iddingsite formation could not have occurred within a short period because no brecciated or shock-molten areas were observed in Y 000593, although it experienced weak shock (5–14 GPa) [71].
- Symplectite formation: The formation of symplectite in olivine indicates that the olivine was oxidized at high temperature, considered here as a shock-induced heat [9]. Symplectites lie parallel to the crystallographic axis (100) of the host olivine and are elongated along the [010] and [013] zones [9]. These symplectites were cross-cut by both sulfate-type iddingsite and Fe-Mg silicate after the siderite formation [16,72].
- Crack formation: Cracks were physically formed in the nakhlite minerals including olivine by the impact event described above. Processes 1 to 3 might have occurred nearly simultaneously. The crack size could be related to the distance between each nakhlite position and the impact site.
- Laihunite formation: Three-fold laihunite with a 3M structure forms along the walls of cracks at 800 °C to 400 °C [16]. Rapid oxidation occurred in olivine cracks by impact-induced hot gas (acid fog) at a short heating duration (e.g., <3 h) [56]. The degree of laihunite development increases closer to the impact site.
- Post-impact hydrothermal fluid reaction: Iddingsite was formed as filling material in cracks by poorly crystalline materials occurring at a maximum temperature of 400 °C. In this phase, the detailed alteration process suggested in the present study is shown in the following processes, as extensively discussed in Section 4.4.
- Oxidative fluid formed at the Martian surface, which caused alteration of the nakhlite host rock from the Martian surface to the subsurface zone. The melting of subsurface ice or permafrost by the heat originating from the shock event can be considered as the origin of this fluid.
- Sulfate dissolution from FeS in mesostasis generated the oxidative and acidic fluid, which formed sulfate minerals such as jarosite.
- The increase in pH by the aqueous alteration of silicate minerals caused the precipitation of FeOOH or the transformation of jarosite to FeOOH. MnOOH was also formed during this stage.
- CO2-rich reductive and alkaline fluid flowed from the subsurface ice by the residual heat. This fluid was incorporated into the nakhlite host rock along with iddingsite from bottom to top. The water reservoir would have been charged with CO2 from the thicker Noachian atmosphere. Further discussion on the origin of this CO2-rich fluid is given in Section 4.6.
- Carbonate was formed in the iddingsite. This fluid could not have reached the region that included laihunite because the coexistence of laihunite and siderite has not been reported. In addition, it is likely that not all of the sulfate veins were overprinted by the HCO3−-rich fluid because coexisting portions of sulfate and laihunite have been observed in Yamato 00 nakhlites [16,61].
- Poorly crystalline clay minerals filled the remaining pores within the iddingsite.
4.6. Discussion of Consistency with Other Nakhlites and Pairs
4.7. Discussion of the Origin of Jarosite and Other Sulfates in Y 000593
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cockell, C.S. Astrobiology: Understanding Life in the Universe; Wiley-Blackwell: Hoboken, NJ, USA, 2015; pp. 307–339. [Google Scholar]
- Murchie, S.L.; Mustard, J.F.; Ehlmann, B.L.; Milliken, R.E.; Bishop, J.L.; McKeown, N.K.; Noe Dobrea, E.Z.; Seelos, F.P.; Buczkowski, D.L.; Wiseman, S.M.; et al. A synthesis of Martian aqueous mineralogy after 1 Mars year of observations from the Mars Reconnaissance Orbiter. J. Geophys. Res. 2009, 114, 711. [Google Scholar] [CrossRef]
