3.1. Symmetrization in Hydrophobicity Profile Comparison (FOD-JS)
T,
O,
H, and
R profiles express different categories of the density of hydrophobicity in the input structure. Each is normalized and has a length of
n. Baseline FOD algorithm employs Kullback–Leibler divergence (D
KL) to quantitatively gauge differences between those profiles. To quickly reiterate
Section 2.1,
O|T symbolizes the D
KL-based distance from
O to
T.
O|R is the coefficient for
O and
R (“random”). Relative Distance (
RD) ties them together by placing the structure on a normalized 0–1 scale. When 0 ≤
RD < 0.5,
O of this structure is closer to
T (accordance with the model, well-ordered hydrophobic core), while 0.5 ≤
RD ≤ 1.0 is the evidence of
O’s closeness to
R (discordance, core’s instability). It is the default, most important, and the most used variant of
RD in the baseline FOD algorithm, branded as
T-O-R. Replacing
R with
H in Equation (5) yields
T-O-H RD which reveals the position of
O with respect to theoretical and intrinsic profiles. It is likewise possible to construct a third (but unused) variant of baseline
RD,
H-O-R.
DKL is not a symmetric measure. This has a few consequences for its application in the comparison of profiles of hydrophobicity. First, it mandates a specific profile order for Equation (4). In the baseline FOD model, it is O|T, O|R, and O|H, which we shall call here the “O first” mode. The three reversed DKL coefficients—namely T|O, R|O, and H|O (“O last”)— present the same profile relation but from the opposite perspective. They are unused in the baseline FOD model because equivalence cannot be expected between them (e.g., O|T ≠ T|O) in real proteins. The purpose of entropy in this context is to measure the difference between two profiles of hydrophobicity. The FOD model gives no special priority to residues with either T ≪ O or T ≫ O during this assessment. Put differently, an excess of hydrophobicity (T ≪ O) is not favored over its deficiency (T ≫ O) as the source of stronger discrepancy and vice versa. However, Equation (3) always favors one of them by assigning higher local divergence to residues exhibiting Q ≪ O in the “O first” mode and Q ≫ O in “O last” mode (Q = T/H/R). The mathematical basis behind this is presented in the next paragraph. However, this begs the question: which approach is more appropriate? First, second, or both? Hydrophobicity profiles often have shallower valleys below the R line compared to taller peaks above it, due to cysteine located at the top of the FOD model’s hydrophobicity scale, for instance. Their uneven treatment by relative entropy is beneficial as it prevents domination of the upper region. The drawback is that it applies to one profile at a time. The need for adherence to one mode (“O first”) also makes the RD calculation that does not involve the O profile cumbersome. For instance, if we wanted to measure the statistical distance between T and R, should we rely on T|R or R|T? This may likewise impede the introduction of new profiles.
The other problem with D
KL is numerical and relates to the handling of two side cases: the value of 0 and values close to 0. When
Pi =
Qi = 0, Equation (3) becomes undefined, but because D
KL(
Pi|
Qi) = D
KL(
Qi|
Pi) = 0 if and only if
Pi =
Qi, we can make an exception to return 0 directly, which is a correct and desirable approach. However, when
Pi = 0 and
Qi > 0, Equation (3) also becomes undefined due to a lack of value of log
20. We could try to avoid it by noticing that
x·log
2x = 0, thus assuming, by convention [
53], that 0·log
20 = 0. It circumvents the problem with the logarithm but introduces another in the form of 0 being reported when
Pi ≠
Qi. Likewise,
Pi > 0 and
Qi = 0 results in division by zero or an assumption of infinity [
53]. The fact that if
Pi → 0 and
Qi > 0 then D
KL(
Pi|
Qi) →0 and if
Pi > 0 and
Qi → 0 then D
KL(
Pi|
Qi) →∞ explains why the “
O first” mode of comparison deems residues with
O higher than the other profile as a source of higher local divergence and vice versa. We verify further down how different it actually is from “
O last”.
