
In Situ EXAFS Study of Sr Adsorption on TiO2(110) under High Ionic Strength Wastewater Conditions

 ,
,  and
and
Abstract
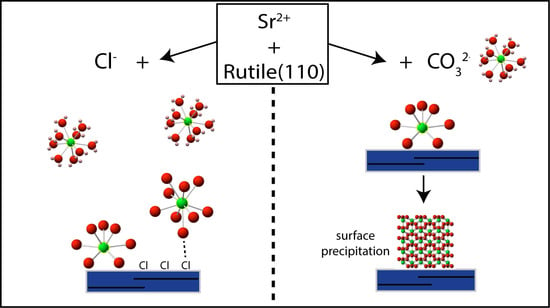
:
1. Introduction
2. Experimental Section
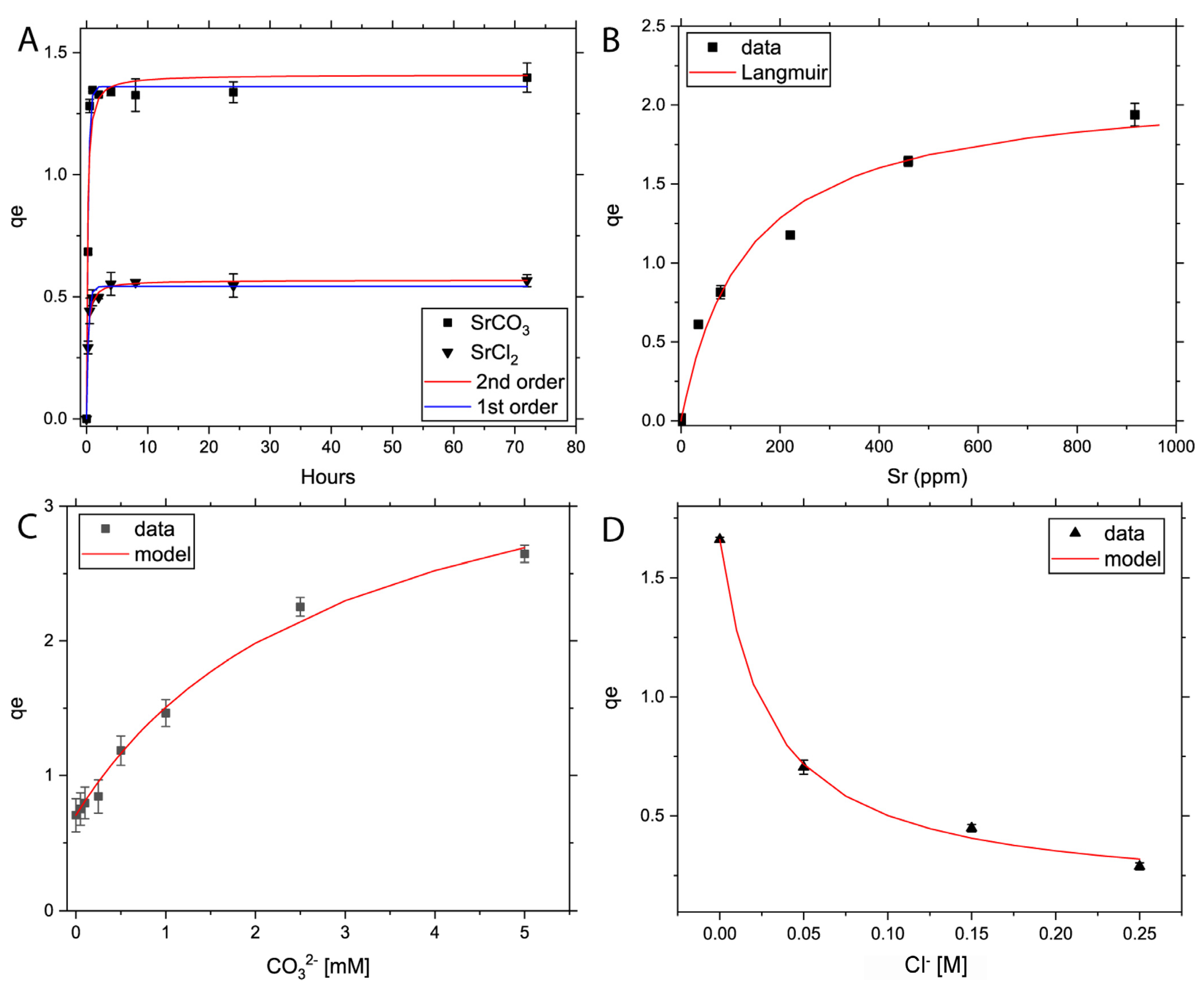
2.1. Powder Adsorption Rates and Isotherm Experiments
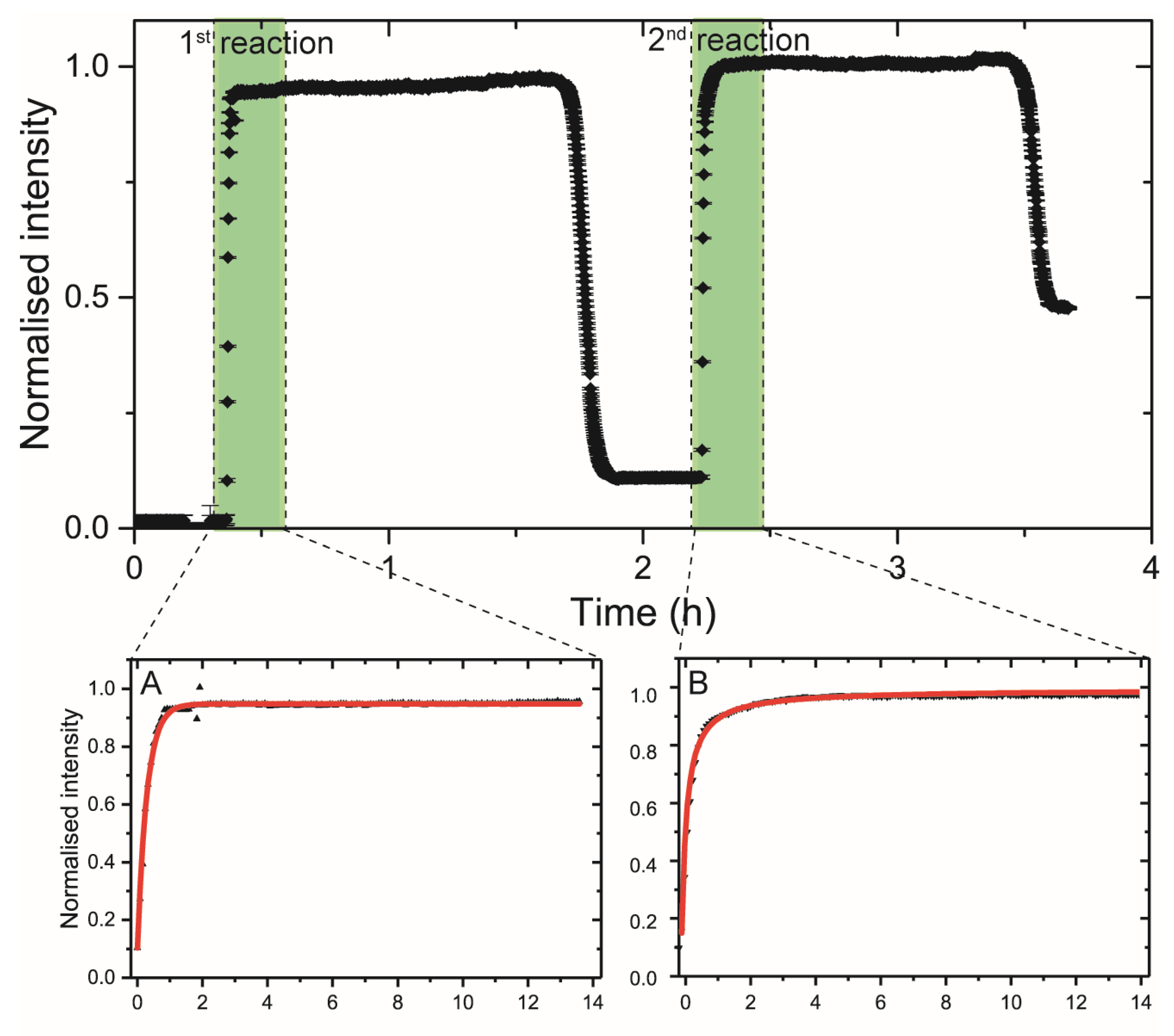
2.2. Ex Situ and In Situ Grazing Incidence EXAFS (GIXAFS) Adsorption Experiments
2.2.1. Ex Situ Batch Experiments
2.2.2. In Situ Single Pass Flow-through Experiment
2.3. Sr K-Edge GIXAFS Analyses
2.4. X-ray Reflectivity (XRR) and Grazing Incidence XRD GI-XRD
2.5. X-ray Photoelectron Spectroscopy (XPS)
3. Results
3.1. Powder Adsorption Rate and Isotherm Experiments
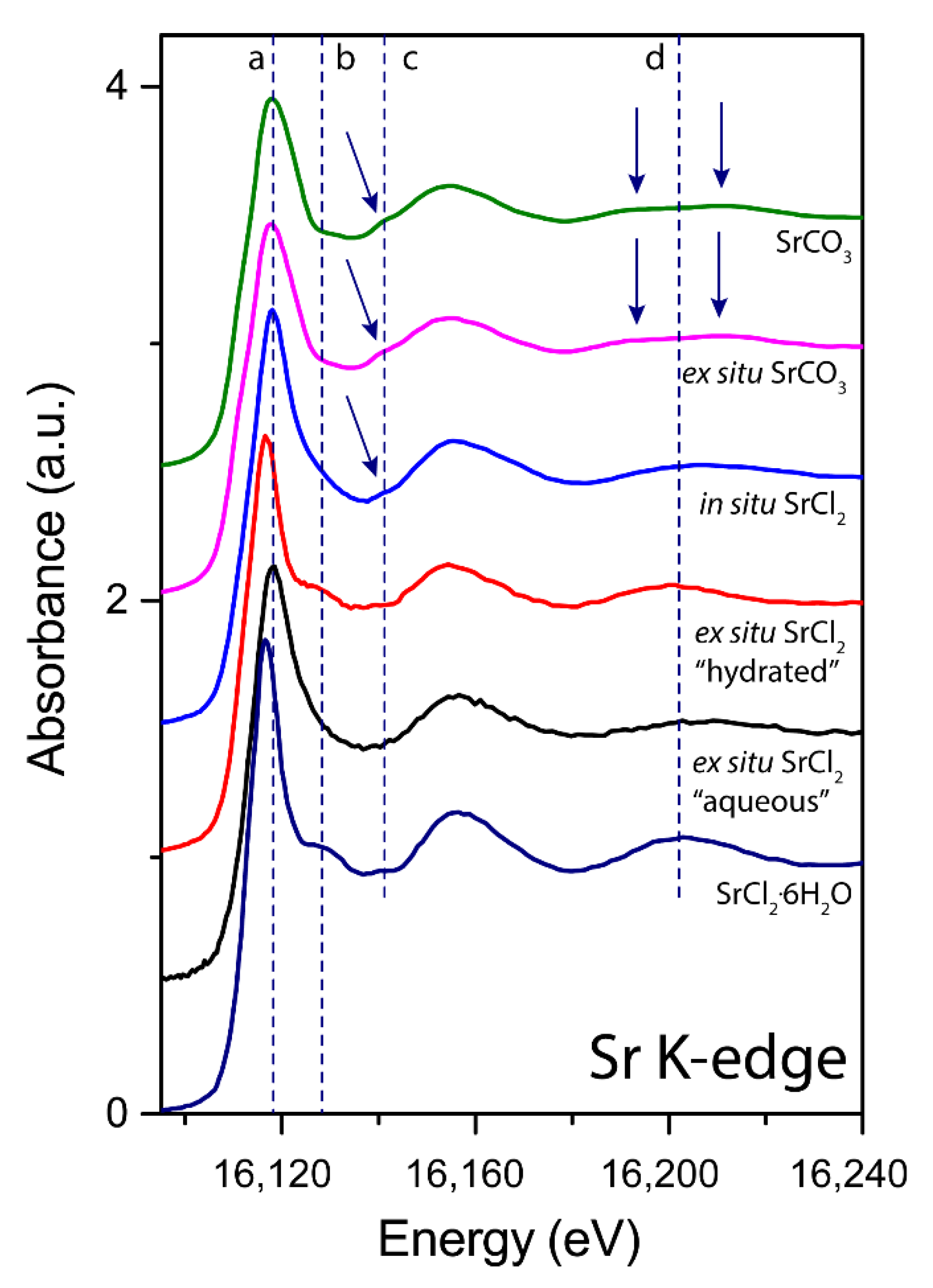