- Ehlmann, B.L.; Mustard, J.F.; Murchie, S.L.; Bibring, J.-P.; Meunier, A.; Fraeman, A.A.; Langevin, Y. Subsurface water and clay mineral formation during the early history of Mars. Nature 2011, 479, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Agee, C.B.; Wilson, N.V.; McCubbin, F.M.; Ziegler, K.; Polyak, V.J.; Sharp, Z.D.; Asmerom, Y.; Nunn, M.H.; Shaheen, R.; Thiemens, M.H.; et al. Unique Meteorite from Early Amazonian Mars: Water-Rich Basaltic Breccia Northwest Africa 7034. Science 2013, 339, 780–785. [Google Scholar] [CrossRef]
- Borg, L.; Drake, M.J. A review of meteorite evidence for the timing of magmatism and of surface or near-surface liquid water on Mars. J. Geophys. Res. Planets 2005, 110, 1678. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.Y.; Nyquist, L.E.; Reese, Y.; Weismann, H. The chronology of the nakhlite, Lafayette: Rb-Sr and Sm-Nd isotopic ages. In Proceedings of the 29th Lunar and Planetary Science Conference Abstracts, Houston, TX, USA, 16–20 March 1998. [Google Scholar]
- Nyquist, L.E.; Bogard, D.D.; Shih, C.Y.; Greshake, A.; Stöffler, D.; Eugster, O. Ages and geologic histories of Martian meteorites. Chronol. Evol. Mars. 2001, 96, 105–164. [Google Scholar]
- Kereszturi, A.; Chatzitheodoridis, E. Searching for the Source Crater of Nakhlite Meteorites. Orig. Life Evol. Biosph. 2016, 46, 455–471. [Google Scholar] [CrossRef]
- Mikouchi, T.; Yamada, I.; Miyamoto, M. Symplectic exsolution in olivine from the Nakhla martian meteorite. Meteorit. Planet. Sci. 2000, 35, 937–942. [Google Scholar] [CrossRef]
- Mikouchi, T.; Makishima, J.; Kurihara, T.; Hoffmann, V.H.; Miyamoto, M. Relative Burial Depth of Nakhlites Revisited. In Proceedings of the 43rd Lunar and Planetary Science Conference Abstracts, Houston, TX, USA, 19–23 March 2012. [Google Scholar]
- Cohen, B.E.; Mark, D.F.; Cassata, W.S.; Lee, M.R.; Tomkinson, T.; Smith, C.L. Taking the pulse of Mars via dating of a plume-fed volcano. Nat. Commun. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udry, A.; Day, J.M.D. 1.34 billion-year-old magmatism on Mars evaluated from the co-genetic nakhlite and chassignite meteorites. Geochim. Cosmochim. Acta 2018, 238, 292–315. [Google Scholar] [CrossRef]
- Mikouchi, T.; Miyahara, M. Comparative cooling rates of Nakhlites as inferred from iron-magnesium and calcium zoning of olivines. In Proceedings of the 33rd Lunar and Planetary Science Conference Abstracts, Houston, TX, USA, 11–15 March 2002. [Google Scholar]
- Mikouchi, T.; Miyamoto, M.; Koizumi, E.; Makishima, J.; McKay, G. Relative Burial Depths of Nakhlites: An Update. In Proceedings of the 37th Annual Lunar and Planetary Science Conference Abstracts, Houston, TX, USA, 13–17 March 2006. [Google Scholar]
- Changela, H.G.; Bridges, J.C. Alteration assemblages in the nakhlites: Variation with depth on Mars. Meteorit. Planet. Sci. 2011, 45, 1847–1867. [Google Scholar] [CrossRef]
- Noguchi, T.; Nakamura, T.; Misawa, K.; Imae, N.; Aoki, T.; Toh, S. Laihunite and jarosite in the Yamato 00 nakhlites: Alteration products on Mars? J. Geophys. Res. 2009, 114, E10004. [Google Scholar] [CrossRef] [Green Version]
- Tomkinson, T.; Lee, M.R.; Mark, D.F.; Smith, C.L. Sequestration of Martian CO2 by mineral carbonation. Nat. Commun. 2013, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Isherwood, R.J.; Jozwiak, L.M.; Jansen, J.C.; Andrews-Hanna, J.C. The volcanic history of Olympus Mons from paleo-topography and flexural modeling. Earth. Planet. Sci. Lett. 2013, 363, 88–96. [Google Scholar] [CrossRef]
