Symmetry Analysis in Mechanistic Studies of Nucleophilic Substitution and β-Elimination Reactions

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
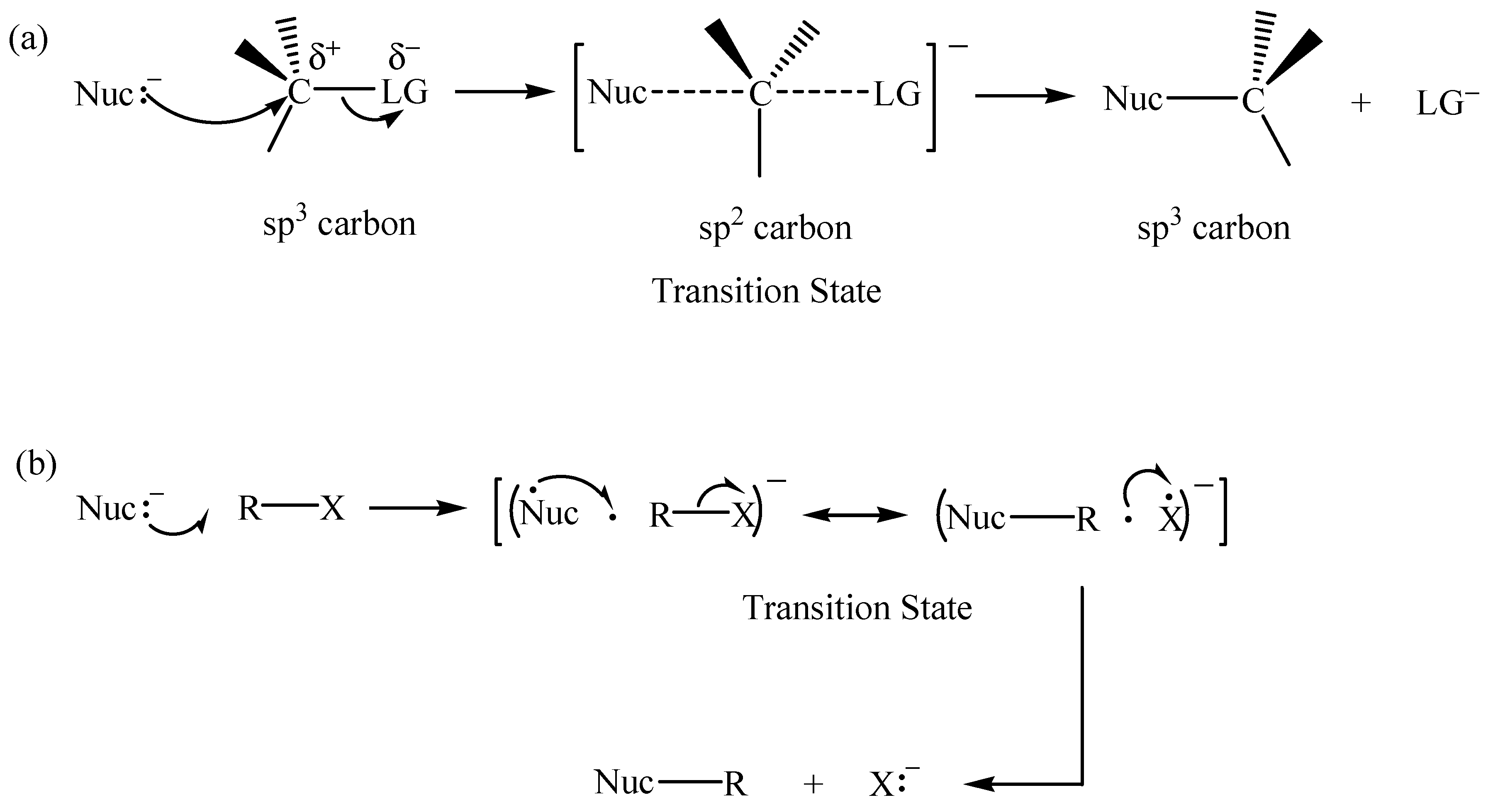
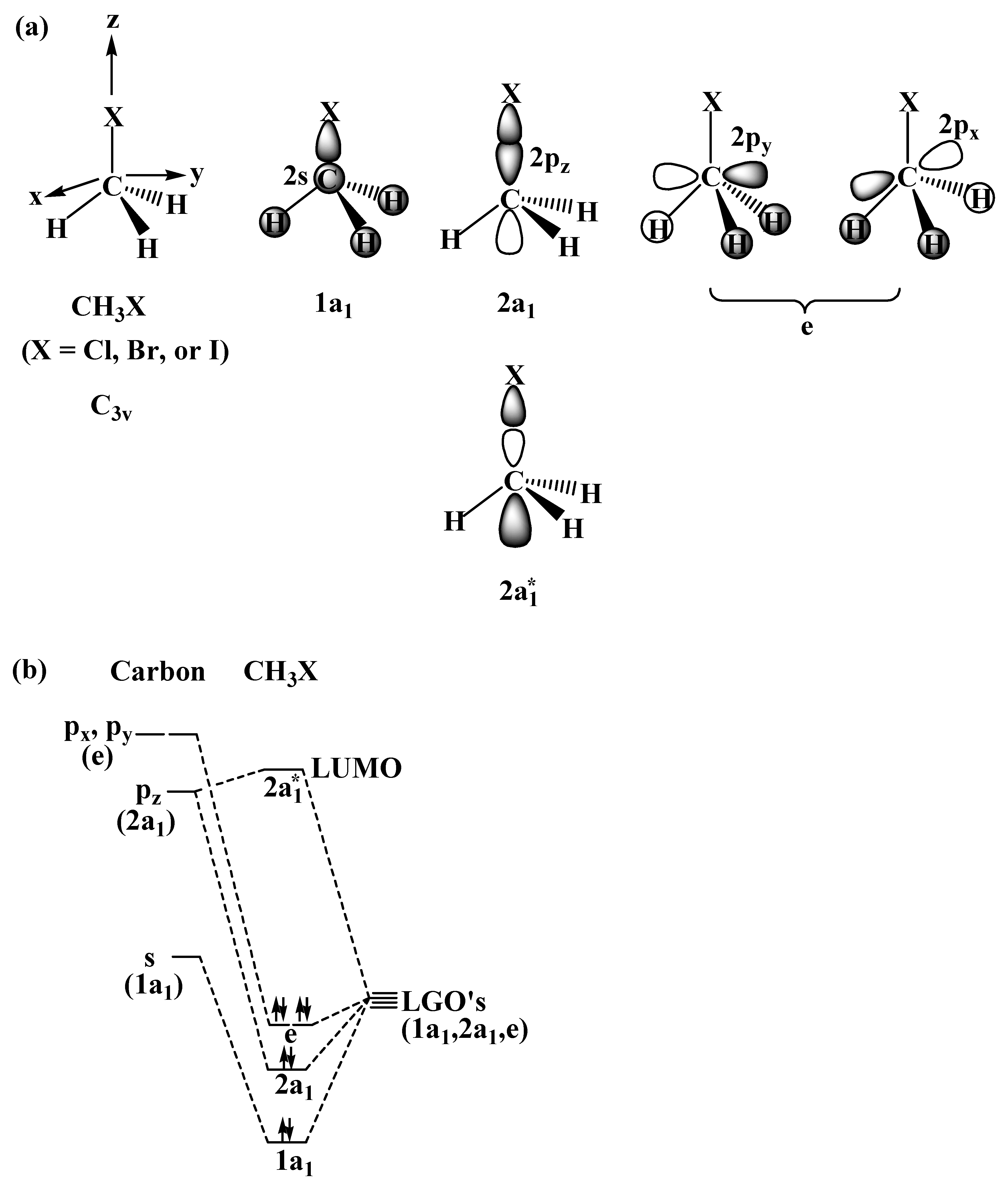
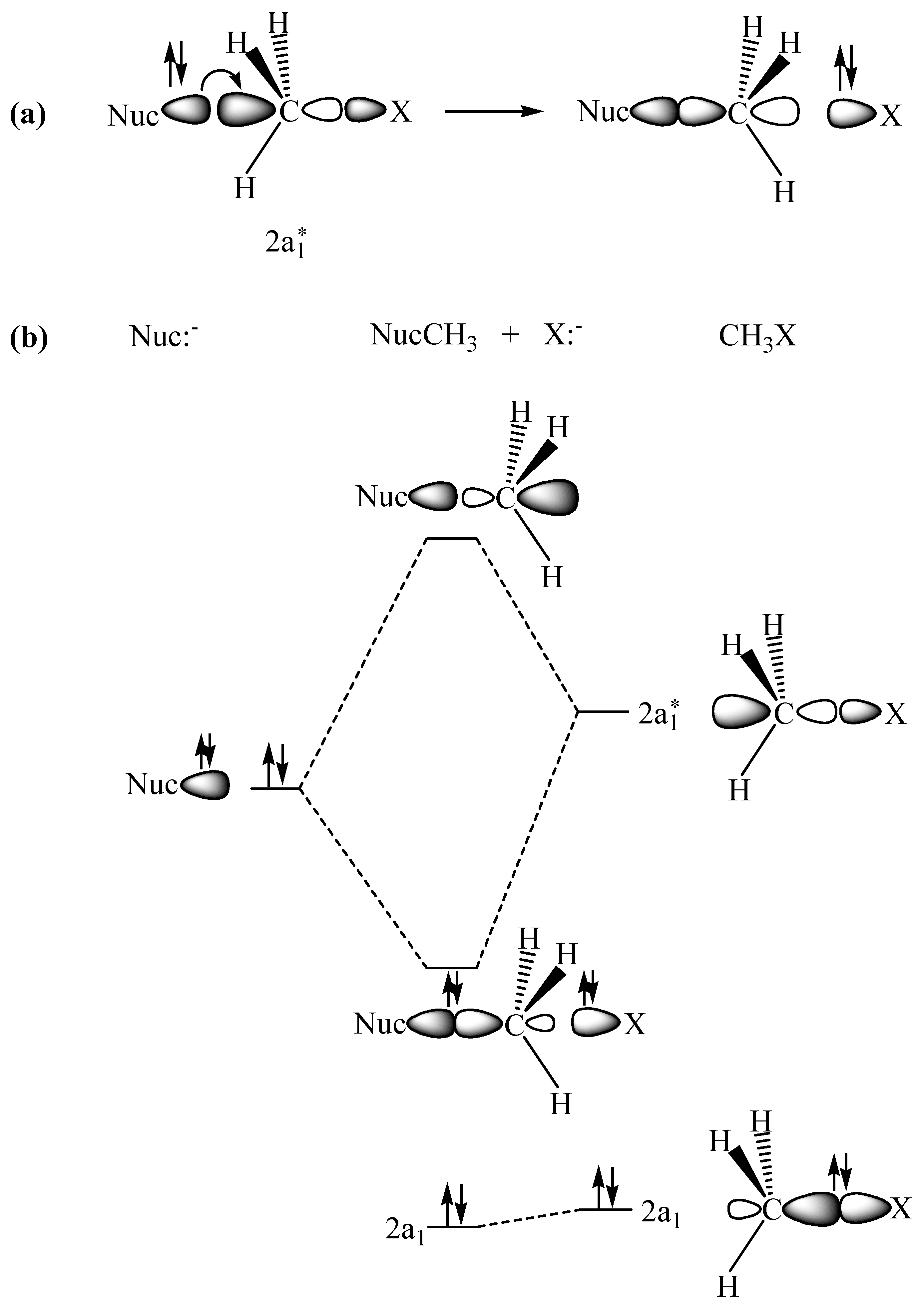
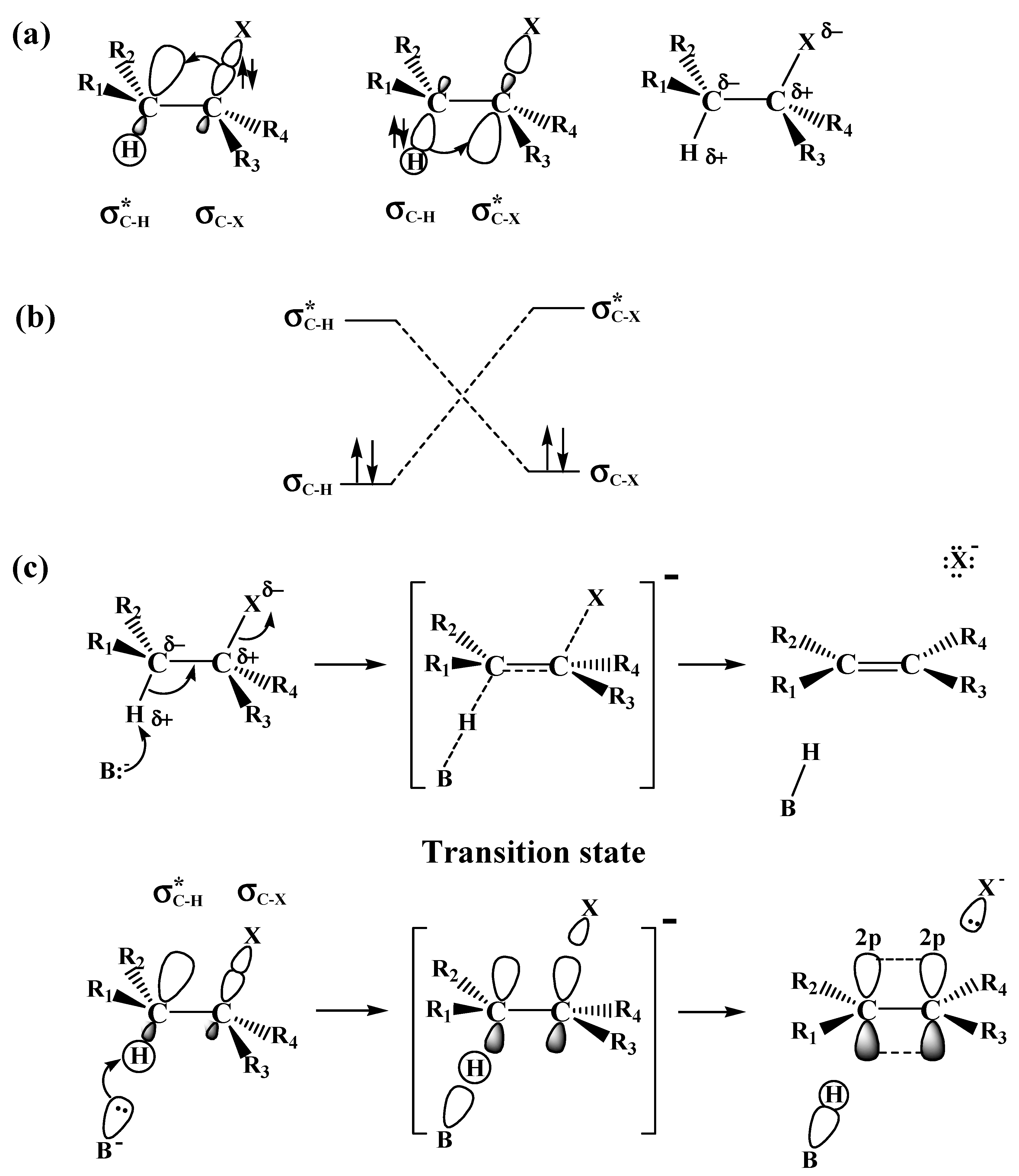
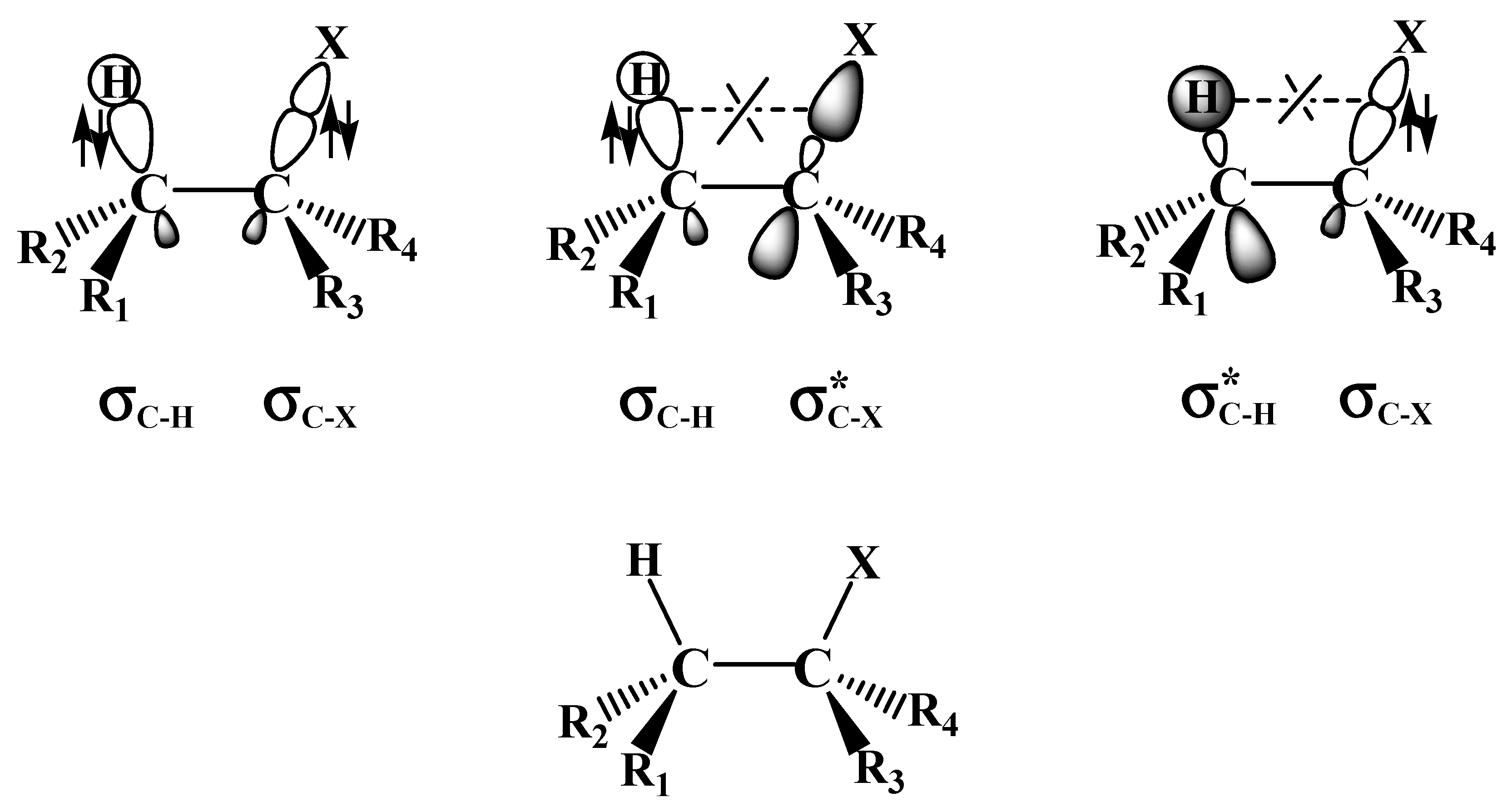
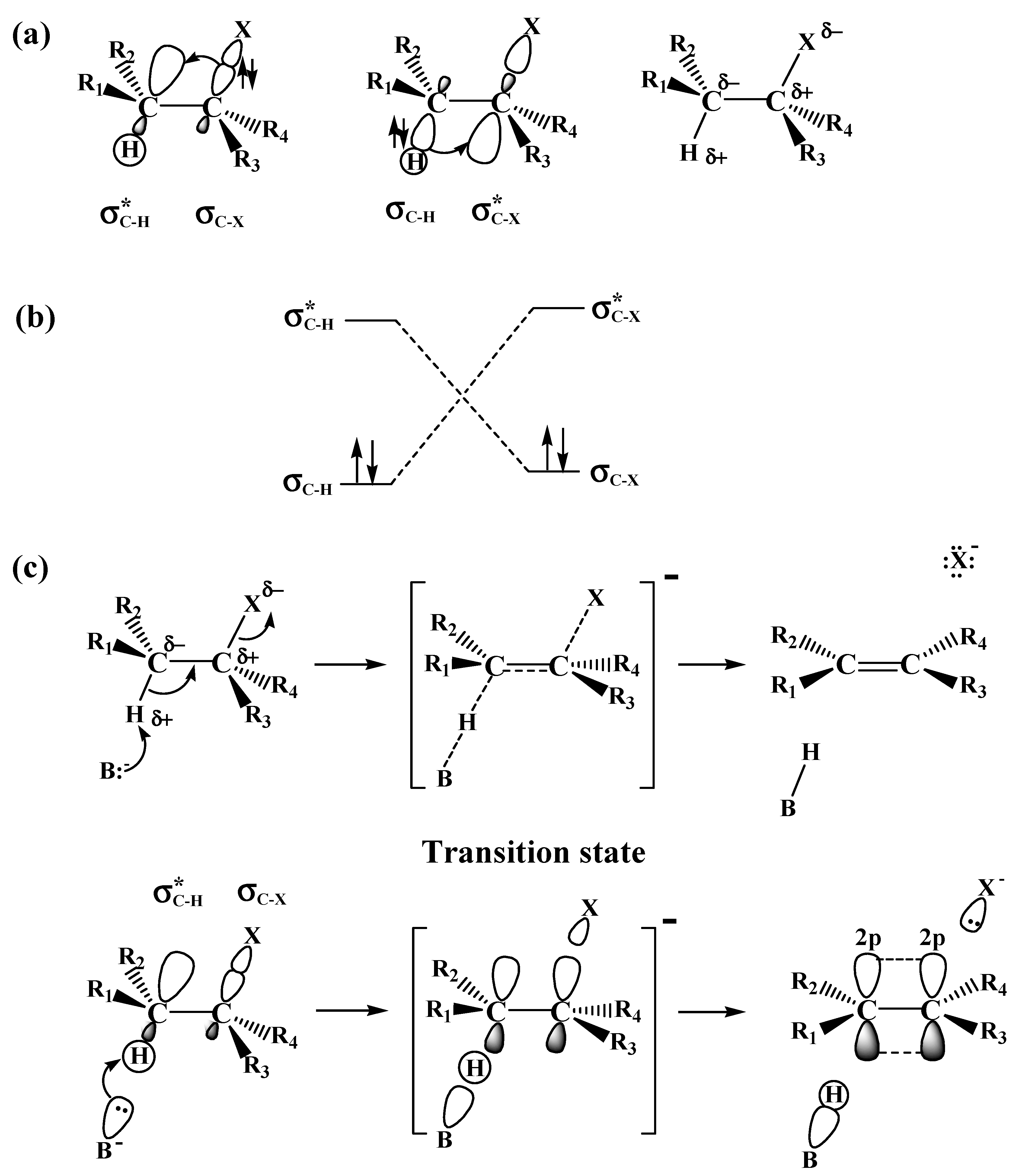
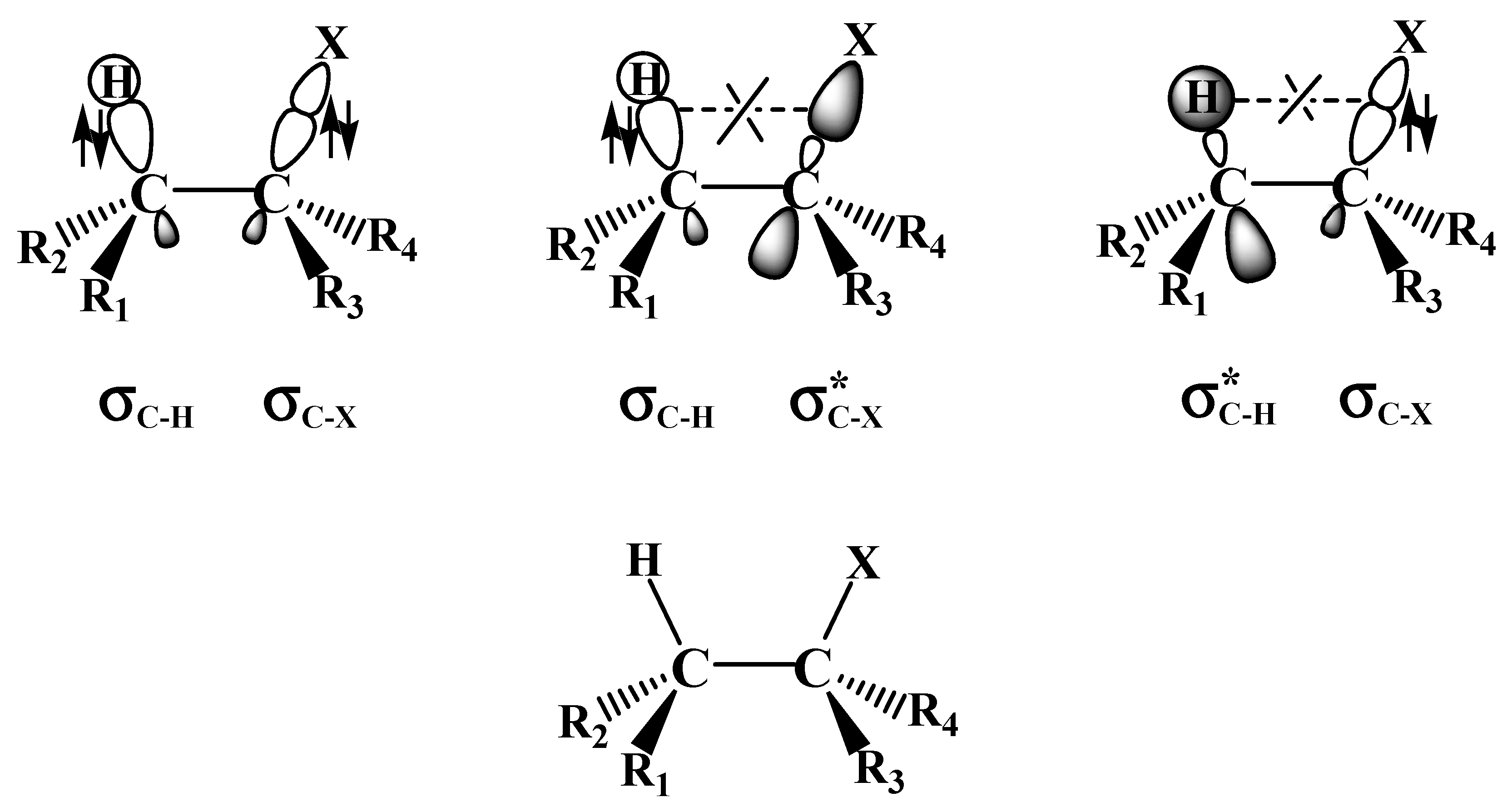
2.1. The SN2 Reaction of Halomethane CH3X (X =Cl, Br, or I)
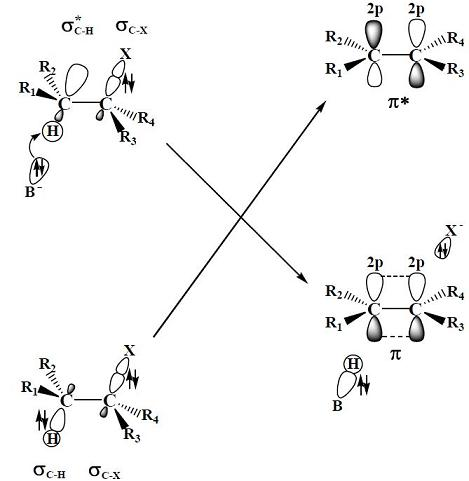
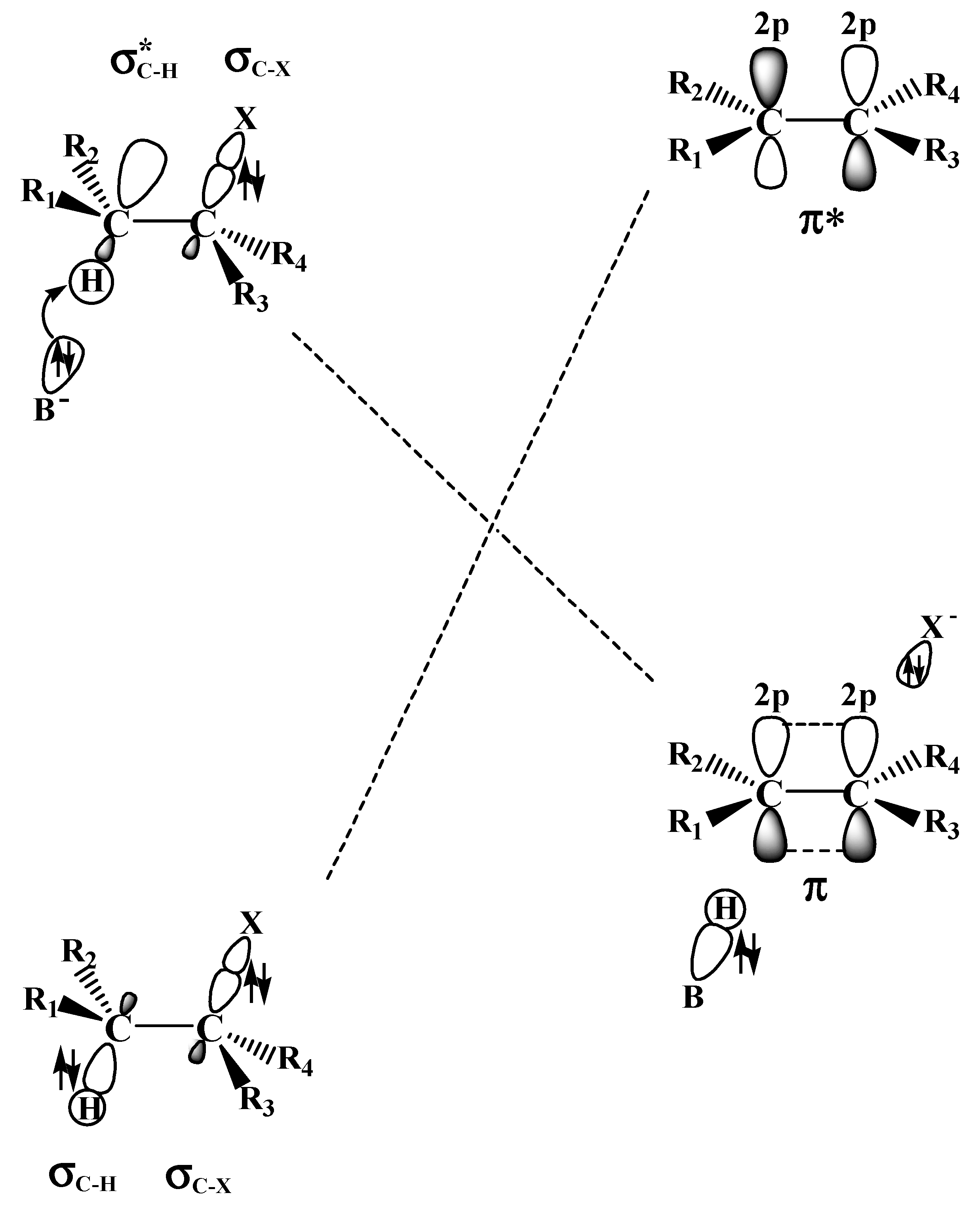
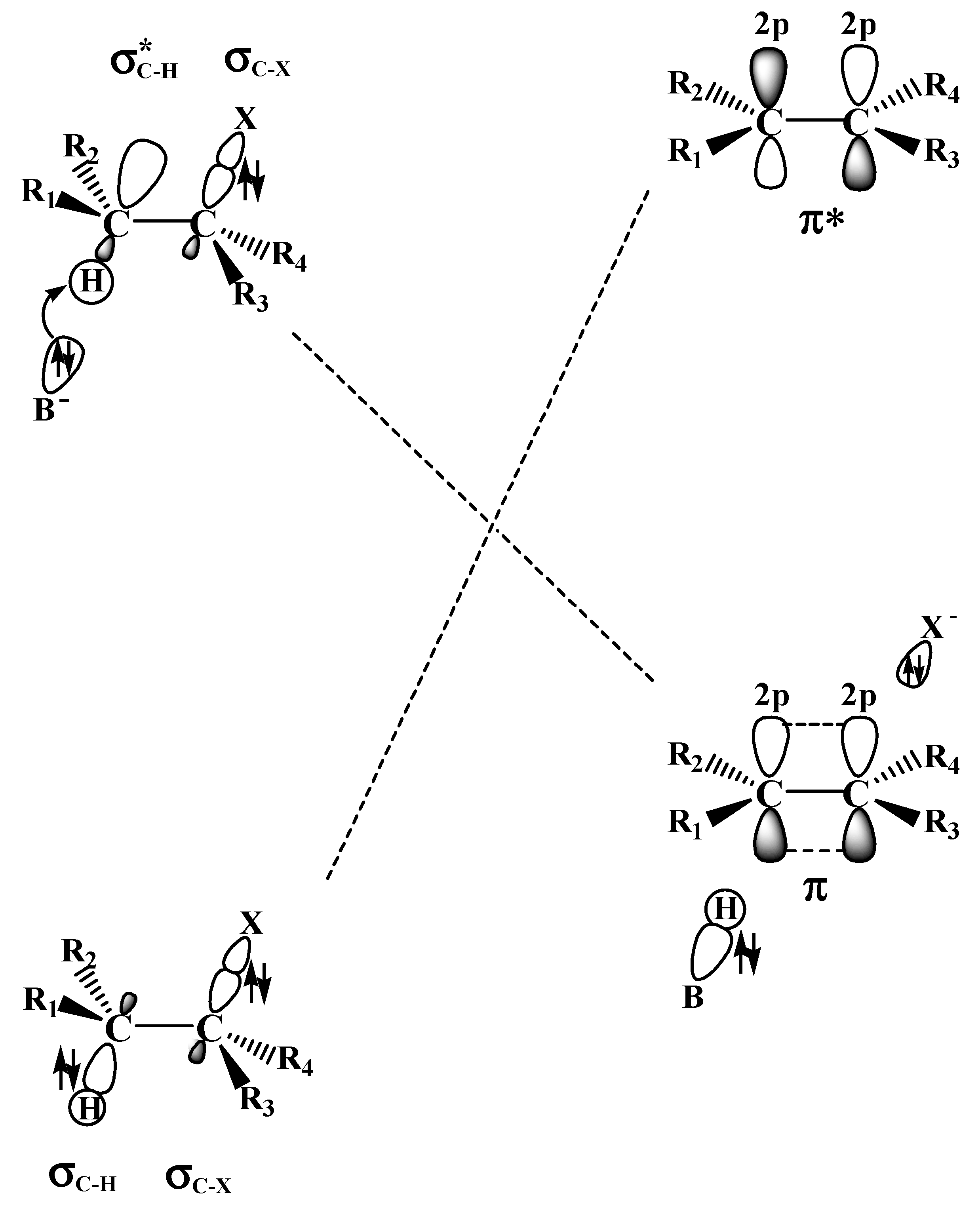
