Effect of Crystal Chemistry Properties on the Distribution Characteristics of H2O and Na+ in Na-Montmorillonite Interlayer Space: Molecular Dynamics Simulation Study

Abstract
1. Introduction
2. Simulation Details
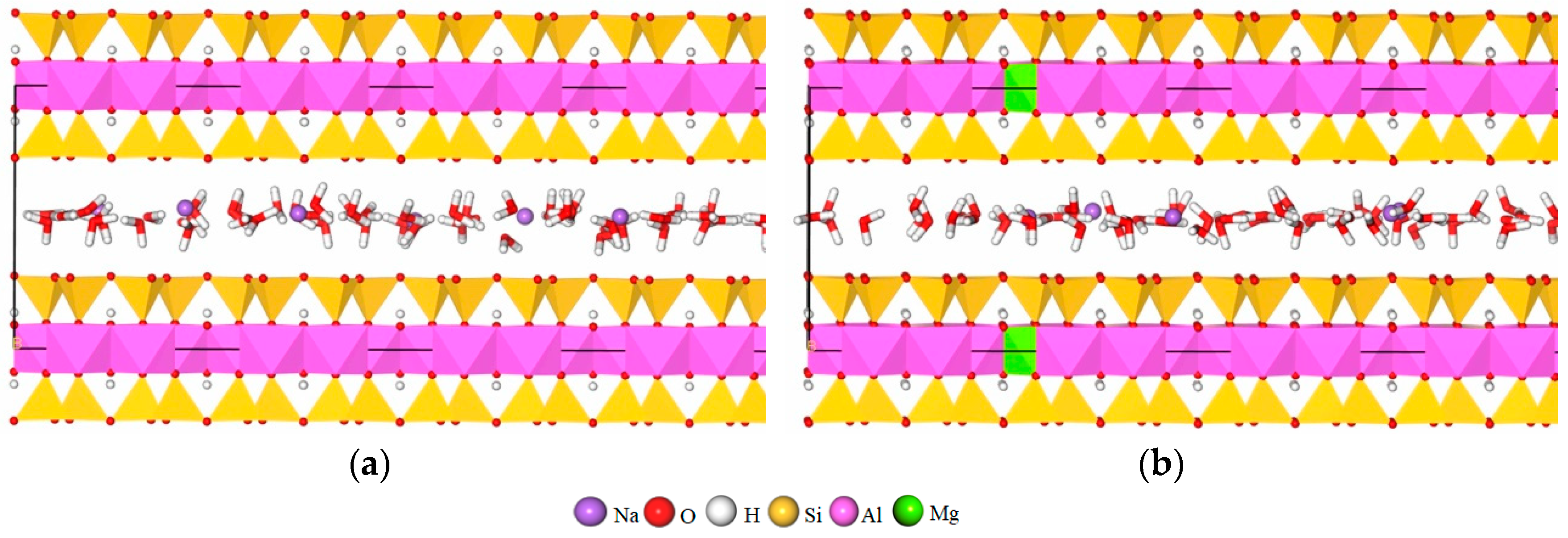
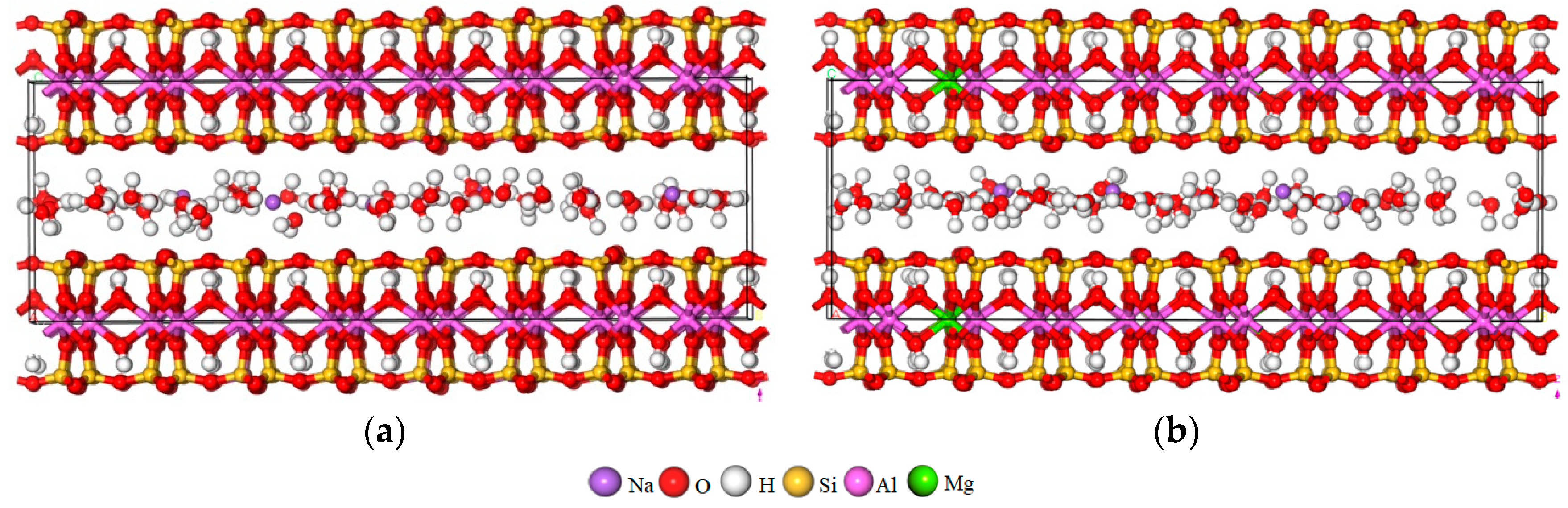
2.1. Model Establishment
2.2. Simulation Parameters
3. Results and Discussion
3.1. Z-Axis Concentration Profiles of Particles in Interlayer of Na-Montmorillonite (Na-MT)
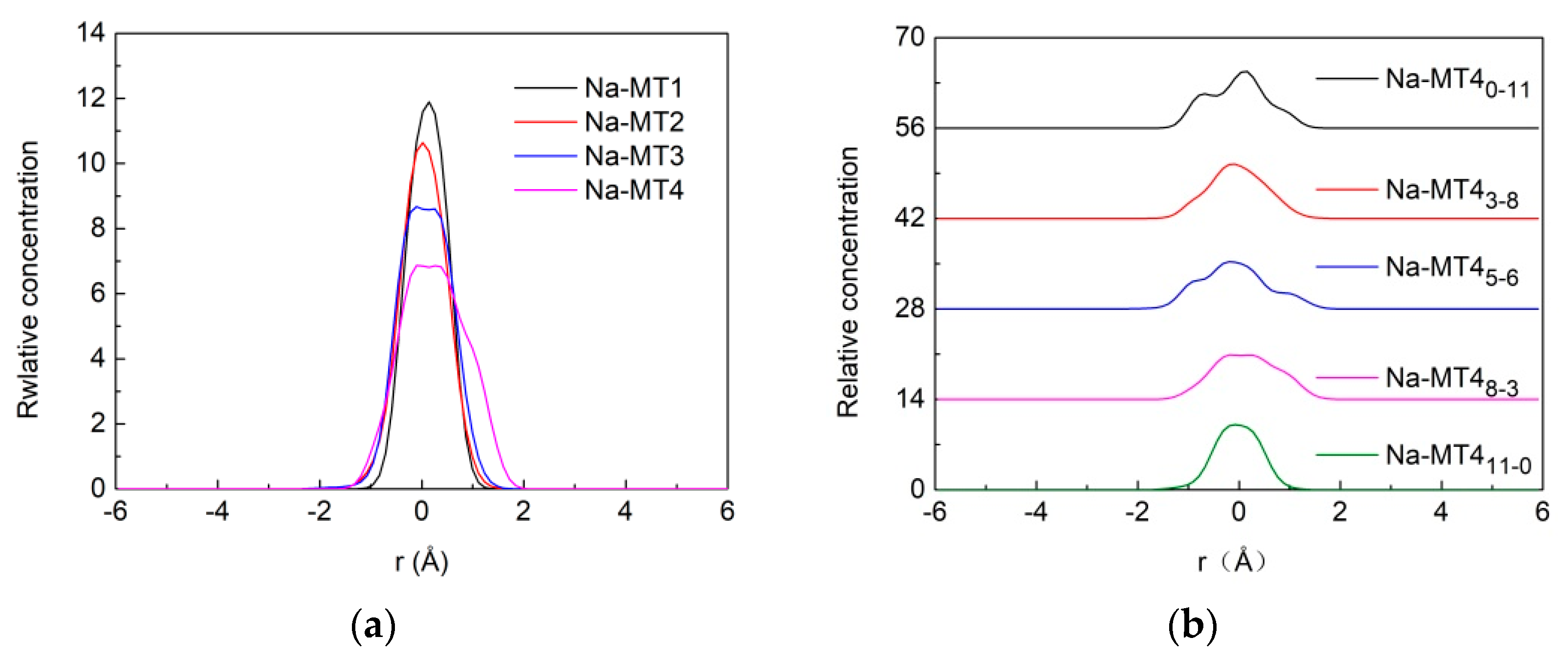
3.1.1. Z-Axis Concentration Profiles of H2O
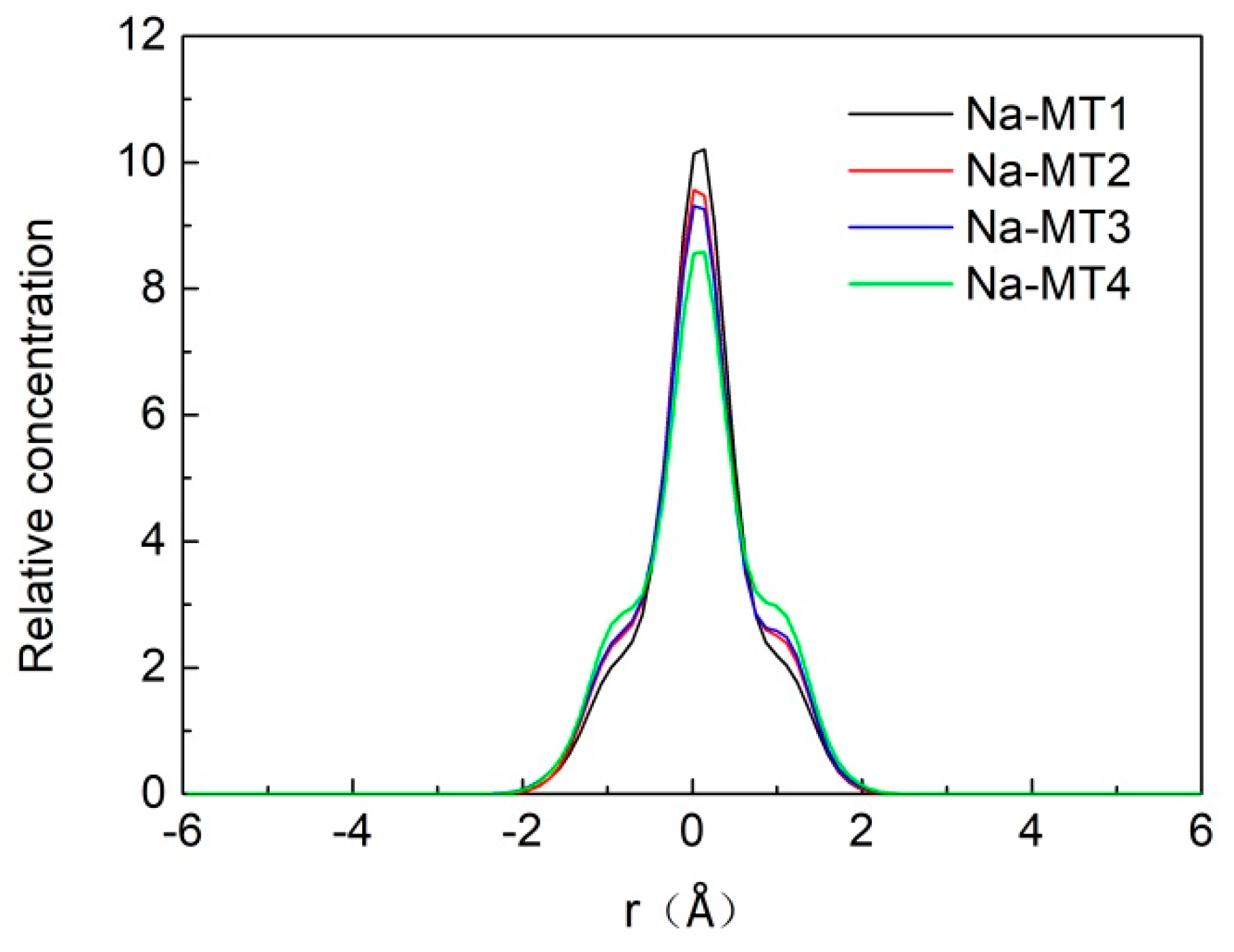
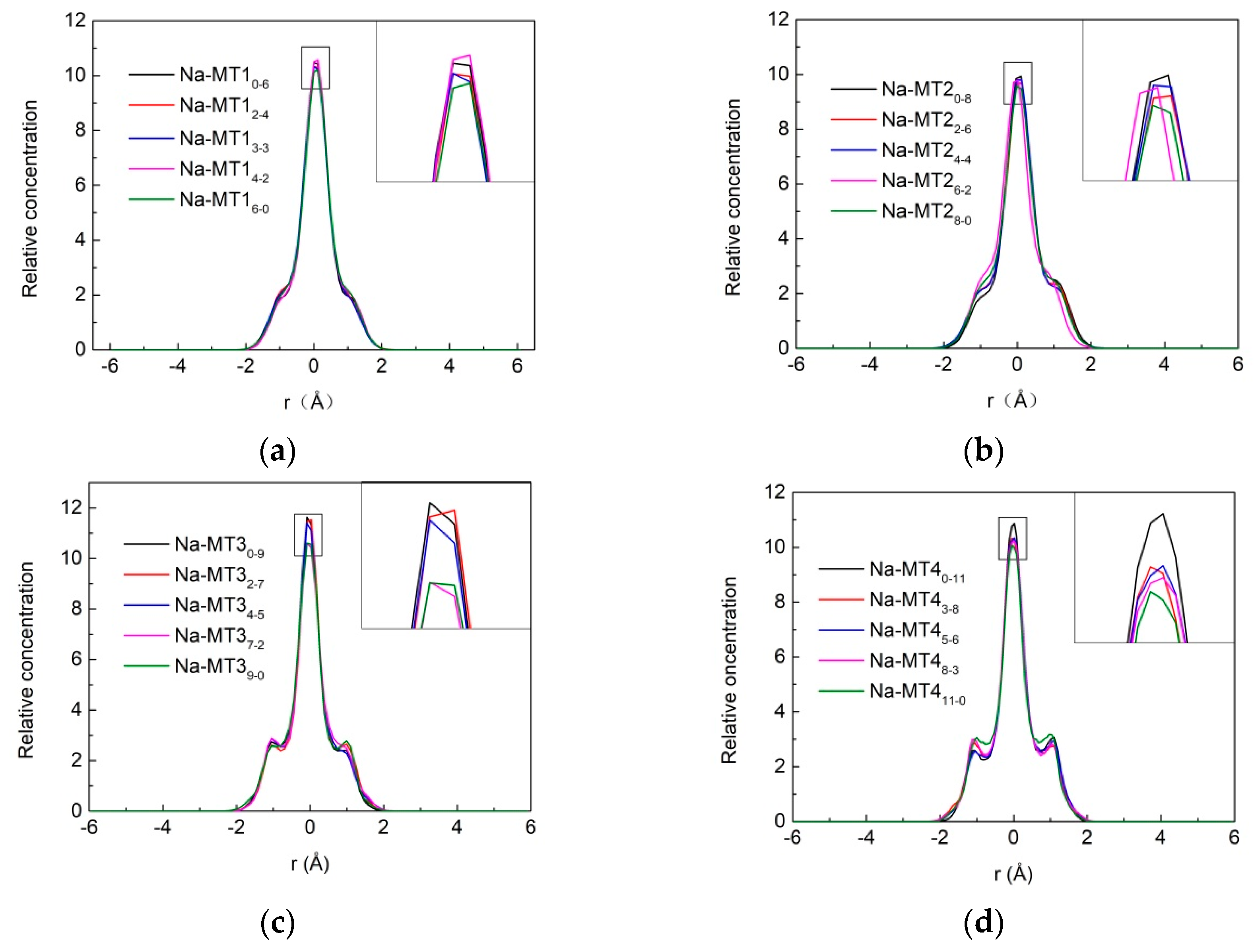
3.1.2. Z-Axis Concentration Profiles of Na+
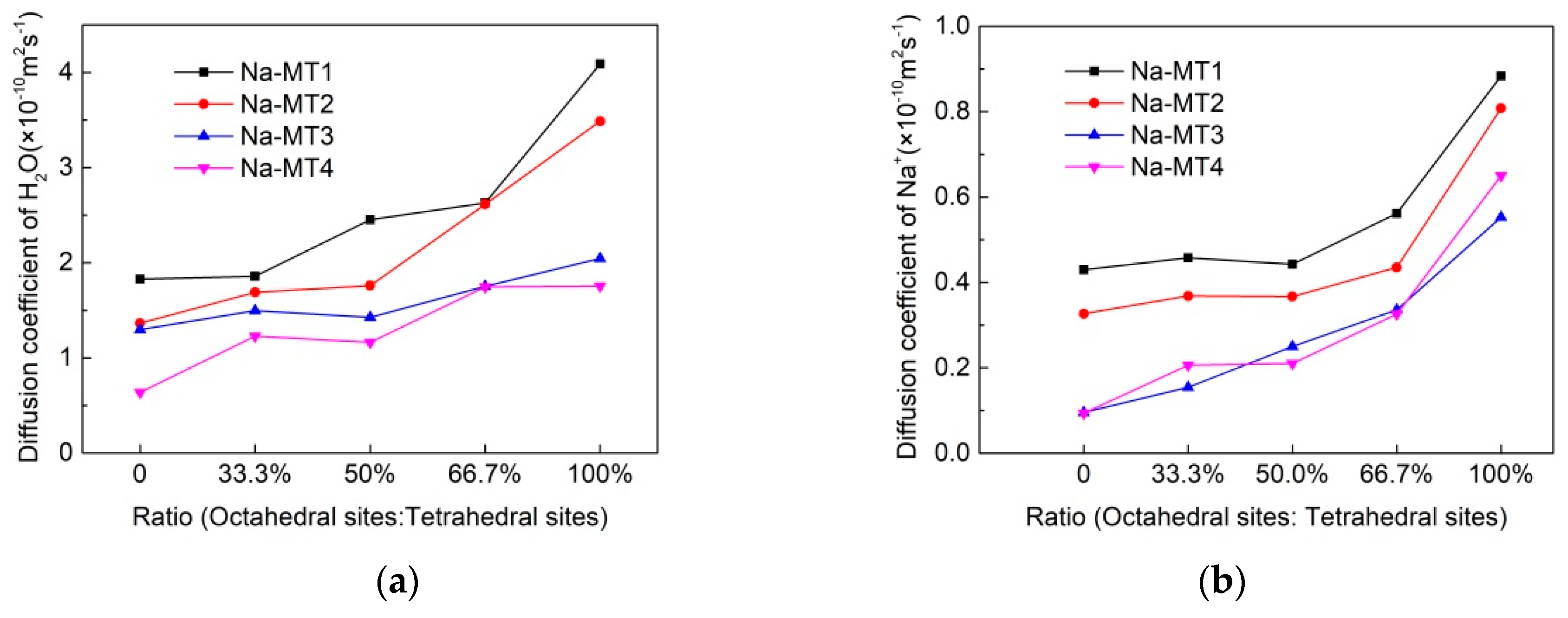
3.2. The Self-Diffusion Coefficient of Particles in Interlayer of Na-MT
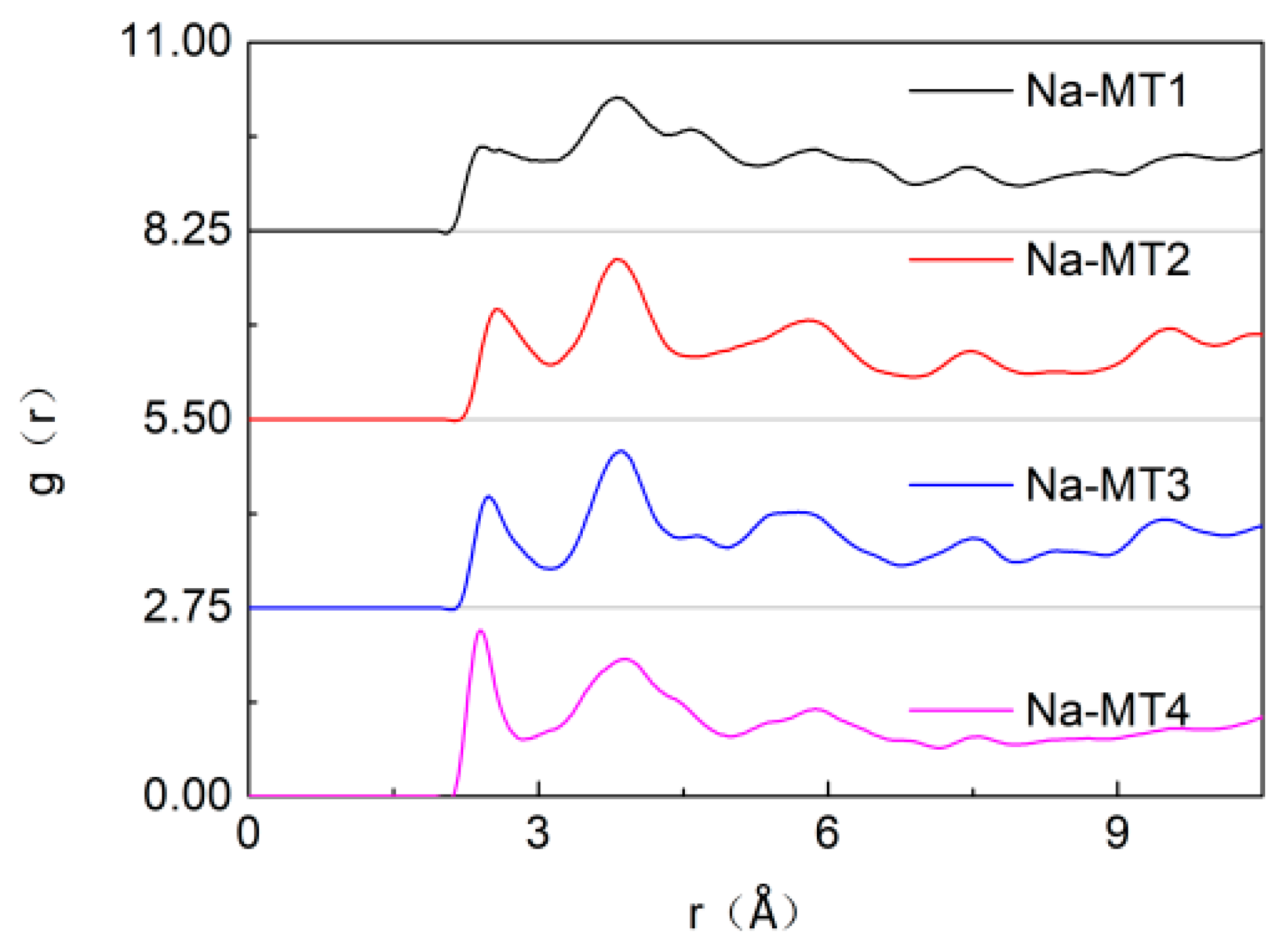
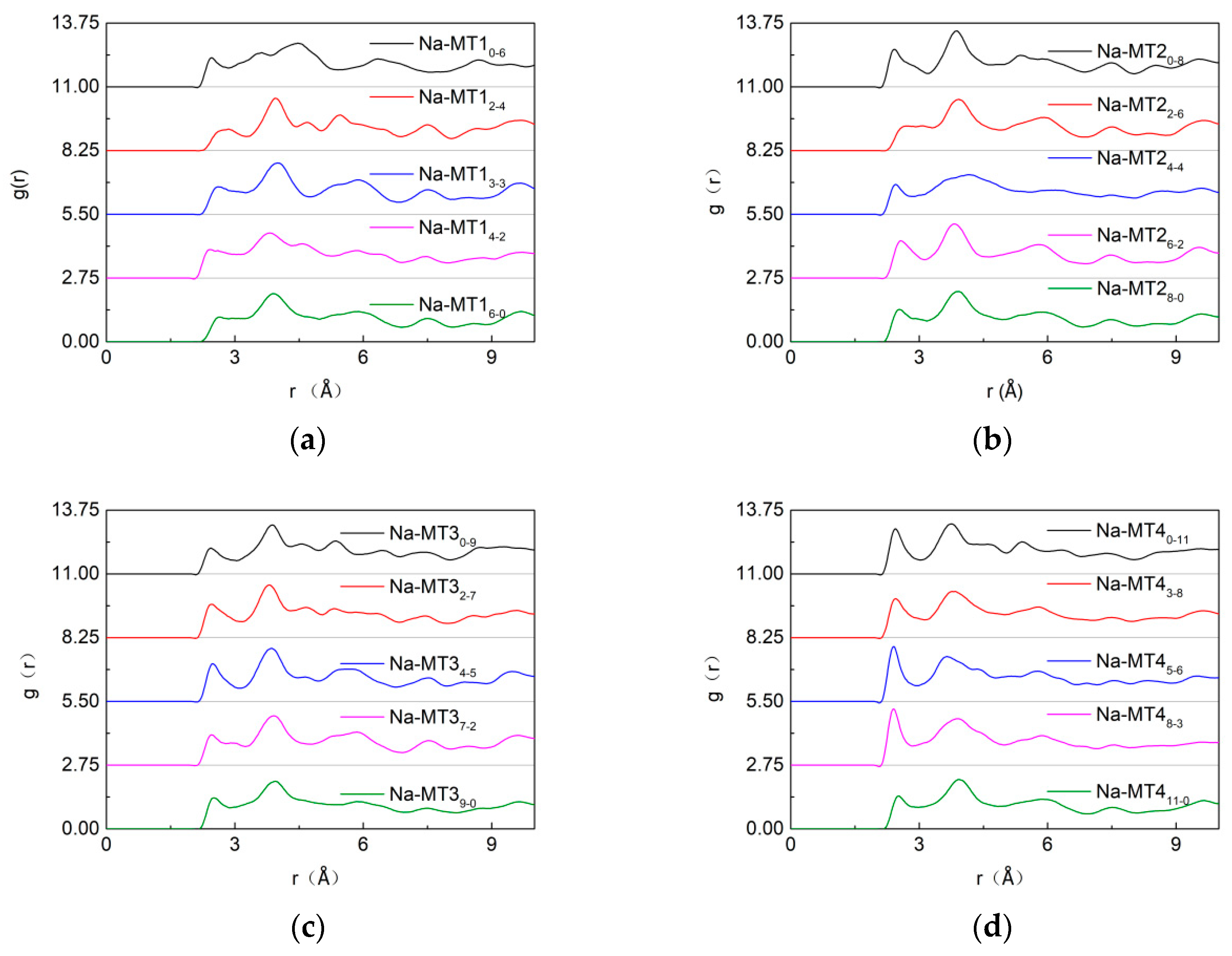
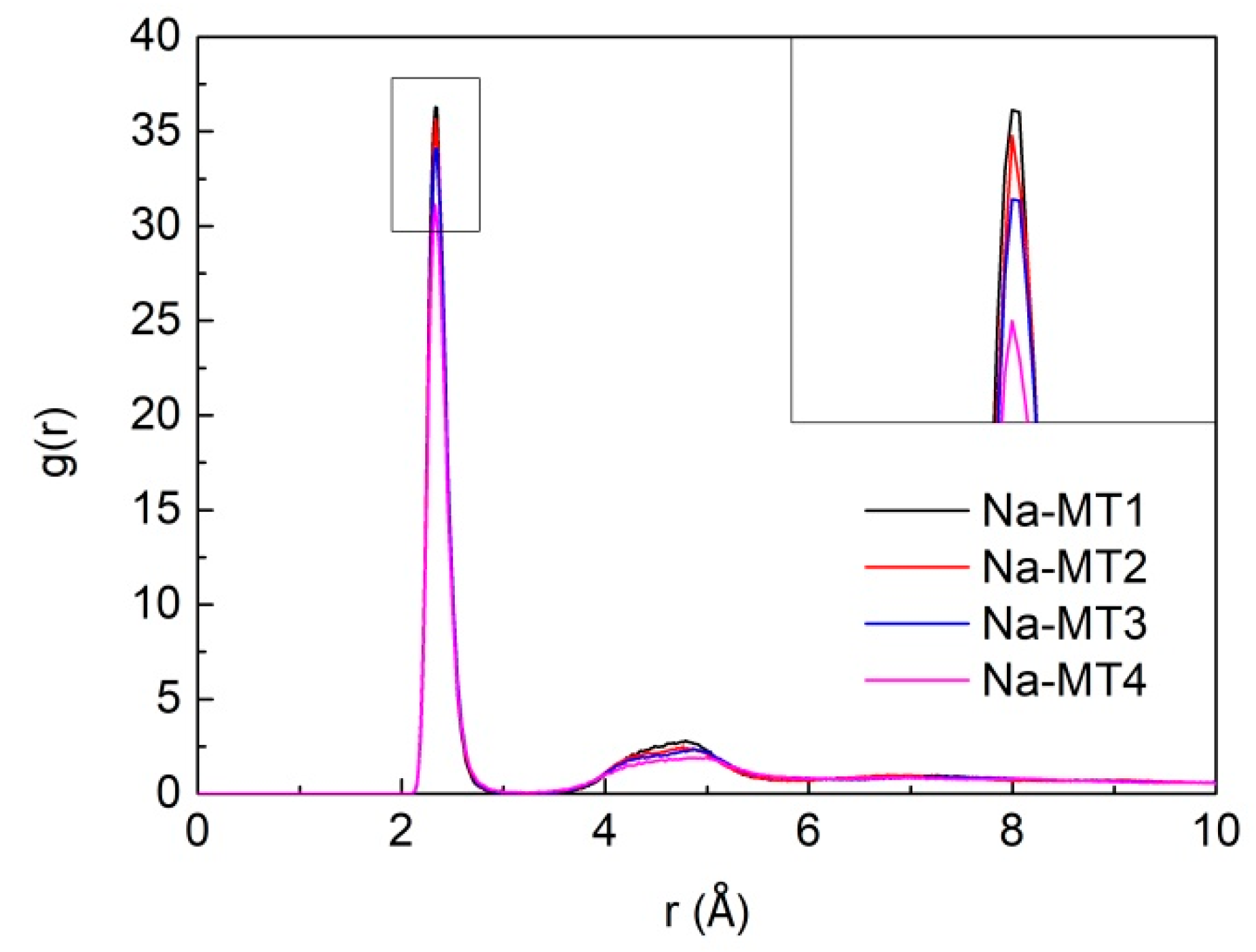
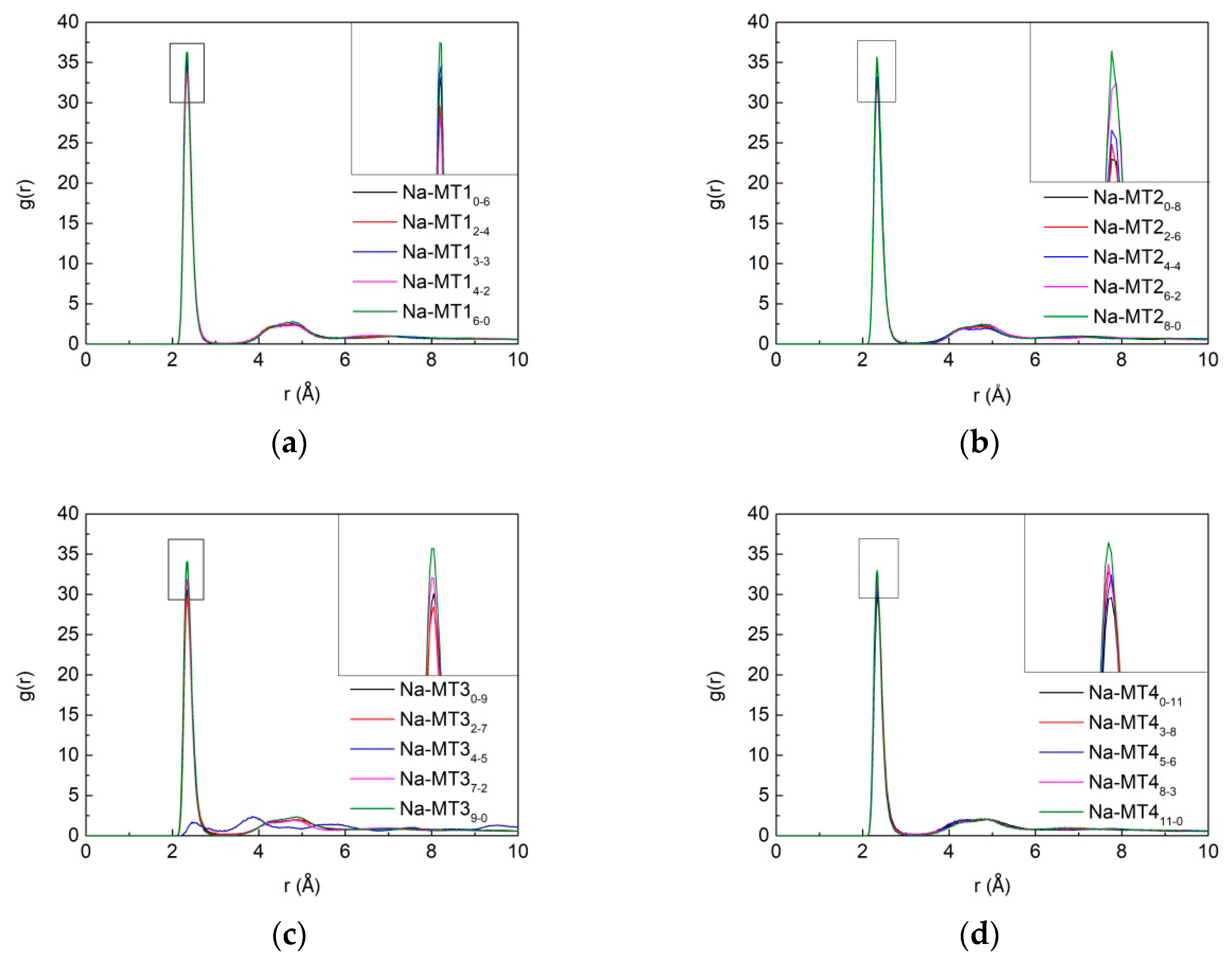
3.3. Radial Distribution Function (RDF) of Particles in Interlayer of Na-MT
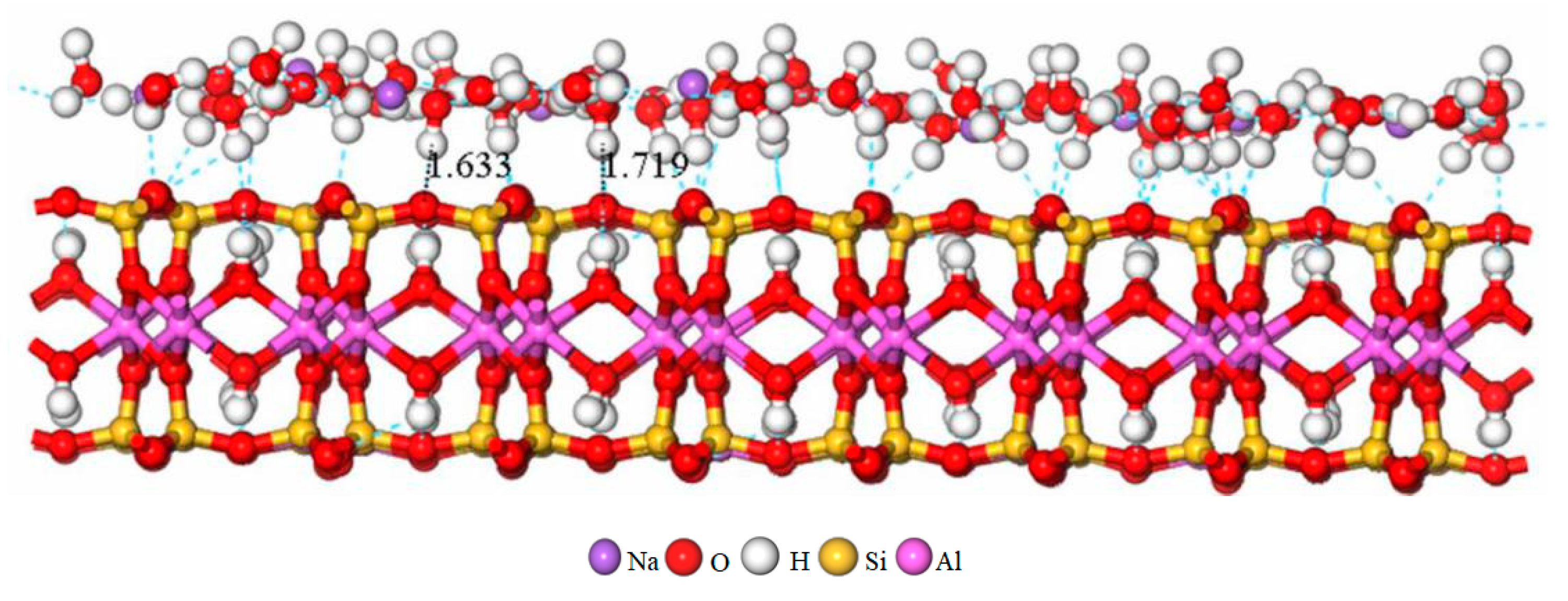
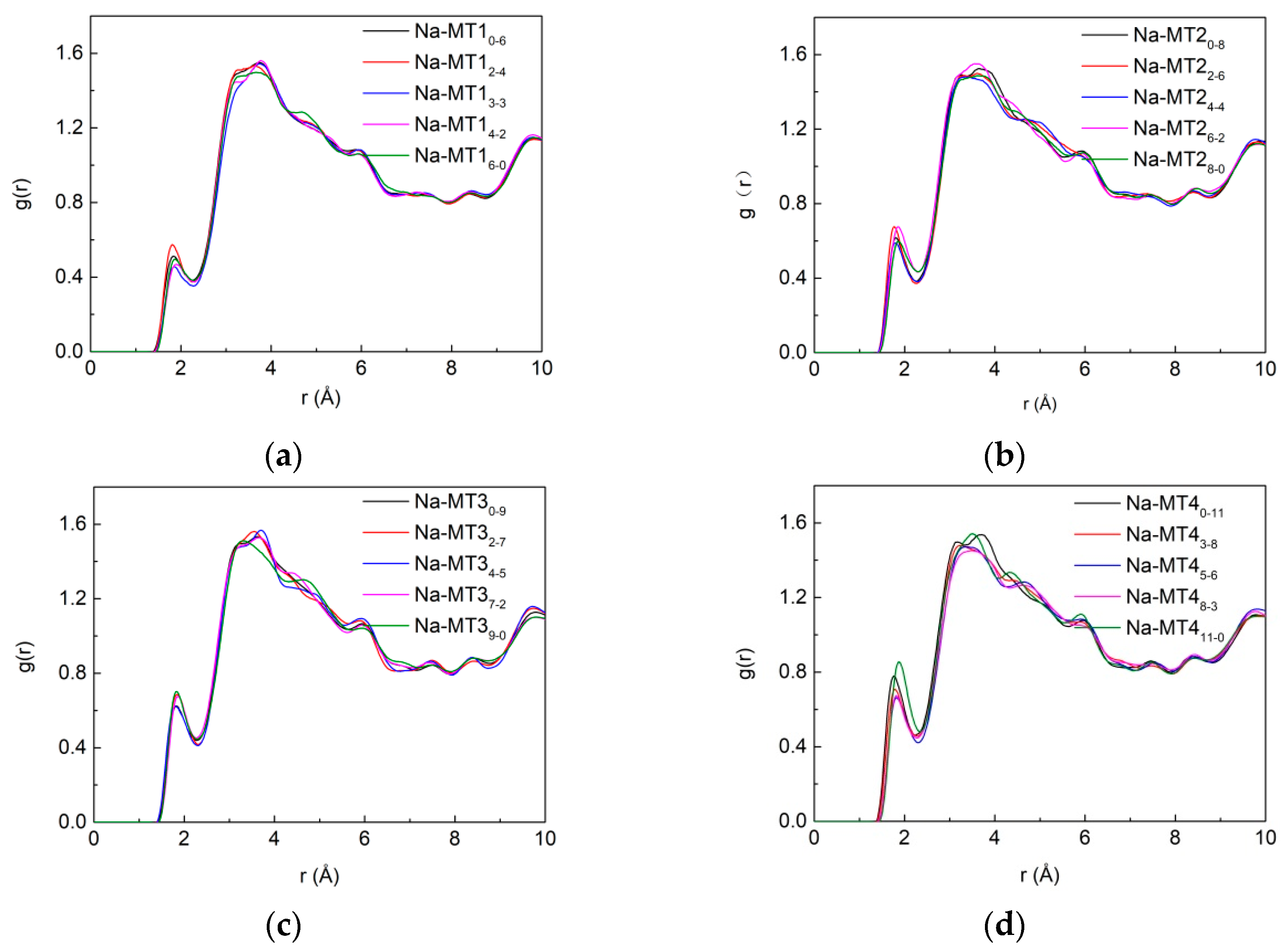
3.3.1. The RDF of Ot-Hw
3.3.2. The RDF of Ot-Na
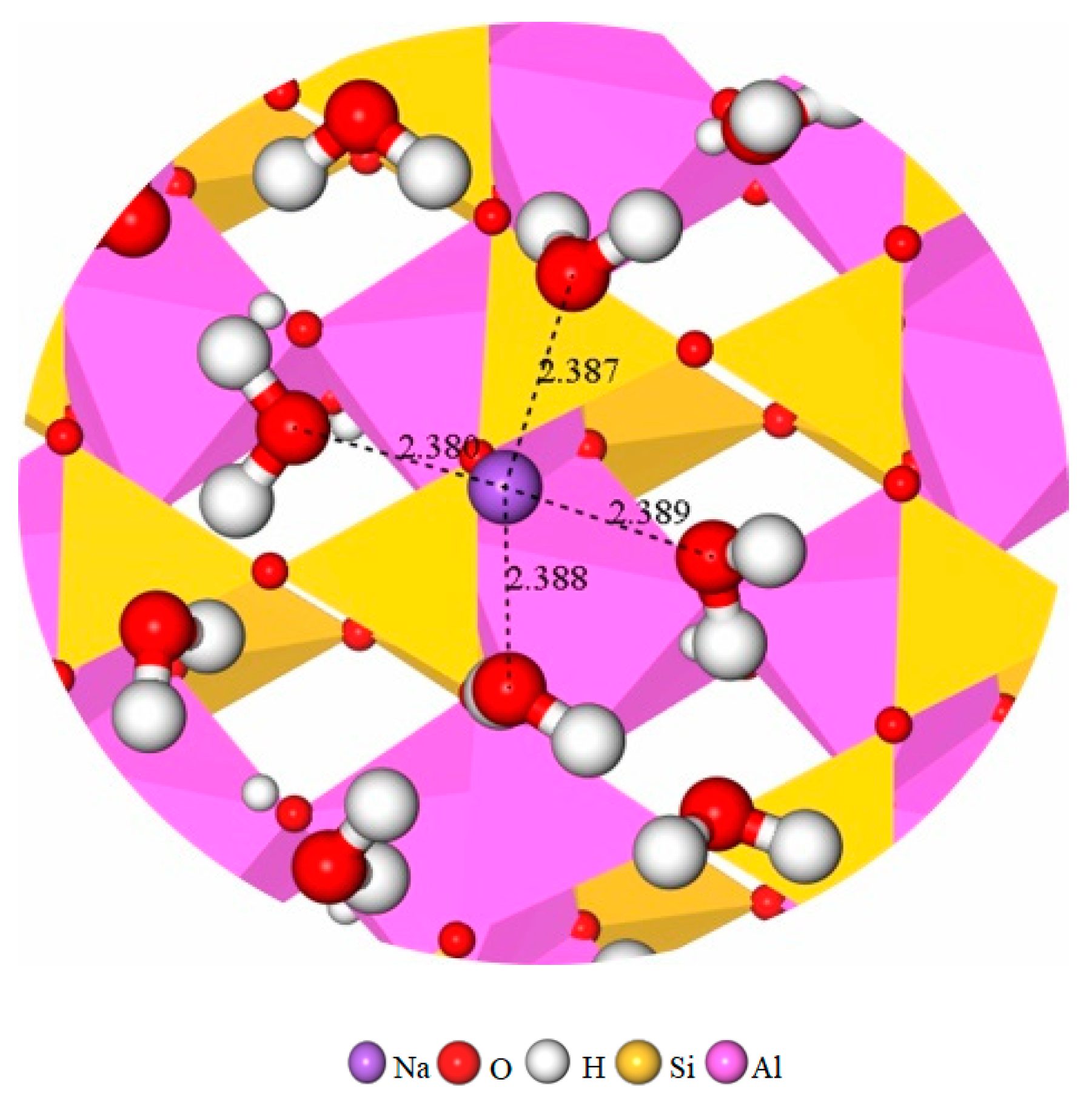
3.3.3. The RDF of Na-Ow
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, W.N.; Hu, H.X.; Li, X.C.; Fang, Z.M. Changes in micromechanical properties of Na-montmorillonite caused by CO2/H2O sorption. Comput. Mater. Sci. 2017, 129, 178–183. [Google Scholar] [CrossRef]
- Churakov, S.V.; Kosakowski, G. An ab initio molecular dynamics study of hydronium complexation in Na-montmorillonite. Philos. Mag. 2010, 90, 2459–2474. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, C.; Yun, J.; Yang, G. Isomorphic Substitutions in Clay Materials and Adsorption of Metal Ions onto External Surfaces: A DFT Investigation. Phys. Chem. 2017, 121, 26722–26732. [Google Scholar] [CrossRef]