3.2. X-ray Absorption near Edge Structure (XANES) Analyses
3.3. GIXAFS Analyses
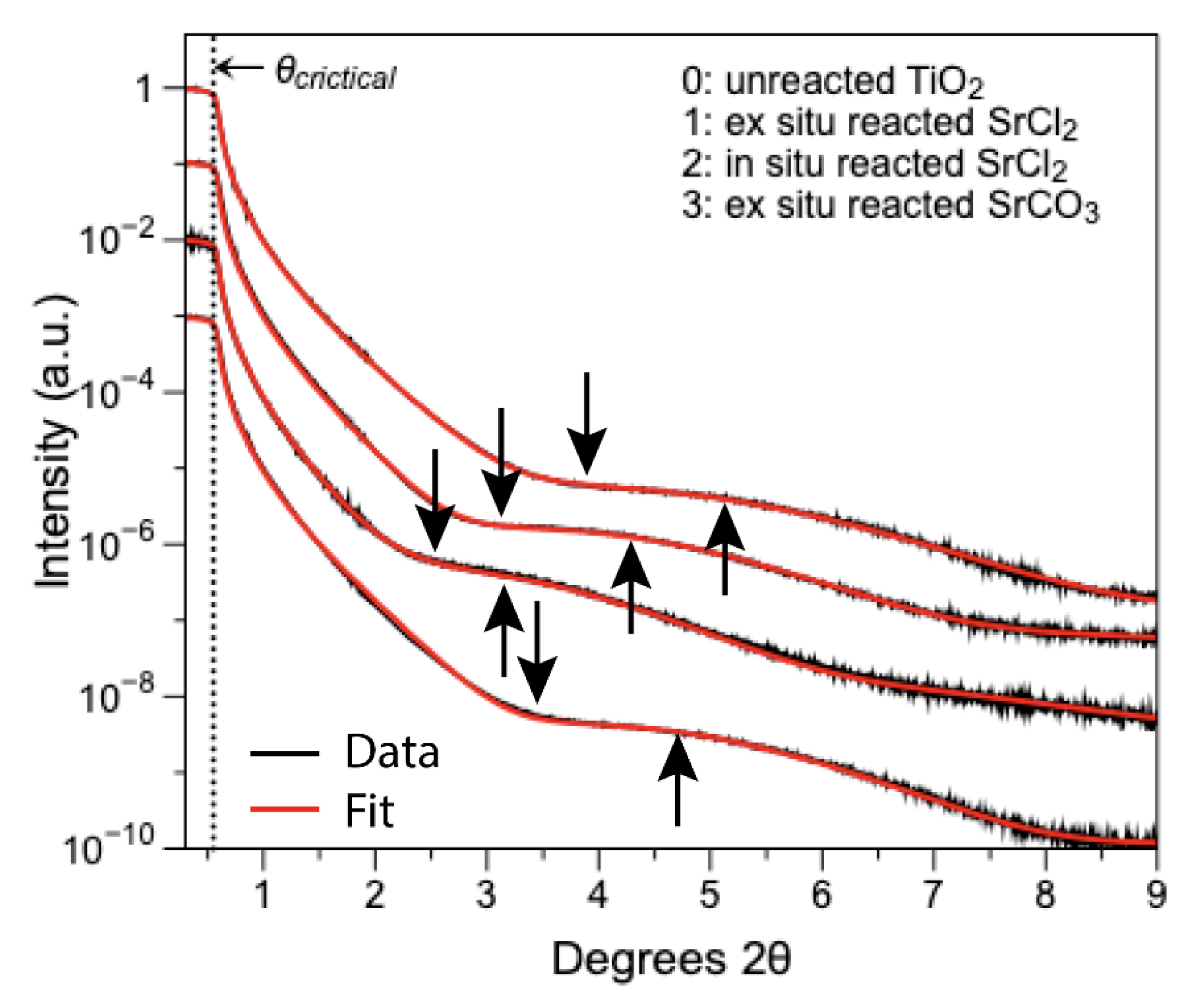
3.4. X-ray Reflectivity and GI-XRD
3.5. X-ray Photoelectron Spectroscopy (XPS)
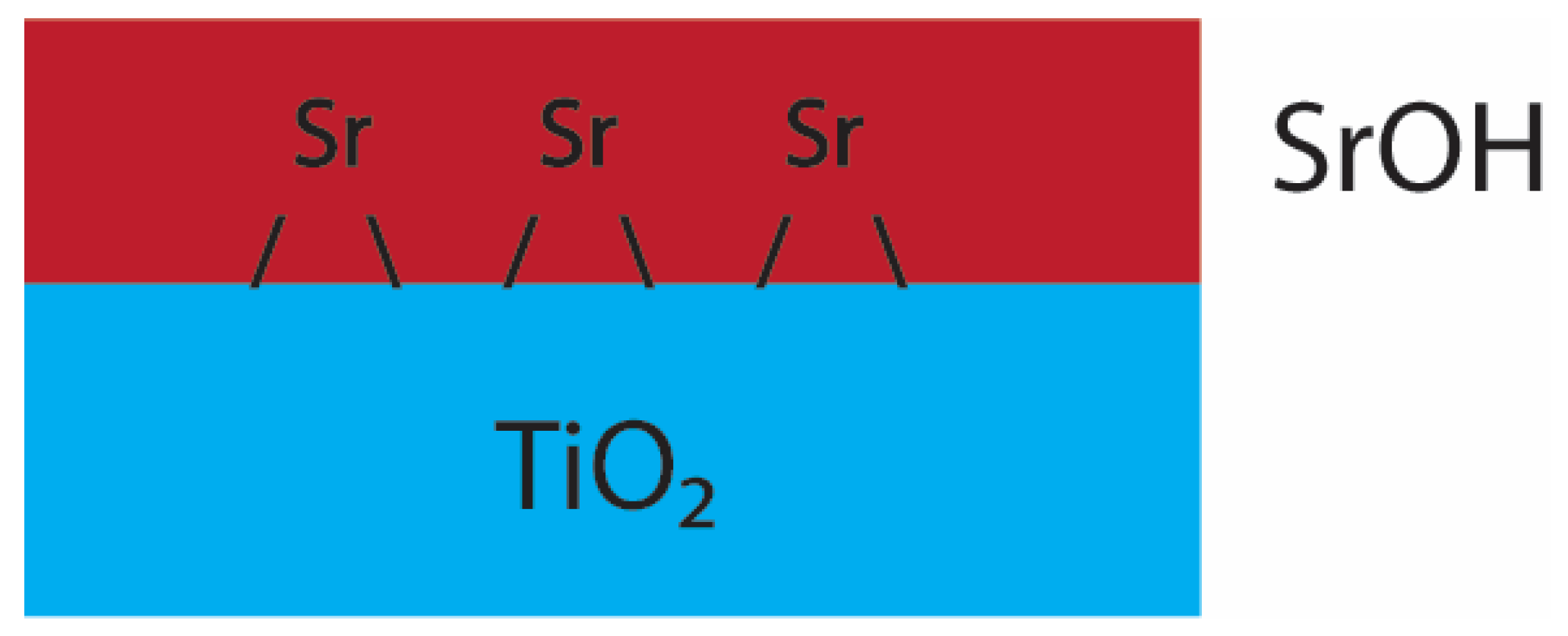
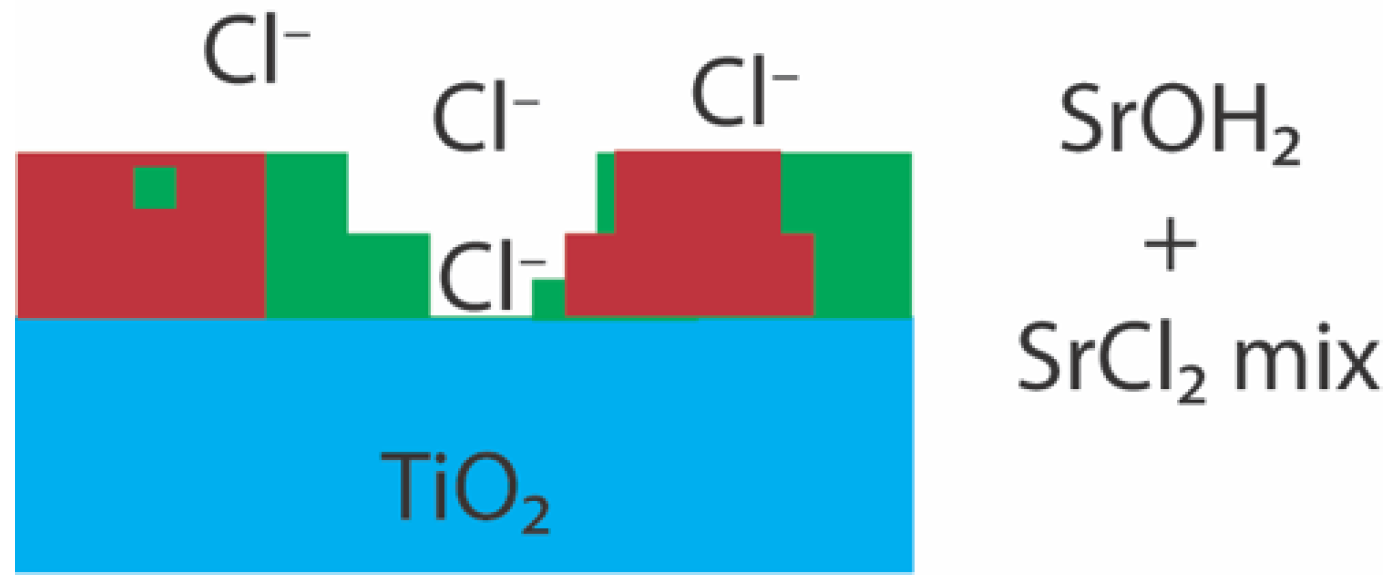
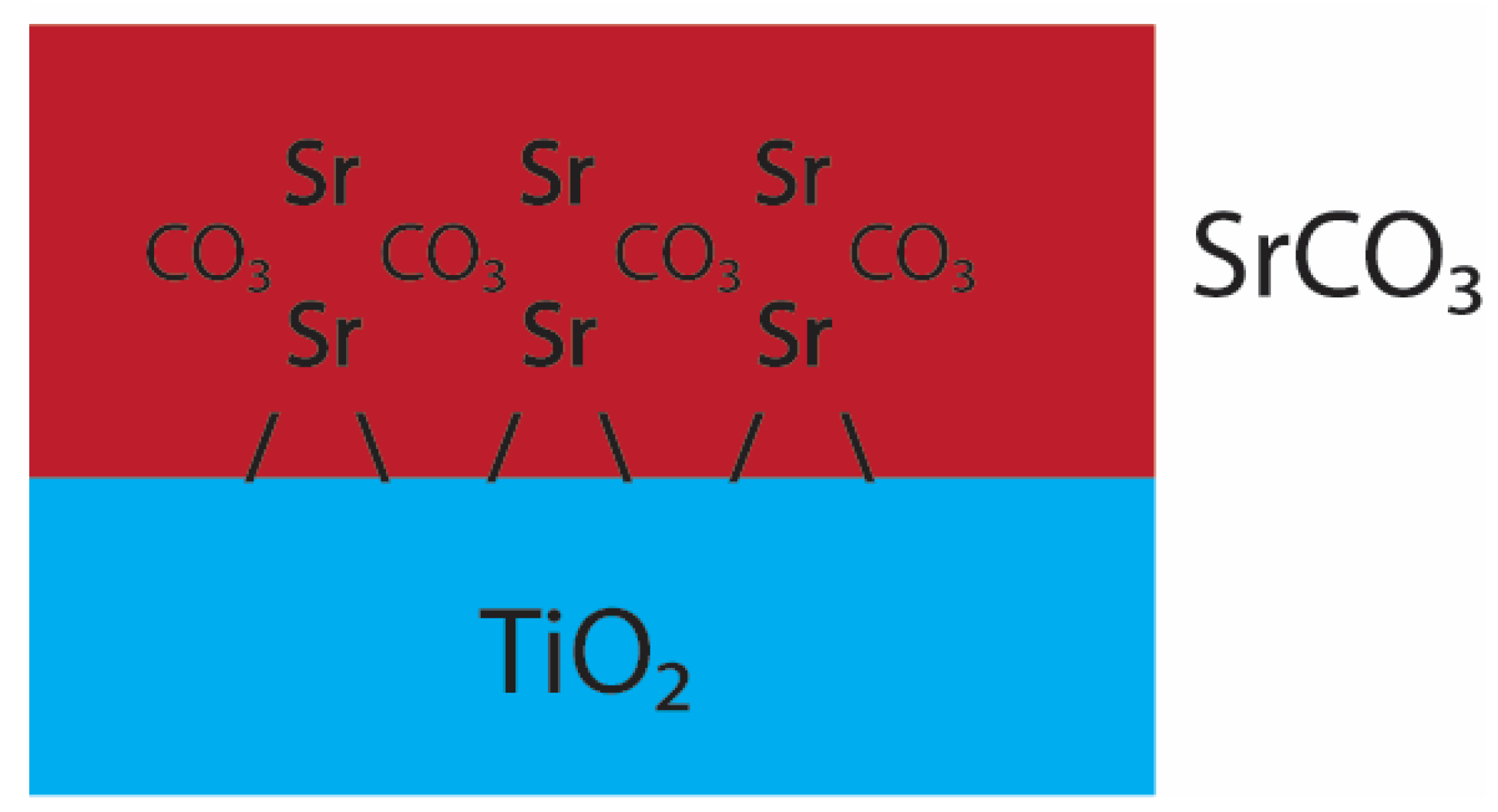
4. Discussion
5. Implications for Sr Wastewater Treatment
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Koarai, K.; Kino, Y.; Takahashi, A.; Suzuki, T.; Shimizu, Y.; Chiba, M.; Osaka, K.; Sasaki, K.; Fukuda, T.; Isogai, E.; et al. 90Sr in teeth of cattle abandoned in evacuation zone: Record of pollution from the Fukushima-Daiichi Nuclear Power Plant accident. Sci. Rep. 2016, 6, 24077. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yonezawa, A.; Kumagai, K.; Sano, M.; Miyake, T. Cs and Sr removal over highly effective adsorbents ETS-1 and ETS-2. J. Mater. Chem. A 2015, 3, 1562–1568. [Google Scholar] [CrossRef]
- Tanaka, K.; Shimada, A.; Hoshi, A.; Yasuda, M.; Ozawa, M.; Kameo, Y. Radiochemical analysis of rubble and trees collected from Fukushima Daiichi Nuclear Power Station. J. Nucl. Sci. Technol. 2014, 51, 1032–1043. [Google Scholar] [CrossRef]
- Kozai, N.; Ohnuki, T.; Arisaka, M.; Watanabe, M.; Sakamoto, F.; Yamasaki, S.; Jiang, M. Chemical states of fallout radioactive Cs in the soils deposited at Fukushima Daiichi Nuclear Power Plant accident. J. Nucl. Sci. Technol. 2012, 49, 473–478. [Google Scholar] [CrossRef]
- Meunier, P.J.; Roux, C.; Seeman, E.; Ortolani, S.; Badurski, J.E.; Spector, T.D.; Cannata, J.; Balogh, A.; Lemmel, E.-M.; Pors-Nielsen, S.; et al. The Effects of Strontium Ranelate on the Risk of Vertebral Fracture in Women with Postmenopausal Osteoporosis. N. Engl. J. Med. 2004, 350, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, K.; Murata, H. Strontium Substitution in Bioactive Calcium Phosphates: A First-Principles Study. J. Phys. Chem. B 2009, 113, 3584–3589. [Google Scholar] [CrossRef]
- Rokita, E.; Hermes, C.; Nolting, H.-F.; Ryczek, J. Substitution of calcium by strontium within selected calcium phosphates. J. Cryst. Growth 1993, 130, 543–552. [Google Scholar] [CrossRef]
- D’Angelo, P.; Migliorati, V.; Sessa, F.; Mancini, G.; Persson, I. XANES Reveals the Flexible Nature of Hydrated Strontium in Aqueous Solution. J. Phys. Chem. B 2016, 120, 4114–4124. [Google Scholar] [CrossRef]
- McKinley, J.P.; Zachara, J.M.; Smith, S.C.; Liu, C. Cation exchange reactions controlling desorption of 90Sr2+ from coarse-grained contaminated sediments at the Hanford site, Washington. Geochim. Cosmochim. Acta 2007, 71, 305–325. [Google Scholar] [CrossRef] [Green Version]
- Ball, J.W.; Nordstrom, D.K. User’s Manual for WATEQ4F, with Revised Thermodynamic Data Base and Text Cases for Calculating Speciation of Major, Trace, and Redox Elements in Natural Waters; U.S. Geological Survey: Denver, CO, USA, 1991; pp. 91–183.