- Hallis, L.J. D/H ratios of the inner Solar System. Phil. Trans. R. Soc. A 2017, 375, 20150390-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leshin, L.A.; Epstein, S.; Stolper, E.M. Hydrogen isotope geochemistry of SNC meteorites. Geochim. Cosmochim. Acta 1996, 60, 2635–2650. [Google Scholar] [CrossRef]
- White, L.M.; Gibson, E.K.; Thomas-Keprta, K.L.; Clemett, S.J.; McKay, D.S. Putative Indigenous Carbon-Bearing Alteration Features in Martian Meteorite Yamato 000593. Astrobiology 2014, 14, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Alexander, C.M.O.; Wang, J.; Simon, J.I.; Jones, J.H. Meteoritic evidence for a previously unrecognized hydrogen reservoir on Mars. Earth Planet. Sci. Lett. 2015, 410, 140–151. [Google Scholar] [CrossRef]
- Hallis, L.J. Alteration assemblages in the Miller Range and Elephant Moraine regions of Antarctica: Comparisons between terrestrial igneous rocks and Martian meteorites. Meteorit. Planet. Sci. 2013, 48, 165–179. [Google Scholar] [CrossRef]
- Hicks, L.J.; Bridges, J.C.; Gurman, S.J. Ferric saponite and serpentine in the nakhlite martian meteorites. Geochim. Cosmochim. Acta 2014, 136, 194–210. [Google Scholar] [CrossRef] [Green Version]
- Bridges, J.C.; Schwenzer, S.P. The nakhlite hydrothermal brine on Mars. Earth Planet. Sci. Lett. 2012, 359–360, 117–123. [Google Scholar] [CrossRef]
- Bridges, J.C.; Grady, M.M. Evaporite mineral assemblages in the nakhlite (Martian) meteorites. Earth Planet. Sci. Lett. 2000, 176, 267–279. [Google Scholar] [CrossRef]
- Gooding, J.L.; Wentworth, S.J.; Zolensky, M.E. Aqueous alteration of the Nakhla meteorite. Meteorit. Planet. Sci. 1991, 26, 135–143. [Google Scholar] [CrossRef]
- Hallis, L.J.; Taylor, G.J. Comparisons of the four Miller Range nakhlites, MIL 03346, 090030, 090032 and 090136: Textural and compositional observations of primary and secondary mineral assemblages. Meteorit. Planet. Sci. 2011, 46, 1787–1803. [Google Scholar] [CrossRef]
- Treiman, A.H.; Barrett, R.A.; Gooding, J.L. Preterrestrial aqueous alteration of the Lafayette (SNC) meteorite. Meteorit. Planet. Sci. 1993, 28, 86–97. [Google Scholar] [CrossRef]
- Okazaki, R.; Nagao, K.; Imae, N.; Kojima, H. Noble gas signatures of Antartic nakhlites, Yamato (Y) 000593, Y000749 and Y000802. Antarct. Meteor. Res. 2003, 16, 58–79. [Google Scholar]
- Misawa, K.; Kojima, H.; Imae, N.; Nakamura, N. The Yamato nakhlite consortium. Antarct. Meteor. Res. 2003, 16, 1–12. [Google Scholar]
- Lee, M.R.; Tomkinson, T.; Mark, D.F.; Stuart, F.M.; Smith, C.L. Evidence for silicate dissolution on Mars from the Nakhla meteorite. Meteorit. Planet. Sci. 2013, 48, 224–240. [Google Scholar] [CrossRef] [Green Version]
- Fendorf, S.E.; Zasoski, R.J. Chromium (III) Oxidation by δ-MnO2. Environ. Sci. Technol. 1992, 26, 79–85. [Google Scholar] [CrossRef]
- Weaver, R.M.; Hochella, M.F., Jr.; Ilton, E.S. Dynamic processes occurring at the CrIIIaq-manganite (γ-MnOOH) interface: Simultaneous adsorption, microprecipitation, oxidation/reduction, and dissolution. Geochim. Cosmochim. Acta 2002, 66, 4119–4132. [Google Scholar] [CrossRef]
- Takeichi, Y.; Inami, N.; Suga, H.; Miyamoto, C.; Ueno, T.; Mase, K.; Takahashi, Y.; Ono, K. Design and performance of a compact scanning transmission X-ray microscope at the Photon Factory. Rev. Sci. Instrum. 2016, 87, 013704. [Google Scholar] [CrossRef]