- Wu, L.M.; Cao, S.Y.; Lv, G.C. Influence of Energy State of Montmorillonite Interlayer Cations on Organic Intercalation. Adv. Mater. Sci. Eng. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Michot, L.J.; Ferrage, E.; Jiménez-Ruiz, M.; Boehm, M.; Delville, A. Anisotropic Features of Water and Ion Dynamics in Synthetic Na- and Ca-Smectites with Tetrahedral Layer Charge. A Combined Quasi-elastic Neutron-Scattering and Molecular Dynamics Simulations Study. Phys. Chem. 2012, 116, 16619–16633. [Google Scholar] [CrossRef]
- Sun, L.L.; Tanskanen, J.T.; Hirvi, J.T.; Kasa, S.; Schatz, T.; Pakkanen, T.A. Molecular dynamics study of montmorillonite crystalline swelling: Roles of interlayer cation species and water content. Chem. Phys. 2015, 455, 23–31. [Google Scholar] [CrossRef]
- Lv, G.; Chang, P.-H.; Xing, X.; Jiang, W.-T.; Jean, J.-S.; Li, Z. Investigation of intercalation of diphenhydramine into the interlayer of smectite by XRD, FTIR, TG-DTG analyses and molecular simulation. Arab. J. Chem. 2017, 10, 855–861. [Google Scholar] [CrossRef]
- Maryan, A.S.; Montazer, M. Natural and organo-montmorillonite as antibacterial nanoclays for cotton garment. Ind. Eng. Chem. 2015, 22, 164–170. [Google Scholar] [CrossRef]
- Zhou, D.J.; Zhang, Z.P.; Tang, J.L.; Wang, F.W.; Liao, L.B. Applied properties of oil-based drilling fluids with montmorillonites modified by cationic and anionic surfactants. Appl. Clay Sci. 2016, 121, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, F.J.; Li, Y.J.; Wu, T.; Sun, D.J.; Zhang, G.C.; Huang, X.H.; Wang, G.P. Effects of Na–montmorillonite particles on the emulsification stability of polymer flooding produced water. Colloids Surf. Physicochem. Eng. Aspects. 2012, 410, 125–129. [Google Scholar] [CrossRef]
- Li, S.Z.; Wu, P.X.; Li, H.L.; Zhu, N.W.; Li, P.; Wu, J.H.; Wang, X.D.; Dang, Z. Synthesis and characterization of organo-montmorillonite supported iron nanoparticles. Appl. Clay Sci. 2010, 50, 330–336. [Google Scholar] [CrossRef]
- Bahranowski, K.; Gaweł, A.; Klimek, A.; Michalik-Zym, A.; Napruszewska, B.D.; Nattich-Rak, M.; Rogowska, M.S.; erwicka, E.M. Influence of purification method of Na-montmorillonite on textural properties of clay mineral composites with TiO2 nanoparticles. Appl. Clay Sci. 2017, 140, 75–80. [Google Scholar] [CrossRef]