2.2. The E2 Reactions of the Chain-Like Haloalkanes
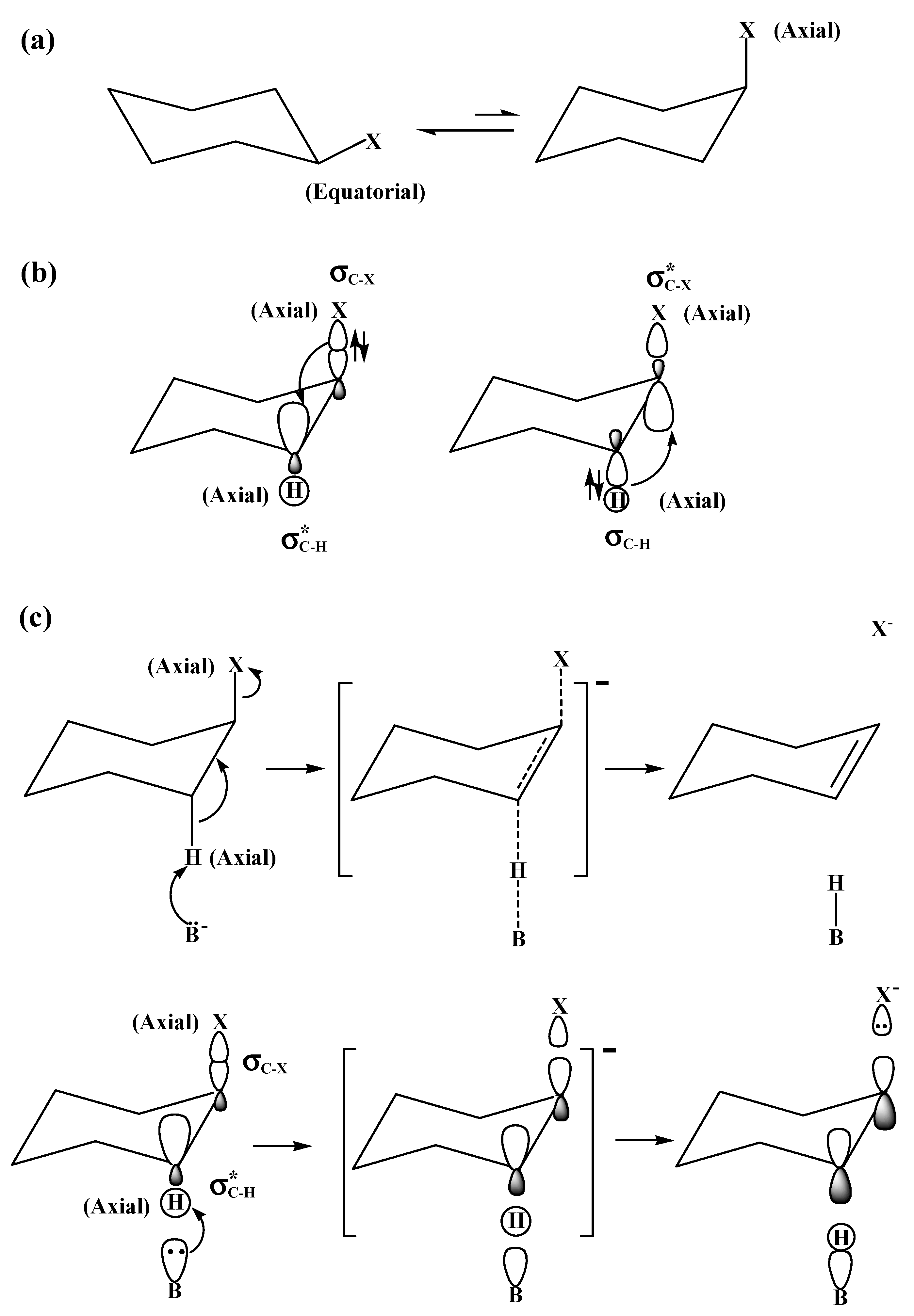
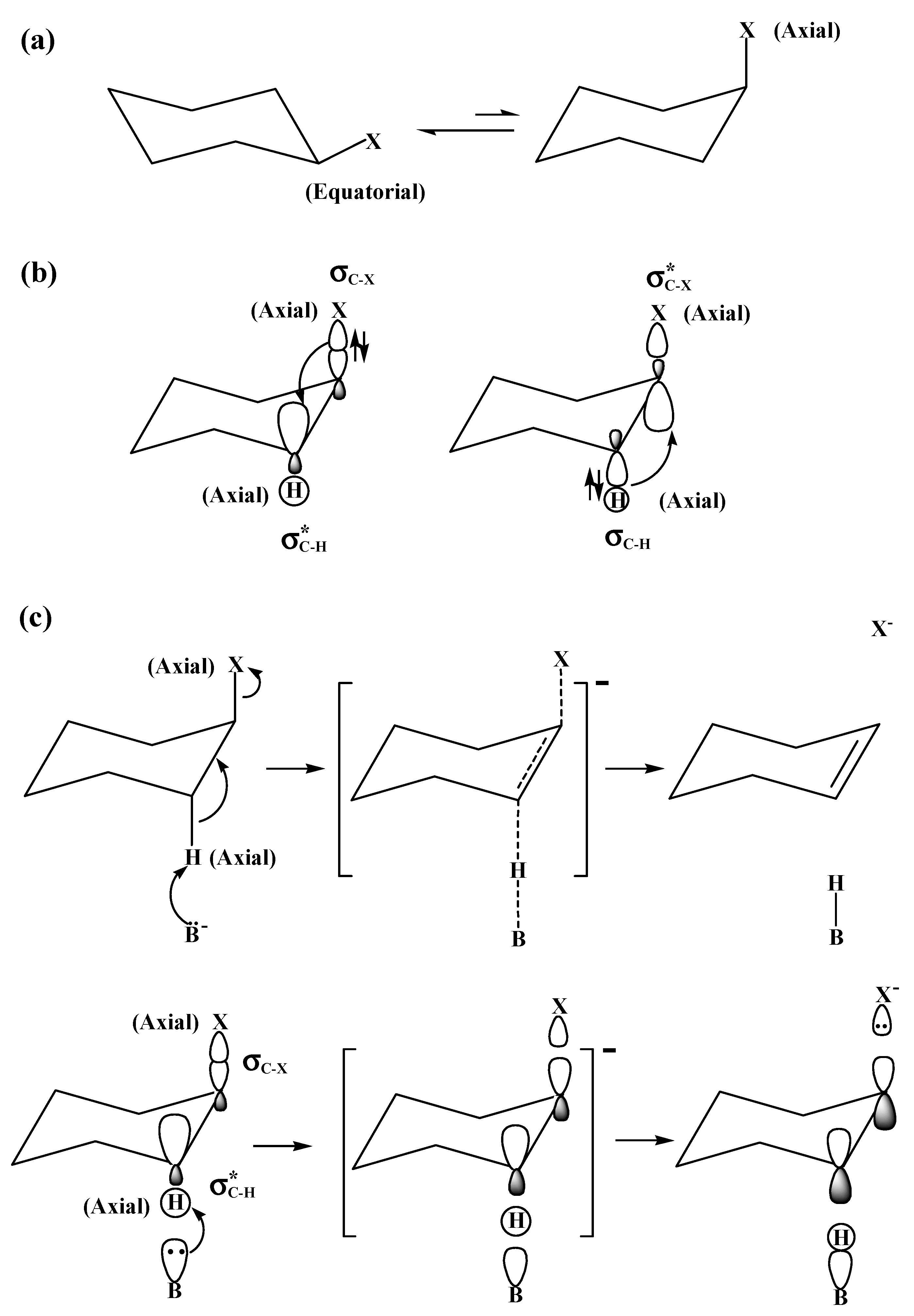
2.3. The E2 Reactions of Halocyclohexanes
3. Conclusions
References and Notes
- Fox, M.A.; Whitesell, J.K. Core Organic Chemistry; Jones and Bartlett Publishers: Boston, MA, USA, 1997; pp. 385–484. [Google Scholar]
- Carey, F.A. Organic Chemistry, 5th ed.; McGraw-Hill: New York, NY, USA, 2003; pp. 326–362. [Google Scholar]
- McMurry, J. Organic Chemistry, 6th ed.; Brooks/Cole: Belmont, CA, USA, 2004; pp. 343–392. [Google Scholar]