- Suga, H.; Kikuchi, S.; Takeichi, Y.; Miyamoto, C.; Miyahara, M.; Mitsunobu, S.; Ohigashi, T.; Mase, K.; Ono, K.; Takahashi, Y. Spatially Resolved Distribution of Fe Species around Microbes at the Submicron Scale in Natural Bacteriogenic Iron Oxides. Microbes Environ. 2017, 32, 283–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikouchi, T.; Koizumi, E.; Monkawa, A.; Miyamoto, A. Mineralogy and petrology of Yamato 000593: Comparison with other Martian nakhlite meteorites. Antarct. Meteor. Res. 2003, 16, 24–57. [Google Scholar]
- Pingitore, N.E., Jr.; Meitzner, G.; Love, K.M. Identification of sulfate in natural carbonates by x-ray absorption spectroscopy. Geochim. Cosmochim. Acta 1995, 59, 2477–2483. [Google Scholar] [CrossRef]
- Franz, H.B.; Kim, S.-T.; Farquhar, J.; Day, J.M.D.; Economos, R.C.; McKeegan, K.D.; Schmitt, A.K.; Irving, A.J.; Hoek, J.; Dottin, J., III. Isotopic links between atmospheric chemistry and the deep sulphur cycle on Mars. Nature 2014, 508, 364–368. [Google Scholar] [CrossRef] [Green Version]
- McCubbin, F.M.; Tosca, N.J.; Smirnov, A.; Nekcasil, H.; Steele, A.; Frie, M.; Lindsley, D.H. Hydrothermal jarosite and hematite in a pyroxene-hosted melt inclusion in martian meteorite Miller Range (MIL) 03346: Implications for magmatic-hydrothermal fluids on Mars. Geochim. Cosmochim. Acta. 2009, 73, 4907–4917. [Google Scholar] [CrossRef]
- Treiman, A.H. The nakhlite meteorites: Augite-rich igneous rocks from Mars. Chem. Erde Geochem. 2005, 65, 203–270. [Google Scholar] [CrossRef]
- Schwartz, J.M.; McCallum, I.S.; Camara, F.; Domeneghetti, C.; Zema, M. Pasamonte Eucrite: Subsolidus Thermal History. In Proceedings of the 33rd Lunar and Planetary Science Conference Abstracts, Houston, TX, USA, 11–15 March 2002. [Google Scholar]
- Schoneveld, L.; Barnes, S.J.; Makkonen, H.V.; Le Vaillant, M.; Paterson, D.J.; Taranovic, V.; Wang, K.Y.; Mao, Y.J. Zoned Pyroxenes as prospectivity indicators for magmatic Ni-Cu sulfide mineralization. Front. Earth Sci. 2020, 8, 256. [Google Scholar] [CrossRef]
- McKay, G.; Le, L.; Mikouchi, T. Al, Ti, and Cr: Complex Zoning in Synthetic and Natural Nakhlite Pyroxenes. In Proceedings of the 31st Symposium on Antarctic Meteorites Abstracts, Tachikawa, Japan, 6 June 2007. [Google Scholar]
- McKay, G.; Mikouchi, T.; Schwandt, C. Additional Complexities in Nakhlite Pyroxenes: A Progress (?) Report. In Proceedings of the 37th Lunar and Planetary Science Conference Abstracts, Houston, TX, USA, 13–17 March 2006. [Google Scholar]
- Ohta, A.; Kagi, H.; Tsuno, H.; Nomura, M.; Okai, T. Speciation study of Cr(VI/III) reacting with humic substances and determination of local structure of Cr binding humic substances using XAFS spectroscopy. Geochem. J. 2012, 46, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Karner, J.M.; Papike, J.J.; Sutton, S.R.; Shearer, C.K.; McKay, G.; Le, L.; Burger, P. Valence state partitioning of Cr between pyroxene-melt: Effects of pyroxene and melt composition and direct determination of Cr valence states by XANES. Application to Martian basalt QUE 94201 composition. Am. Mineral. 2007, 92, 2002–2005. [Google Scholar] [CrossRef]
- Eeckhout, S.G.; Bolfan-Casanova, N.; Mccammon, C.; Klemme, S.; Amiguet, E. XANES study of the oxidation state of Cr in lower mantle phases: Periclase and magnesium silicate perovskite. Am. Mineral. 2007, 92, 966–972. [Google Scholar] [CrossRef]
- Fang, Z.; Qin, L.; Liu, W.; Yao, T.; Chen, X.; Wei, S. Absence of hexavalent chromium in marine carbonates: Implications for chromium isotopes as paleoenvironment proxy. Natl. Sci. Rev. 2020, 1–8. [Google Scholar] [CrossRef]