- Holmboe, M.; Wold, S.; Jonsson, M. Porosity investigation of compacted bentonite using XRD profile modeling. J. Contam. Hydrol. 2012, 128, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Viennet, J.-C.; Hubert, F.; Ferrage, E.; Tertre, E.; Legout, A.; Turpault, M.-P. Investigation of clay mineralogy in a temperate acidic soil of a forest using X-ray diffraction profile modeling: Beyond the HIS and HIV description. Geoderma 2015, 241, 75–86. [Google Scholar] [CrossRef]
- Ferrage, E.; Tournassat, C.; Rinnert, E.; Lanson, B. Influence of pH on the interlayer cationic composition and hydration state of Ca-montmorillonite: Analytical chemistry, chemical modelling and XRD profile modelling study. Geochim. Cosmochim. Acta 2005, 69, 2797–2812. [Google Scholar] [CrossRef][Green Version]
- Yi, H.; Jia, F.F.; Zhao, Y.L.; Wang, W.; Song, S.X.; Li, H.Q.; Liu, C. Surface wettability of montmorillonite (0 0 1) surface as affected by surface charge and exchangeable cations: A molecular dynamic study. Appl. Surf. Sci. 2018, 459, 148–154. [Google Scholar] [CrossRef]
- Yang, N.N.; Liu, S.Y.; Yang, X.N. Molecular simulation of preferential adsorption of CO2 over CH4 in Na-montmorillonite clay material. Appl. Surf. Sci. 2015, 356, 1262–1271. [Google Scholar] [CrossRef]
- Suter, J.L.; Kabalan, L.; Khader, M.; Coveney, P.V. Ab initio molecular dynamics study of the interlayer and micropore structure of aqueous montmorillonite clays. Geochim. Cosmochim. Acta 2015, 169, 17–29. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Yi, H.; Jia, F.F.; Li, H.L.; Peng, C.S.; Song, S.X. A novel method for determining the thickness of hydration shells on nanosheets: A case ofmontmorillonite in water. Powder Technol. 2017, 307, 74–79. [Google Scholar] [CrossRef]
- Yi, H.; Zhang, X.; Zhao, Y.L.; Liu, L.Y.; Song, S.X. Molecular dynamics simulations of hydration shell on montmorillonite (001) in water. Surf. Interface Anal. 2016, 48, 976–980. [Google Scholar] [CrossRef]