- Parkman, R.H.; Charnock, J.M.; Livens, F.R.; Vaughan, D.J. A study of the interaction of strontium ions in aqueous solution with the surfaces of calcite and kaolinite. Geochim. Cosmochim. Acta 1998, 62, 1481–1492. [Google Scholar] [CrossRef]
- Mucci, A.; Morse, J.W. The incorporation of Mg2+ and Sr2+ into calcite overgrowths: Influences of growth rate and solution composition. Geochim. Cosmochim. Acta 1983, 47, 217–233. [Google Scholar] [CrossRef]
- Chorover, J.; Choi, S.; Rotenberg, P.; Serne, R.J.; Rivera, N.; Strepka, C.; Thompson, A.; Mueller, K.T.; O’Day, P.A. Silicon control of strontium and cesium partitioning in hydroxide-weathered sediments. Geochim. Cosmochim. Acta 2008, 72, 2024–2047. [Google Scholar] [CrossRef]
- Chiang, P.N.; Wang, M.K.; Huang, P.M.; Wang, J.J.; Chiu, C.Y. Cesium and strontium sorption by selected tropical and subtropical soils around nuclear facilities. J. Environ. Radioact. 2010, 101, 472–481. [Google Scholar] [CrossRef]
- Zhao, Y.; Shao, Z.; Chen, C.; Hu, J.; Chen, H. Effect of environmental conditions on the adsorption behavior of Sr(II) by Na-rectorite. Appl. Clay Sci. 2014, 87, 1–6. [Google Scholar] [CrossRef]
- Marinin, D.V.; Brown, G.N. Studies of sorbent/ion-exchange materials for the removal of radioactive strontium from liquid radioactive waste and high hardness groundwaters. Waste Manag. 2000, 20, 545–553. [Google Scholar] [CrossRef]
- Zachara, J.M.; Ainsworth, C.C.; Brown, G.E., Jr.; Catalano, J.G.; McKinley, J.P.; Qafoku, O.; Smith, S.C.; Szecsody, J.E.; Traina, S.J.; Warner, J.A. Chromium speciation and mobility in a high level nuclear waste vadose zone plume. Geochim. Cosmochim. Acta 2004, 68, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Hu, J.; Shao, D.; Li, J.; Wang, X. Adsorption behavior of multiwall carbon nanotube/iron oxide magnetic composites for Ni(II) and Sr(II). J. Hazard. Mater. 2009, 164, 923–928. [Google Scholar] [CrossRef]
- Chegrouche, S.; Mellah, A.; Barkat, M. Removal of strontium from aqueous solutions by adsorption onto activated carbon: Kinetic and thermodynamic studies. Desalination 2009, 235, 306–318. [Google Scholar] [CrossRef]
- Zhang, A.; Kuraoka, E.; Kumagai, M. Removal of Pd(II), Zr(IV), Sr(II), Fe(III), and Mo(VI) from simulated high level liquid waste by extraction chromatography utilizing the 17 icroporous silica-based polymeric materials. Sep. Purif. Technol. 2006, 50, 35–44. [Google Scholar] [CrossRef]
- Duff, M.C.; Hunter, D.B.; Hobbs, D.T.; Fink, S.D.; Dai, Z.; Bradley, J.P. Mechanisms of Strontium and Uranium Removal from High-Level Radioactive Waste Simulant Solutions by the Sorbent Monosodium Titanate. Environ. Sci. Technol. 2004, 38, 5201–5207. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, H.; Igarashi, K. Reaction of Strontium Carbonate with Anatase and Rutile. J. Am. Ceram. Soc. 1986, 69, 310–314. [Google Scholar] [CrossRef]
- Sverjensky, D.A. Prediction of the speciation of alkaline earths adsorbed on mineral surfaces in salt solutions. Geochim. Cosmochim. Acta 2006, 70, 2427–2453. [Google Scholar] [CrossRef]
- Ridley, M.K.; Machesky, M.L.; Kubicki, J.D. Experimental Study of Strontium Adsorptio on Anatase Nanoparticles as a Function of Size with a Density Functional Theory and CD Model Interpretation. Langmuir 2015, 31, 703–713. [Google Scholar] [CrossRef]
- Behrens, E.A.; Sylvester, P.; Clearfield, A. Assessment of a Sodium Nonatitanate and Pharmacosiderite-Type Ion Exchangers for Strontium and Cesium Removal from DOE Waste Simulants. Environ. Sci. Technol. 1998, 32, 101–107. [Google Scholar] [CrossRef]
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Zhang, Z.; Fenter, P.; Cheng, L.; Sturchio, N.C.; Bedzyk, M.J.; Předota, M.; Bandura, A.; Kubicki, J.D.; Lvov, S.N.; Cummings, P.T.; et al. Ion Adsorption at the Rutile−Water Interface: Linking Molecular and Macroscopic Properties. Langmuir 2004, 20, 4954–4969. [Google Scholar] [CrossRef]