- Reingold, I.D. Organic Chemistry: Am Introduction Emphasizing Biological Connections; Houghton Mifflin Company: Boston, MA, USA, 2002; pp. 310–320. [Google Scholar]
- Brown, W.H.; Foote, C.S.; Iverson, B.L.; Anslyn, E.V. Organic Chemistry, 5th ed.; Brooks/Cole: Belmont, CA, USA, 2009; pp. 321–373. [Google Scholar]
- Askham, F.R.; Stanley, G.G.; Marques, E.C. A new type of transition-metal dimer based on a Hexaphosphine Ligand system: Co2(CO)4(eHTP)2+. J. Am. Chem. Soc. 1985, 107, 7423–7431. [Google Scholar] [CrossRef]
- Harcourt, R.D. Increased-valence structures and hypervalent molecules. Int. J. Quantum Chem. 1996, 60, 553–566. [Google Scholar] [CrossRef]
- Pross, A. The single electron shift as a fundamental process in organic chemistry: The relationship between polar and electron-transfer pathways. Acc. Chem. Res. 1985, 18, 212–219. [Google Scholar] [CrossRef]
- Shaik, S.S. The lego way: Curve crossing diagrams as general models in physical organic chemistry. Pure Appl. Chem. 1991, 63, 195–204. [Google Scholar] [CrossRef]
- Shaik, S.S.; Schlegel, H.B.; Wolfe, S. Theoretical Aspects of Physical Organic Chemistry: The SN2 Mechanism; Wiley: New York, NY, USA, 1992. [Google Scholar]
- Sun, X. A Study of the SN2 mechanism by symmetry rules and qualitative molecular orbital theory. Chem. Educator 2003, 8, 303–306. [Google Scholar]