- Titiloye, J.O.; Skipper, N.T. Monte Carlo and molecular dynamics simulations of methane in potassium montmorillonite clay hydrates at elevated pressures and temperatures. J. Colloid Interface Sci. 2005, 282, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Voora, V.K.; Al-Saidi, W.A.; Jordan, K.D. Density functional theory study of pyrophyllite and M-montmorillonites (M = Li, Na, K, Mg, and Ca): Role of dispersion interactions. J. Phys. Chem. A 2011, 115, 9695–9703. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.B.; Lv, G.C.; Zhu, W.S.; He, C.; Liao, L.B.; Mei, L.F.; Li, Z.H.; Li, G.W. The binding energy between the interlayer cations and montmorillonite layers and its influence on Pb2+ adsorption. Appl. Clay Sci. 2015, 112, 117–122. [Google Scholar] [CrossRef]
- Mei, L.F.; Tao, H.S.; He, C.; Xin, X.B.; Liao, L.B.; Wu, L.M.; Lv, G.C. Cd2+ Exchange for Na+and K+ in the Interlayer of Montmorillonite: Experiment and Molecular Simulation. J. Nanomater. 2015, 2015. [Google Scholar] [CrossRef]
- Wu, L.M.; Liao, L.B.; Lv, G.C. Influence of interlayer cations on organic intercalation of montmorillonite. J. Colloid Interface Sci. 2015, 454, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.H.; Lu, X.C.; Boek, E.S. Changes in the Interlayer Structure and Thermodynamics of Hydrated Montmorillonite under Basin Conditions: Molecular Simulation Approaches. Clays Clay Miner. 2016, 64, 503–511. [Google Scholar] [CrossRef]
- Akinwunmi, B.; Sun, L.L.; Hirvi, J.T.; Kasa, S.; Pakkanen, T.A. Influence of temperature on the swelling pressure of bentonite clay. Chem. Phys. 2019, 516, 177–181. [Google Scholar] [CrossRef]
- Peng, J.F.; Yi, H.; Song, S.X.; Zhan, W.Q.; Zhao, Y.L. Driving force for the swelling of montmorillonite as affected by surface charge and exchangeable cations: A molecular dynamic study. Results Phys. 2019, 12, 113–117. [Google Scholar] [CrossRef]
- Qiu, J.; Li, G.Q.; Liu, D.L.; Jiang, S.; Wang, G.F.; Chen, P.; Zhu, X.N.; Yao, G.; Liu, X.D.; Lyu, X.J. Effect of Layer Charge Density on Hydration Properties of Montmorillonite: Molecular Dynamics Simulation and Experimental Study. Int. J. Mol. Sci. 2019, 20, 3991–3997. [Google Scholar] [CrossRef]