- van Veelen, A.; Copping, R.; Law, G.T.W.; Smith, A.J.; Bargar, J.R.; Rogers, J.; Shuh, D.K.; Wogelius, R.A. Uranium uptake onto Magnox sludge minerals studied using EXAFS. Mineral. Mag. 2012, 76, 3095–3104. [Google Scholar] [CrossRef]
- van Veelen, A.; Preedy, O.; Qi, J.; Law, G.T.W.; Morris, K.; Mosselmans, J.F.W.; Ryan, M.P.; Evans, N.D.M.; Wogelius, R.A. Uranium and technetium interactions with wüstite [Fe1–xO] and portlandite [Ca(OH)2] surfaces under geological disposal facility conditions. Mineral. Mag. 2014, 78, 1097–1113. [Google Scholar] [CrossRef]
- van Veelen, A.; Bargar, J.R.; Law, G.T.W.; Brown, G.E.; Wogelius, R.A. Uranium Immobilization and Nanofilm Formation on Magnesium-Rich Minerals. Environ. Sci. Technol. 2016, 50, 3435–3443. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, D.L.; Appelo, C. User’s Guide to PHREEQC (Version 2): A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S. Geological Survey: Denver, CO, USA, 1999.
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Downward, L.; Booth, C.H.; Lukens, W.W.; Bridges, F. A Variation of the F-Test for Determining Statistical Relevance of Particular Parameters in EXAFS Fits. AIP Conf. Proc. 2007, 882, 129–131. [Google Scholar]
- Bots, P.; van Veelen, A.; Mosselmans, J.F.W.; Muryn, C.; Wogelius, R.A.; Morris, K. Neptunium (V) and Uranium (VI) Reactions at the Magnetite (111) Surface. Geosciences 2019, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Anné, J.; Wogelius, R.A.; Edwards, N.P.; van Veelen, A.; Buckley, M.; Sellers, W.I.; Bergmann, U.; Sokaras, D.; Alonso-Mori, R.; Harvey, V.L.; et al. Morphological and chemical evidence for cyclic bone growth in a fossil hyaena. J. Anal. At. Spectrom. 2018, 33, 2062–2069. [Google Scholar] [CrossRef]
- Anné, J.; Wogelius, R.A.; Edwards, N.P.; van Veelen, A.; Ignatyev, K.; Manning, P.L. Chemistry of bone remodelling preserved in extant and fossil Sirenia. Metallomics 2016, 8, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.A.; van Veelen, A.; Morris, K.; Mosselmans, J.F.W.; Wogelius, R.A.; Burton, N.A. Uranium (VI) Adsorbate Structures on Portlandite [Ca(OH)2] Type Surfaces Determined by Computational Modelling and X-ray Absorption Spectroscopy. Minerals 2021, 11, 1241. [Google Scholar] [CrossRef]
- Anné, J.; Edwards, N.P.; Wogelius, R.A.; Tumarkin-Deratzian, A.R.; Sellers, W.I.; van Veelen, A.; Bergmann, U.; Sokaras, D.; Alonso-Mori, R.; Ignatyev, K.; et al. Synchrotron imaging reveals bone healing and remodelling strategies in extinct and extant vertebrates. J. R. Soc. Interface 2014, 11, 20140277. [Google Scholar] [CrossRef] [Green Version]
- Edwards, N.P.; van Veelen, A.; Anne, J.; Manning, P.L.; Bergmann, U.; Sellers, W.I.; Egerton, V.M.; Sokaras, D.; Alonso-Mori, R.; Wakamatsu, K.; et al. Elemental characterization of melanin in feathers via synchrotron X-ray imaging and absorption spectroscopy. Sci. Rep. 2016, 6, 34002. [Google Scholar] [CrossRef]
- Thorpe, C.L.; Lloyd, J.R.; Law, G.T.W.; Burke, I.T.; Shaw, S.; Bryan, N.D.; Morris, K. Strontium sorption and precipitation behaviour during bioreduction in nitrate impacted sediments. Chem. Geol. 2012, 306, 114–122. [Google Scholar] [CrossRef]
- Anné, J.; Edwards, N.P.; van Veelen, A.; Egerton, V.M.; Manning, P.L.; Mosselmans, J. Fredrick, W.; Parry, S.; Sellers, W.I.; Buckley, M.; et al. Visualisation of developmental ossification using trace element mapping. J. Anal. At. Spectrom. 2017, 32, 967–974. [Google Scholar] [CrossRef]
- Agron, P.A.; Busing, W.R. Calcium and Strontium Dichloride Hexahydrates by Neutron Diffraction. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1986, 42, 141–143. [Google Scholar] [CrossRef]