Avoidance of 0 in the observed profile is the reason why every effective atom must be located within a 9 Å of at least one other effective atom in the input. While this requirement should be satisfied by all valid and (mostly) complete protein structures, it is nonetheless an unnecessary obstacle, making it harder to experiment with residue selections or values of c. To prevent 0 from appearing in H (and possibly in O), we had to increase at one point the hydrophobicity parameter of lysine to 0.001 and use enough significant digits in the textual output of the algorithm to avoid issues with rounding.
DKL’s lack of an upper bound means that a small profile change close to 0, undetectable by humans and irrelevant from the perspective of the overall stability of the hydrophobic core, can boost local divergence, assigning undue importance to affected residues. It may be detrimental to the optimization of protein structures (i.e., folding simulation or prediction of complex formation), which is carried out with FOD by finding the state of the system that corresponds to the lowest statistical distance between T and O.
From the perspective of a programmer, an inadvertent profile switch during a call to a function that calculates DKL (e.g., T swapped with O) produces perfectly valid code and results that are disastrous for conclusions regarding the status of the hydrophobic core. Because such mistake may go unnoticed until much later, extra care is needed during access to this part of the FOD library. Finally, plots of the DKL series are inherently unintuitive with their mix of positive and negative values.
All above problems can be solved or strongly diminished by “promoting” D
KL to D
JS, the so-called Jensen–Shannon divergence [
54]. D
JS is a symmetric variant of Kullback–Leibler entropy, averaging the statistical distance from
Pi and
Qi to their average (
a):
Like D
KL, D
JS(
Pi|
Qi) = 0 if and only if
Pi = Qi and D
JS(
P|
Q) = 0 if and only if
P ≡ Q. It also has a few other useful properties [
55,
56,
57,
58]. The first is its aforementioned symmetry: D
JS(
Pi|
Qi) = D
JS(
Qi|
Pi) and D
JS(
P|
Q) = D
JS(
Q|
P). The second is its non-negativity and its upper bound: 0 ≤ D
JS(
Pi|
Qi) ≤ 1 and 0 ≤ D
JS(
P|
Q) ≤ 1. The third is that its square root is a proper metric—it satisfies triangle inequality. Finally, owing to being a sum of two factors, it can gracefully handle appearance of 0 anywhere in the compared distributions. To verify that, once again we need to assume that 0·log
20 = 0 [
53]. We can then rewrite Equation (6) by incorporating Equation (3) directly in it:
Now, without loss of generality, if we assume that
Pi = 0 and
Qi > 0, we obtain:
Setting Pi = 0 thus yields a divergence equal to half of Qi—the mean of Qi and 0. If we replace the second half of that sum with 0 when Qi = 0, 0 will be returned automatically for DJS(0|0), thus requiring only two special cases to properly manage all three variants with 0. It also means that, unlike the unbound DKL, Equation (6) cannot reach values higher than half the maximum of P and Q, making it predictable and placing its human-readable plot in the same (or scaled down) value range as its input distributions.