- Shen, W.; Li, L.; Zhou, H.J.; Zhou, Q.; Chen, M.; Zhu, J.X. Effects of charge density on the hydration of siloxane surface of montmorillonite: A molecular dynamics simulation study. Appl. Clay Sci. 2018, 159, 10–15. [Google Scholar] [CrossRef]
- Wungu, T.D.K.; Diño, W.A.; Dipojono, H.K.; Kasai, H. Effect of Lithium Absorption at Tetrahedral Site and Isomorphic Substitution on Montmorillonite Properties: A Density Functional Theory Study. Jpn. J. Appl. Phys. 2011, 50, 055701. [Google Scholar]
- Zhang, X.; Yi, H.; Bai, H.Y.; Zhao, Y.L.; Min, F.F.; Song, S.X. Correlation of montmorillonite exfoliation with interlayer cations in the preparation of two-dimensional nanosheets. Rsc. Adv. 2017, 7, 41471–41478. [Google Scholar] [CrossRef]
- Chang, F.R.C.; Skipper, N.T.; Sposito, G. Computer Simulation of Interlayer Molecular Structure in Sodium Montmorillonite Hydrates. Langmuir 1995, 11, 2734–2741. [Google Scholar] [CrossRef]
- Tambach, T.J.; Bolhuis, P.G.; Hensen, E.J.M.; Smit, B. Hysteresis in Clay Swelling Induced by Hydrogen Bonding: Accurate Prediction of Swelling States. Langmuir 2006, 22, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, L. Molecular insight into competitive adsorption of methane and carbon dioxide in montmorillonite: Effect of clay structure and water content. Fuel 2019, 239, 32–43. [Google Scholar] [CrossRef]
- Ferrage, E. Investigation of smectite hydration properties by modeling experimental X-ray diffraction patterns: Part I. Montmorillonite hydration properties. Am. Mineral. 2005, 90, 1358–1374. [Google Scholar] [CrossRef]
- Zhang, X.; Yi, H.; Zhao, Y.L.; Min, F.F.; Song, S.X. Study on the differences of Na- and Ca-montmorillonites in crystalline swelling regime through molecular dynamics simulation. Adv. Powder Technol. 2016, 27, 779–785. [Google Scholar] [CrossRef]
- Yang, N.N.; Yang, X.N. Molecular simulation of swelling and structure for Na-Wyoming montmorillonite in supercritical CO2. Mol. Sim. 2011, 37, 1063–1070. [Google Scholar] [CrossRef]