- Grady, M.M.; Anand, M.; Gilmore, M.A.; Watson, J.S.; Wright, I.P. Alteration of the nakhlite lava pile: Was water on the surface, seeping down, or at depth, percolating up? Evidence (such as it is) from carbonates. In Proceedings of the 28th Lunar and Planetary Science Conference Abstracts, Houston, TX, USA, 12–16 March 2007. [Google Scholar]
- Morris, R.V.; Ruff, S.W.; Gellert, R.; Ming, D.W.; Arvidson, R.E.; Clark, B.C.; Golden, D.C.; Siebach, K.; Klingelhöfer, G.; Schröder, C.; et al. Identification of Carbonate-Rich Outcrops on Mars by the Spirit Rover. Science 2010, 329, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Mittlefehldt, D.W. ALH84001, a cumulate orthopyroxenite member of the Martian meteorite clan. Meteorit. Planet. Sci. 1994, 29, 214–221. [Google Scholar]
- Bridges, J.C.; Hicks, L.J.; Treiman, A.H. Carbonates on Mars. In Volatiles in the Martian Crust; Elsevier: Amsterdam, The Netherlands, 2019; Chapter 5; pp. 89–118. [Google Scholar] [CrossRef]
- Banfield, J.F.; Veblen, D.R.; Jones, B.F. Transmission electron microscopy of subsolidus oxidation and weathering of olivine. Contrib. Miner. Petrol. 1990, 106, 110–123. [Google Scholar] [CrossRef]
- Kondoh, S.; Kitamura, M.; Morimoto, N. Synthetic laihunite (□xFe2+2-3x Fe3+2xSiO4), an oxidation product of olivine. Am. Mineral. 1985, 70, 737–746. [Google Scholar]
- Tomioka, N.; Morlok, A.; Koike, C.; Köhler, M.; Grady, M. Laihunite in planetary materials: An FTIR and TEM study of oxidized synthetic and meteoritic Fe-rich olivine. J. Mineral. Petrol. Sci. 2012, 107, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.P.; Mojzsis, S.J.; Coath, C.D. Sulfur isotopic compositions of individual sulfides in Martian meteorites ALH84001 and Nakhla: Implications for crust–regolith exchange on Mars. Earth Planet. Sci. Lett. 2000, 184, 23–35. [Google Scholar] [CrossRef]
- Chiriță, P. Aqueous Oxidation of Iron Monosulfide (FeS) by Molecular Oxygen. Miner. Process. Extr. Metall. Rev. 2016, 37, 305–310. [Google Scholar] [CrossRef]
- Farquhar, J.; Thiemens, M.H. Oxygen cycle of the Martian atmosphere-regolith system: Δ17O of secondary phases in Nakhla and Lafayette. J. Geophys. Res. Planets 2000, 105, 11991–11997. [Google Scholar] [CrossRef]
- Greenwood, J.P.; Riciputi, L.R.; McSween, H.Y., Jr.; Taylor, L.A. Modified sulfur isotopic compositions of sulfides in the nakhlites and Chassigny. Geochim. Cosmochim. Acta 2000, 64, 1121–1131. [Google Scholar] [CrossRef]
- Suga, H.; Sago, N.; Miyahara, M.; Ohigashi, T.; Inagaki, Y.; Yamaguchi, A.; Ohtani, E. Chemical speciation analyses of alteration products in nakhlites Y 000593. In Proceedings of the Eighth Symposium on Polar Science Abstracts, Tachikawa, Japan, 5 December 2017. [Google Scholar]
- Shiraishi, N.; Suga, H.; Miyahara, M.; Ohigashi, T.; Inagaki, Y.; Yamaguchi, A.; Tomioka, N.; Kodama, Y.; Ohtani, E. Elucidation of aqueous alteration of Yamato 000749 based on multi-probe microscopy. In Proceedings of the Tenth Symposium on Polar Science Abstracts, Tachikawa, Japan, 5 December 2019. [Google Scholar]
- Madden, M.E.E.; Bodnar, R.J.; Rimstidt, J.D. Jarosite as an indicator of water-limited chemical weathering on Mars. Nature 2004, 431, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Miyahara, M.; Suga, H.; Yamaguchi, A.; Wakabayashi, D.; Yamashita, S.; Takeichi, Y.; Takahashi, Y.; Ohtani, E. The discovery of Mn-precipitates in nakhlites Yamato 000802. In Proceedings of the 11th Symposium on Polar Science Abstracts, Tachikawa, Japan, 3 December 2020. [Google Scholar]
- Hollister, L.S.; Gancarz, A.J. Conpositional sextor-zoning in clinopyroxene from the narce area, Italy. Am. Mineral. 1971, 56, 959–979. [Google Scholar]