- O’Day, P.A.; Newville, M.; Neuhoff, P.S.; Sahai, N.; Carroll, S.A. X-Ray Absorption Spectroscopy of Strontium(II) Coordination. J. Colloid Interface Sci. 2000, 222, 184–197. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, J.P.R. Crystal structures of aragonite, strontianite and witherite. Am. Mineral. 1971, 56, 758–767. [Google Scholar]
- Dupin, J.-C.; Gonbeau, D.; Vinatier, P.; Levasseur, A. Systematic XPS studies of metal oxides, hydroxides and peroxides. Phys. Chem. Chem. Phys. 2000, 2, 1319–1324. [Google Scholar] [CrossRef]
- Stipp, S.L.; Hochella, M.F., Jr. Structure and bonding environments at the calcite surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy electron diffraction (LEED). Geochim. Cosmochim. Acta 1991, 55, 1723–1736. [Google Scholar] [CrossRef]
- Li, J.; Li, S.; Liu, F.; Alim, M.A.; Chen, G. The origin of varistor property of SrTiO3-based ceramics. J. Mater. Sci. Mater. Electron. 2003, 14, 483–486. [Google Scholar] [CrossRef]
- Jung, W.; Tuller, H.L. Investigation of surface Sr segregation in model thin film solid oxide fuel cell perovskite electrodes. Energy Environ. Sci. 2012, 5, 5370–5378. [Google Scholar] [CrossRef] [Green Version]
- Kosmulski, M. pH-dependent surface charging and points of zero charge. IV. Update and new approach. J. Colloid Interface Sci. 2009, 337, 439–448. [Google Scholar] [CrossRef]
- Kosmulski, M. The significance of the difference in the point of zero charge between rutile and anatase. Adv. Colloid Interface Sci. 2002, 99, 255–264. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Shell | CN | R (Å) | σ2 | S02 | ΔE0 (eV) | R | |
|---|---|---|---|---|---|---|---|---|
| (A) SrCO3 standard | Sr–O | 3 ± 0.2 | 2.54 (2) | 0.005 (2) | 0.91 (2) | −1.17 | ±1.37 | 0.009 |
| Sr–O | 6 ± 0.2 | 2.69 (2) | 0.007 (1) | |||||
| Sr–C | 3 ± 0.3 | 3.03 (2) | 0.005 (2) | |||||
| Sr–C | 2 ± 1.0 | 3.56 (9) | 0.016 (13) | |||||
| Sr–Sr | 6 ± 0.5 | 4.14 (2) | 0.013 (3) | |||||
| Sr–Sr/O | 2 ± 0.3 | 4.27 (3) | 0.009 (3) | |||||
| (B) ex situ SrCO3 | Sr–O | 8.8 ± 1.1 | 2.64 (3) | 0.012 (1) | 1.08 (6) | −0.82 | ±3.13 | 0.022 |
| Sr–C | 5.5 ±1.2 | 3.11 (3) | 0.011 (3) | |||||
| (C) in situ SrCl2 | Sr–O | 3 ± 0.2 | 2.47 (2) | 0.005 (1) | 0.95 (3) | −6.36 | ±1.07 | 0.009 |
| Sr–O | 5.1 ± 0.2 | 2.61 (1) | 0.004 (01) | |||||
| Sr-Ti | 1.1 ± 0.3 | 3.26 (3) | 0.016 (8) | |||||
| (D) ex situ SrCl2 hydrated | Sr–O | 7.7 ± 0.4 | 2.69 (1) | 0.010 (1) | 0.95 (6) | 5.03 | ±1.16 | 0.021 |
| Sr–Cl | 2.5 ± 0.5 | 3.10 (3) | 0.012 (4) | |||||
| (E) ex situ SrCl2 aqueous | Sr–O | 7.4 ± 0.9 | 2.61 (2) | 0.007 (1) | 0.93 (4) | 2.47 | ±1.05 | 0.013 |
| Sr–Ti | 2.7 ± 05 | 3.36 (4) | 0.011 (5) | |||||
| (F) SrCl2–6H2O standard | Sr–O | 8.6 ± 0.6 | 2.66 (2) | 0.010 (1) | 0.89 (5) | 5.03 | ±1.55 | 0.009 |
| Sr–Cl | 2.4 ± 0.4 | 2.99 (1) | 0.003 (1) | |||||
| Sr–Sr | 2.3 ± 1.1 | 3.83 (3) | 0.008 (4) | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Veelen, A.; Francisco, P.C.M.; Edwards, N.P.; Mosselmans, J.F.W.; Sato, T.; Wogelius, R.A. In Situ EXAFS Study of Sr Adsorption on TiO2(110) under High Ionic Strength Wastewater Conditions. Minerals 2021, 11, 1386. https://doi.org/10.3390/min11121386
van Veelen A, Francisco PCM, Edwards NP, Mosselmans JFW, Sato T, Wogelius RA. In Situ EXAFS Study of Sr Adsorption on TiO2(110) under High Ionic Strength Wastewater Conditions. Minerals. 2021; 11(12):1386. https://doi.org/10.3390/min11121386
Chicago/Turabian Stylevan Veelen, Arjen, Paul C. M. Francisco, Nicholas P. Edwards, Julian Frederick W. Mosselmans, Tsutomu Sato, and Roy A. Wogelius. 2021. "In Situ EXAFS Study of Sr Adsorption on TiO2(110) under High Ionic Strength Wastewater Conditions" Minerals 11, no. 12: 1386. https://doi.org/10.3390/min11121386
APA Stylevan Veelen, A., Francisco, P. C. M., Edwards, N. P., Mosselmans, J. F. W., Sato, T., & Wogelius, R. A. (2021). In Situ EXAFS Study of Sr Adsorption on TiO2(110) under High Ionic Strength Wastewater Conditions. Minerals, 11(12), 1386. https://doi.org/10.3390/min11121386