- Qiu, J.; Li, G.Q.; Liu, D.L.; Jiang, S.; Wang, G.F.; Chen, P.; Zhu, X.N.; Cao, X.Q.; Lyu, X.J. Effect of Layer Charge Characteristics on the Distribution Characteristics of H2O and Ca(2+) in Ca-Montmorillonites Interlayer Space: Molecular Dynamics Simulation. Materials 2019, 12, 2318. [Google Scholar] [CrossRef]
- Vao-soongnern, V.; Pipatpanukul, C.; Horpibulsuk, S. A combined X-ray absorption spectroscopy and molecular dynamic simulation to study the local structure potassium ion in hydrated montmorillonite. J. Mater. Sci. 2015, 50, 7126–7136. [Google Scholar] [CrossRef]
- Zheng, Y.; Zaoui, A. Mechanical behavior in hydrated Na-montmorillonite clay. Phys. A 2018, 505, 582–590. [Google Scholar] [CrossRef]
- Underwood, T.; Erastova, V.; Greenwell, H.C. Ion Adsorption at Clay-mineral Surfaces: The Hofmeister Series For Hydrated Smectite Minerals. Clays Clay Miner. 2016, 64, 472–487. [Google Scholar] [CrossRef]
- Zheng, Y.; Zaoui, A. How water and counterions diffuse into the hydrated montmorillonite. Solid State Ionics 2011, 203, 80–85. [Google Scholar] [CrossRef]
- Liu, T.; Chen, Y.Q. A molecular dynamics study of the swelling patterns of Na/Cs-montmorillonites and the hydration of interlayer cations. Chin. Phys. B 2013, 22, 027103. [Google Scholar] [CrossRef]
- Zhou, Q.; Lu, X.C.; Liu, X.D.; Zhang, L.H.; He, H.P.; Zhu, J.X.; Yuan, P. Hydration of methane intercalated in Na-smectites with distinct layer charge: Insights from molecular simulations. J. Colloid Interface Sci. 2011, 355, 237–242. [Google Scholar] [CrossRef]
- Yan, K.F.; Li, X.S.; Xu, C.G.; Lv, Q.N.; Ruan, X.K. Molecular dynamics simulation of the intercalation behaviors of methane hydrate in montmorillonite. J. Mol. Model. 2014, 20, 2311. [Google Scholar] [CrossRef]
- Randall, T.C.; Liang, J.J.; Andrey, G.K. Molecular Models of Hydroxide, Oxyhydroxide, and Clay Phases and the Development of a General Force Field. J. Phys. Chem. 2004, 108, 1255–1266. [Google Scholar]