- Noda, N.; Imamura, S.; Sekine, Y.; Kurisu, M.; Fukushi, K.; Terada, N.; Uesugi, S.; Numako, C.; Takahashi, Y.; Hartmann, J. Highly Oxidizing Aqueous Environments on Early Mars Inferred from Scavenging Pattern of Trace Metals on Manganese Oxides. J. Geophys. Res. Planets 2019, 124, 1282–1295. [Google Scholar] [CrossRef]
- Lanza, N.L.; Fischer, W.W.; Wiens, R.; Grotzinger, J.; Ollila, A.M.; Cousin, A.; Anderson, R.B.; Clark, B.C.; Gellert, R.; Mangold, N.; et al. High manganese concentrations in rocks at Gale crater, Mars. Geophys. Res. Lett. 2014, 41, 5755–5763. [Google Scholar] [CrossRef] [Green Version]
- Lanza, N.L.; Wiens, R.C.; Arvidson, R.E.; Clark, B.C.; Fischer, W.W.; Gellert, R.; Grotzinger, J.P.; Hurowitz, J.A.; McLennan, S.M.; Morris, R.V.; et al. Oxidation of manganese in an ancient aquifer, Kimberley formation, Gale crater, Mars. Geophys. Res. Lett. 2016, 43, 7398–7407. [Google Scholar] [CrossRef] [Green Version]
- Durocher, J.L.; Schindler, M. Iron-hydroxide, iron-sulfate and hydrous-silica coatings in acid-mine tailings facilities: A comparative study of their trace-element composition. Appl. Geochem. 2011, 26, 1337–1352. [Google Scholar] [CrossRef]
- Brookins, D.G. Eh-pH Diagrams for Geochemistry; Springer: Berlin, Germany, 1988; pp. 94–96. [Google Scholar]
- Fritz, J.; Artemieva, N.; Greshake, A. Ejection of Martian meteorites. Meteorit. Planet. Sci. 2005, 40, 1393–1411. [Google Scholar] [CrossRef]
- Lee, M.R.; Chatzitheodoridis, E. Formation of berthierine in the Martian meteorite Nakhla be replacement of aluminosilicate glass. In Proceedings of the 78th Annual Meeting of the Meteoritical Society Abstracts, Berkeley, CA, USA, 27–31 July 2015. [Google Scholar]
- Shiraishi, N.; Suga, H.; Miyahara, M.; Ohigashi, T.; Inagaki, Y.; Yamaguchi, A.; Tomioka, N.; Kodama, Y.; Ohtani, E. Aqueous alteration of Yamato 000749 based on multi-probe microscopic observation. In Proceedings of the Ninth Symposium on Polar Science Abstracts, Tachikawa, Japan, 4 December 2018. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suga, H.; Suzuki, K.; Usui, T.; Yamaguchi, A.; Sekizawa, O.; Nitta, K.; Takeichi, Y.; Ohigashi, T.; Takahashi, Y. A New Constraint on the Physicochemical Condition of Mars Surface during the Amazonian Epoch Based on Chemical Speciation for Secondary Minerals in Martian Nakhlites. Minerals 2021, 11, 514. https://doi.org/10.3390/min11050514
Suga H, Suzuki K, Usui T, Yamaguchi A, Sekizawa O, Nitta K, Takeichi Y, Ohigashi T, Takahashi Y. A New Constraint on the Physicochemical Condition of Mars Surface during the Amazonian Epoch Based on Chemical Speciation for Secondary Minerals in Martian Nakhlites. Minerals. 2021; 11(5):514. https://doi.org/10.3390/min11050514
Chicago/Turabian StyleSuga, Hiroki, Keika Suzuki, Tomohiro Usui, Akira Yamaguchi, Oki Sekizawa, Kiyofumi Nitta, Yasuo Takeichi, Takuji Ohigashi, and Yoshio Takahashi. 2021. "A New Constraint on the Physicochemical Condition of Mars Surface during the Amazonian Epoch Based on Chemical Speciation for Secondary Minerals in Martian Nakhlites" Minerals 11, no. 5: 514. https://doi.org/10.3390/min11050514
APA StyleSuga, H., Suzuki, K., Usui, T., Yamaguchi, A., Sekizawa, O., Nitta, K., Takeichi, Y., Ohigashi, T., & Takahashi, Y. (2021). A New Constraint on the Physicochemical Condition of Mars Surface during the Amazonian Epoch Based on Chemical Speciation for Secondary Minerals in Martian Nakhlites. Minerals, 11(5), 514. https://doi.org/10.3390/min11050514