- Huheey, J.E.; Keiter, E.A.; Keiter, R.L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th ed.; Harper Collins: New York, NY, USA, 1993; pp. 53–63; 413–433; A-13–A-20. [Google Scholar]
- Gimarc, B.M.; Khan, S.A. The shapes and other properties of non-transition elements complexes. 2. AB4. J. Am. Chem. Soc. 1978, 100, 2340–2345. [Google Scholar] [CrossRef]
- Gimarc, B.M. The shapes and other properties of non-transition elements complexes. 3. AB5. J. Am. Chem. Soc. 1978, 100, 2346–2353. [Google Scholar] [CrossRef]
- Gimarc, B.M.; Liebman, J.F.; Hohn, M. The shapes and other properties of non-transition elements complexes. 1. AB6. J. Am. Chem. Soc. 1978, 100, 2334–2339. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J.; Nibbering, N.M.M.; Ziegler, T. Theoretical investigation on base-induced 1,2-Eliminations in a model system F- + CH3CH2F. The role of the base as a catalyst. J. Am. Chem. Soc. 1993, 115, 9160–9173. [Google Scholar] [CrossRef]
- Ensing, B.; Laio, A.; Gervasio, F.L.; Parrinello, M.; Klein, M.L. A minimum free energy reaction path for the E2 reaction between fluoro ethane and a floride ion. J. Am. Chem. Soc. 2004, 126, 9492–9493. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, R. Advance Organic Chemistry: Reaction Mechanisms; Harcourt: San Diego, CA, USA, 2002; pp. 129–167. [Google Scholar]
Supplemental Materials–Symmetry Analysis and Ligand Group Orbitals (LGO’s) for Various Functionalized Molecules
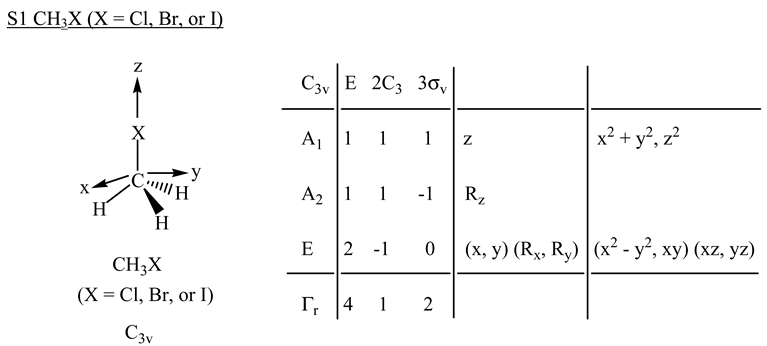
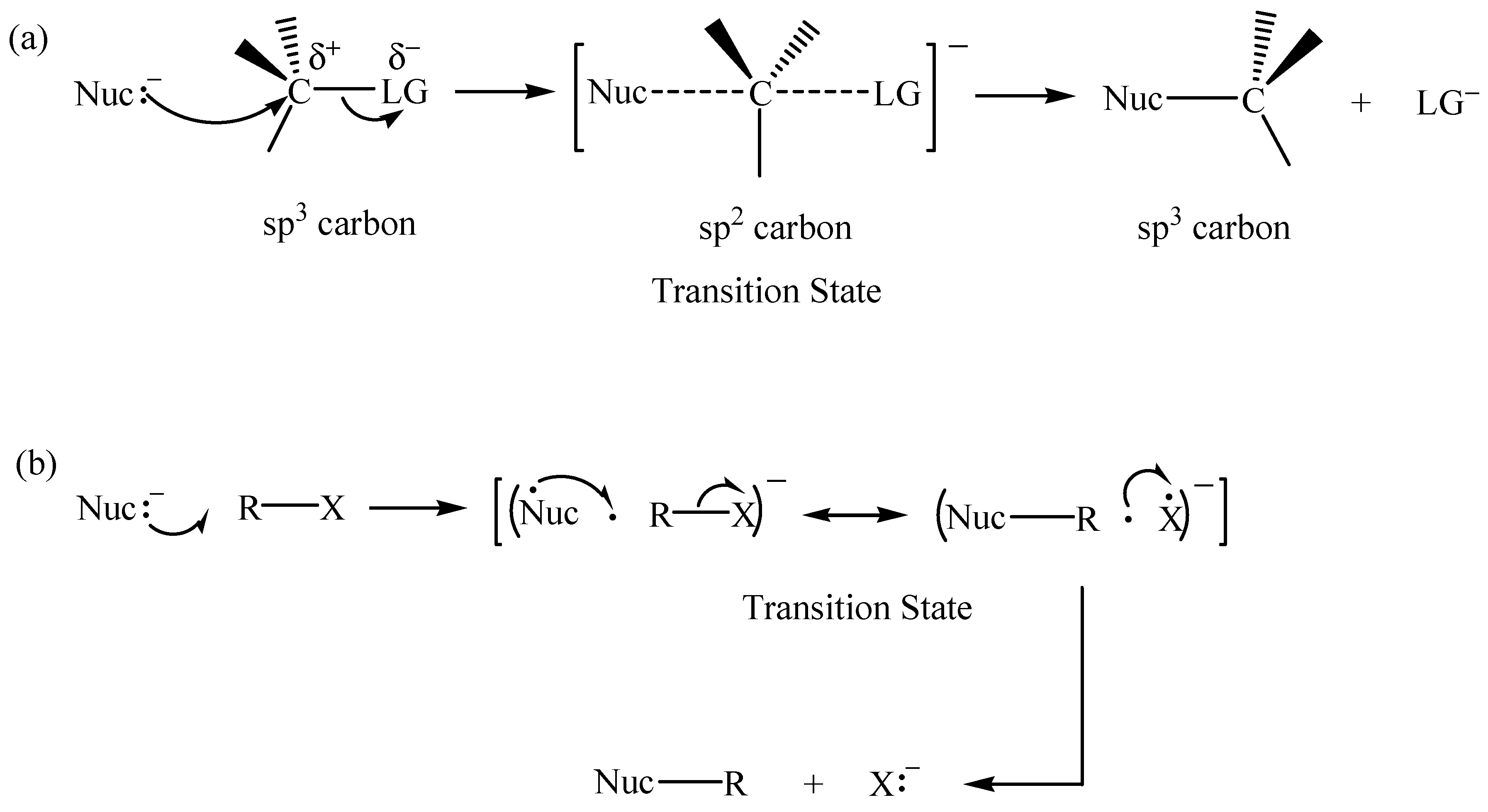
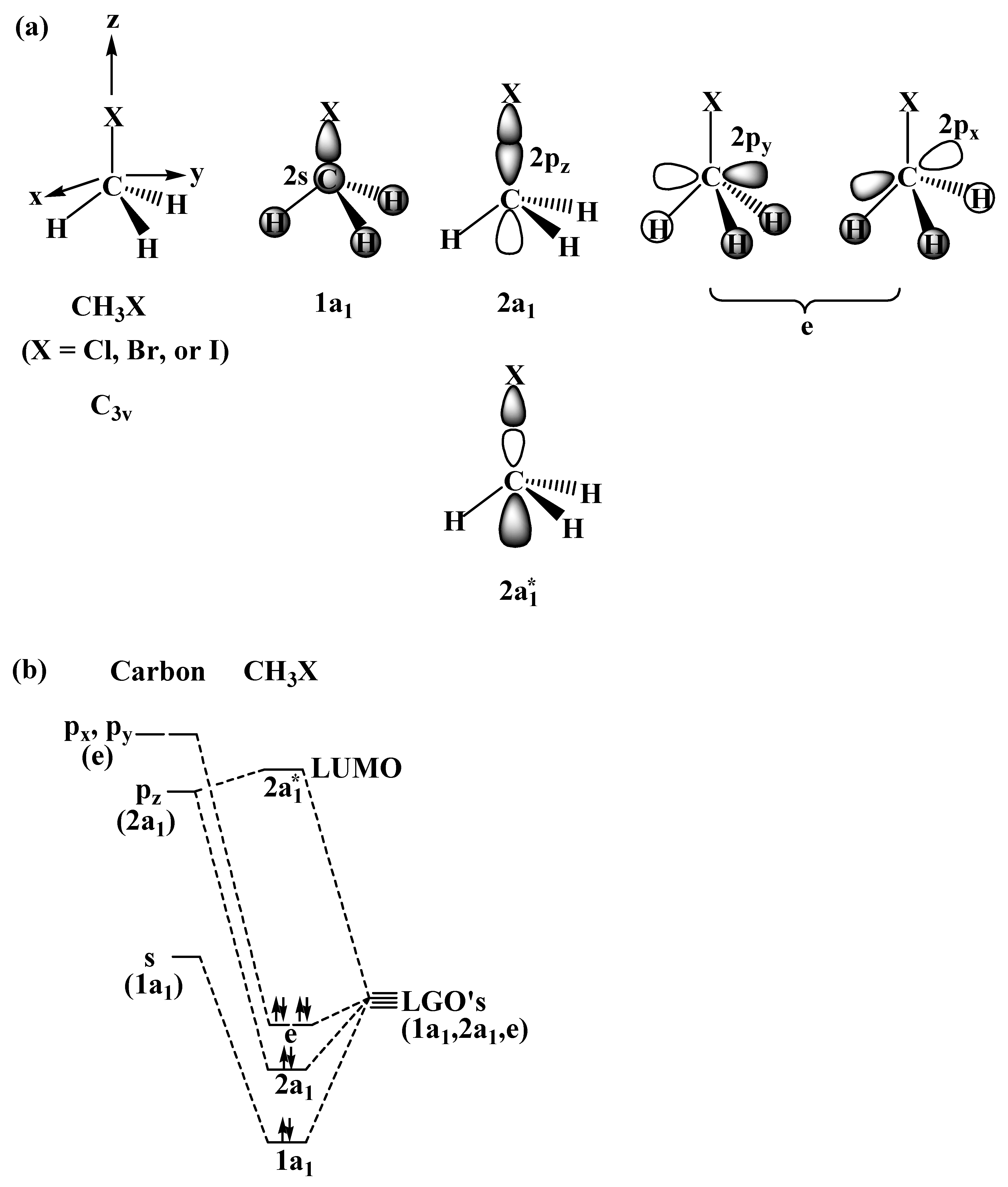
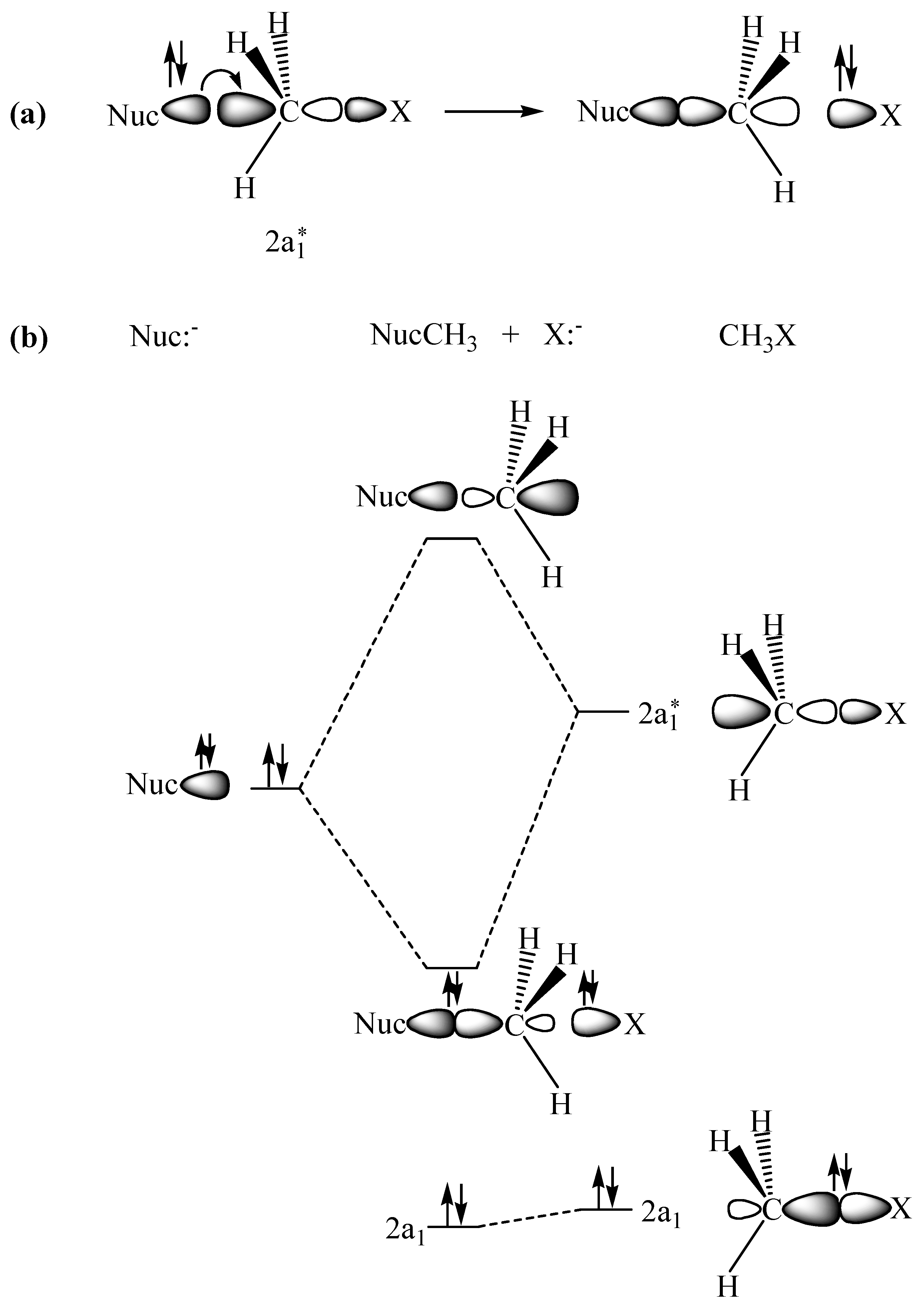
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, X. Symmetry Analysis in Mechanistic Studies of Nucleophilic Substitution and β-Elimination Reactions. Symmetry 2010, 2, 201-212. https://doi.org/10.3390/sym2010201
Sun X. Symmetry Analysis in Mechanistic Studies of Nucleophilic Substitution and β-Elimination Reactions. Symmetry. 2010; 2(1):201-212. https://doi.org/10.3390/sym2010201
Chicago/Turabian StyleSun, Xiaoping. 2010. "Symmetry Analysis in Mechanistic Studies of Nucleophilic Substitution and β-Elimination Reactions" Symmetry 2, no. 1: 201-212. https://doi.org/10.3390/sym2010201
APA StyleSun, X. (2010). Symmetry Analysis in Mechanistic Studies of Nucleophilic Substitution and β-Elimination Reactions. Symmetry, 2(1), 201-212. https://doi.org/10.3390/sym2010201