- Meleshyn, A.; Tunega, D. Adsorption of phenanthrene on Na-montmorillonite: A model study. Geoderma 2011, 169, 41–46. [Google Scholar] [CrossRef]
- Wang, J.F.; Gutierrez, M. Molecular simulations of clay minerals: A study considering the change of cell size and shape. Acta Geotech. 2010, 5, 151–167. [Google Scholar] [CrossRef]
- Seppälä, A.; Puhakka, E.; Olin, M. Effect of layer charge on the crystalline swelling of Na+, K+ and Ca2+ montmorillonites: DFT and molecular dynamics studies. Clay Miner. 2018, 51, 197–211. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Na-MT1 | 0-6 | 2-4 | 3-3 | 4-2 | 6-0 |
| Oct:Td substitutions ratios | 0 | 33.3% | 50.0% | 66.7% | 100.0% |
| Na-MT2 | 0-8 | 2-6 | 4-4 | 6-2 | 8-0 |
| Oct:Td substitutions ratios | 0 | 25.0% | 50.0% | 75.0% | 100.0% |
| Na-MT3 | 0-9 | 2-7 | 4-5 | 7-2 | 9-0 |
| Oct:Td substitutions ratios | 0 | 22.2% | 44.4% | 77.8% | 100.0% |
| Na-MT4 | 0-11 | 3-8 | 5-6 | 8-3 | 11-0 |
| Oct:Td substitutions ratios | 0 | 27.3% | 45.5% | 72.7% | 100.0% |
| Na-MT1 | 0-6 | 2-4 | 3-3 | 4-2 | 6-0 |
| Volume/Å3 | 9195.10 | 9165.03 | 9198.71 | 9140.91 | 9218.54 |
| c/Å | 12.30 | 12.28 | 12.34 | 12.21 | 12.31 |
| β/° | 99.7 | 100.3 | 100.5 | 99.2 | 99.2 |
| Na-MT2 | 0-8 | 2-6 | 4-4 | 6-2 | 8-0 |
| Volume/Å3 | 9143.39 | 9176.34 | 9176.06 | 9038.18 | 9168.08 |
| c/Å | 12.24 | 12.27 | 12.26 | 12.12 | 12.30 |
| β/° | 99.9 | 99.7 | 99.3 | 100.5 | 100.6 |
| Na-MT3 | 0-9 | 2-7 | 4-5 | 7-2 | 9-0 |
| Volume/Å3 | 9056.51 | 9037.13 | 9084.02 | 9138.52 | 9156.57 |
| c/Å | 12.11 | 12.11 | 1214 | 12.21 | 12.22 |
| β/° | 99.5 | 100.2 | 99.5 | 99.4 | 98.8 |
| Na-MT4 | 0-11 | 3-8 | 5-6 | 8-3 | 11-0 |
| Volume/Å3 | 9073.53 | 9132.79 | 9146.43 | 9165.33 | 9129.02 |
| c/Å | 12.16 | 12.23 | 12.22 | 12.24 | 12.17 |
| β/° | 100.1 | 100.1 | 99.3 | 99.1 | 98.6 |
| Species | Type | 0 | 33.3% | 50.0% | 66.7% | 100% |
|---|---|---|---|---|---|---|
| H2O | Na-MT1 | 1.8282 ± 0.0000 | 1.8582 ± 0.0003 | 2.4531 ± 0.0002 | 2.6298 ± 0.0000 | 4.0896 ± 0.0002 |
| Na-MT2 | 1.3651 ± 0.0002 | 1.6890 ± 0.0000 | 1.7600 ± 0.0002 | 2.6150 ± 0.0002 | 3.4874 ± 0.0002 | |
| Na-MT3 | 1.2979 ± 0.0004 | 1.4970 ± 0.0002 | 1.4260 ± 0.0002 | 1.7523 ± 0.0003 | 2.0442 ± 0.0002 | |
| Na-MT4 | 0.6381 ± 0.0001 | 1.2300 ± 0.0002 | 1.1637 ± 0.0002 | 1.7482 ± 0.0003 | 1.7549 ± 0.0001 | |
| Na+ | Na-MT1 | 0.4298 ± 0.0001 | 0.4578 ± 0.0001 | 0.4428 ± 0.0002 | 0.5617 ± 0.0001 | 0.8835 ± 0.0000 |
| Na-MT2 | 0.3268 ± 0.0000 | 0.3683 ± 0.0002 | 0.3670 ± 0.0001 | 0.4352 ± 0.0003 | 0.8083 ± 0.0000 | |
| Na-MT3 | 0.0958 ± 0.0002 | 0.1545 ± 0.0003 | 0.2500 ± 0.0002 | 0.3360 ± 0.0005 | 0.5530 ± 0.0002 | |
| Na-MT4 | 0.0940 ± 0.0000 | 0.2060 ± 0.0002 | 0.2103 ± 0.0002 | 0.3258 ± 0.0003 | 0.6498 ± 0.0003 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, J.; Liu, D.; Jiang, S.; Chen, G.; Wang, Y.; Li, G.; Yao, G.; Wu, P.; Zhu, X.; Wang, G.; et al. Effect of Crystal Chemistry Properties on the Distribution Characteristics of H2O and Na+ in Na-Montmorillonite Interlayer Space: Molecular Dynamics Simulation Study. Minerals 2020, 10, 162. https://doi.org/10.3390/min10020162
Qiu J, Liu D, Jiang S, Chen G, Wang Y, Li G, Yao G, Wu P, Zhu X, Wang G, et al. Effect of Crystal Chemistry Properties on the Distribution Characteristics of H2O and Na+ in Na-Montmorillonite Interlayer Space: Molecular Dynamics Simulation Study. Minerals. 2020; 10(2):162. https://doi.org/10.3390/min10020162
Chicago/Turabian StyleQiu, Jun, Dongliang Liu, Shan Jiang, Guowei Chen, Yueting Wang, Guoqing Li, Geng Yao, Peng Wu, Xiangnan Zhu, Guifang Wang, and et al. 2020. "Effect of Crystal Chemistry Properties on the Distribution Characteristics of H2O and Na+ in Na-Montmorillonite Interlayer Space: Molecular Dynamics Simulation Study" Minerals 10, no. 2: 162. https://doi.org/10.3390/min10020162
APA StyleQiu, J., Liu, D., Jiang, S., Chen, G., Wang, Y., Li, G., Yao, G., Wu, P., Zhu, X., Wang, G., & Lyu, X. (2020). Effect of Crystal Chemistry Properties on the Distribution Characteristics of H2O and Na+ in Na-Montmorillonite Interlayer Space: Molecular Dynamics Simulation Study. Minerals, 10(2), 162. https://doi.org/10.3390/min10020162