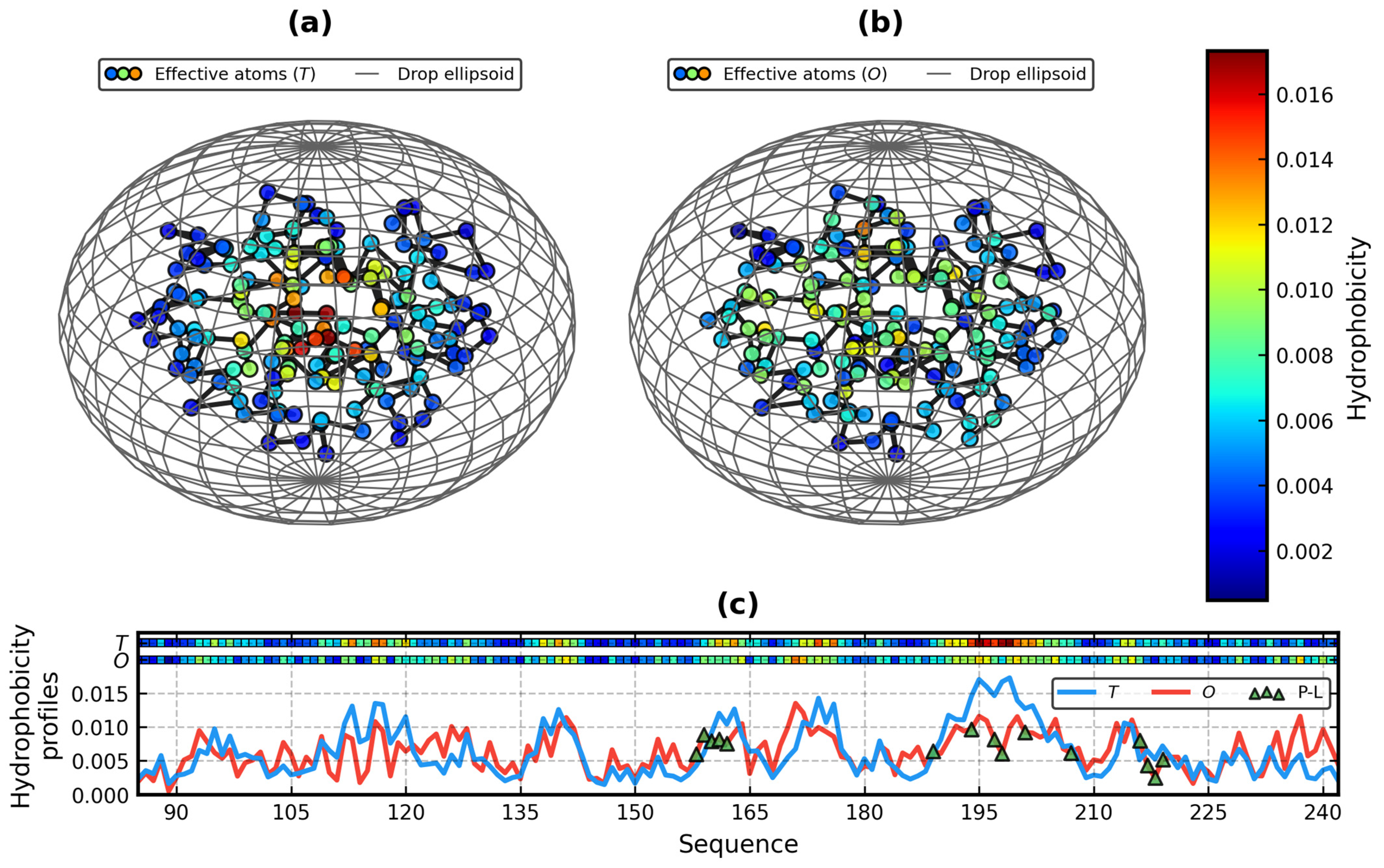
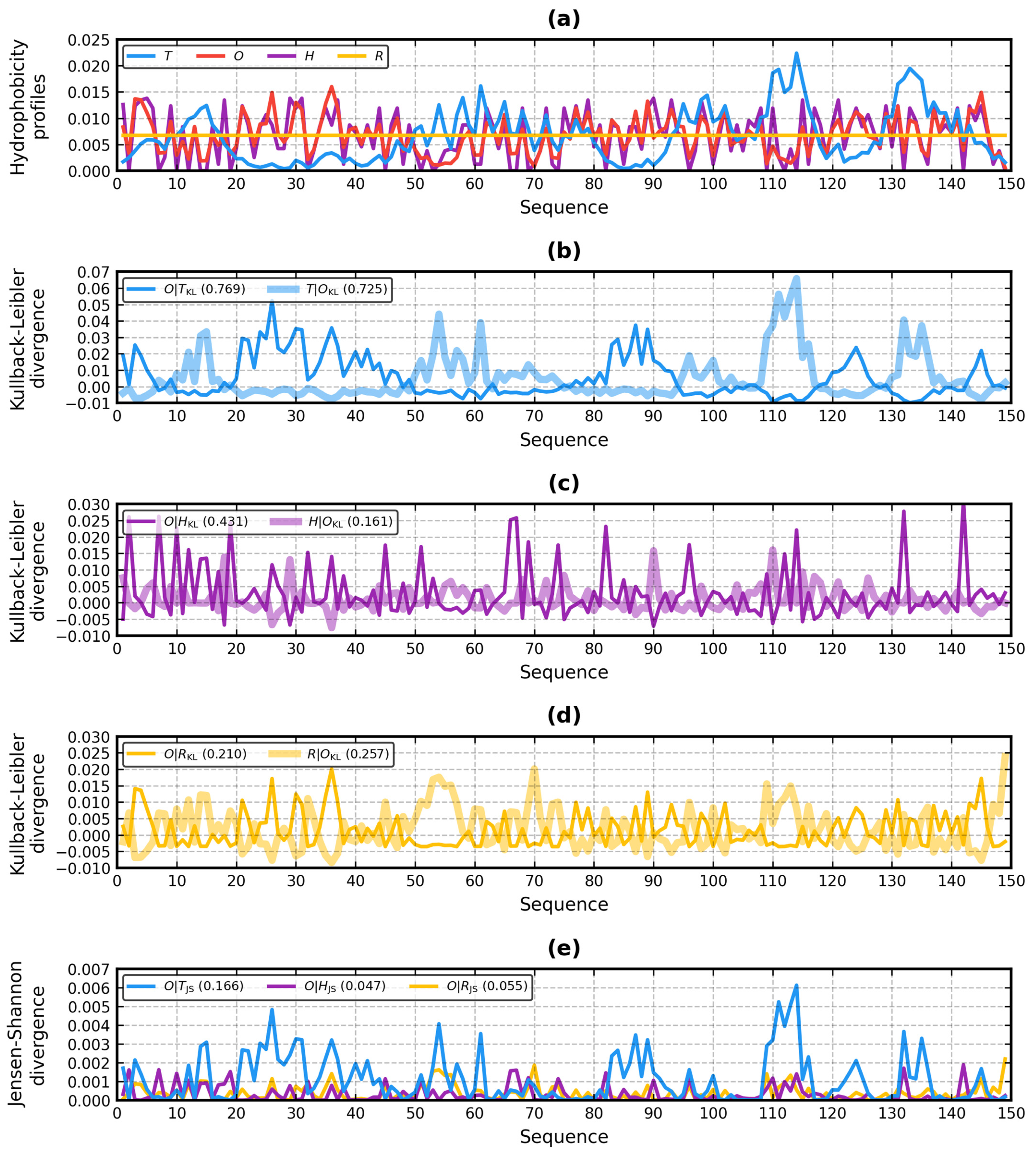
Figure 2 presents the results of the application of the above theory in practice. It displays a comparison between
T,
O,
H, and
R profiles in 1DIV:A via D
KL and D
JS.
Because hydrophobicity profiles of the FOD model are now comparable using two measures of entropy, we had to introduce new symbols in this paper to tell them apart: O|TKL, O|TJS, T-O-RKL, T-O-RJS, and others. Their meaning should be obvious.
Due to two domains and non-globular conformation, 1DIV:A is highly discordant with the model, as confirmed by
T-O-RKL = 0.785 and
T-O-HKL = 0.641. Furthermore, when one looks closely at
Figure 2b–d, it becomes clear that the “
O first” D
KL series—D
KL(
O|T), D
KL(
O|R), and D
KL(
O|H)—indeed reports higher local divergence for residues where
O is above the other profile. It is very noticeable in the G107-L117 region where a prominent
T peak contributes a tiny, negative bend to
O|T, while in the N20-T40 region,
T is below
O yet it achieves top values of D
KL(
O|T) (
Figure 2b). Both views are reversed in the “
O last” D
KL series—D
KL(
T|O), D
KL(
R|O), and D
KL(
H|O). It resulted in these values:
O|TKL = 0.769,
T|OKL = 0.725,
O|RKL = 0.21,
R|OKL = 0.257,
O|HKL = 0.431, and
H|OKL = 0.161.
T and
O, which disagree the most in 1DIV:A (
Figure 2a), exhibit smallest arithmetic difference between their alternative D
KL perspectives with
O|R and
R|O closely behind them in this sense. However,
O|H is nearly three times higher than
H|O. An explanation can be found again in Equation (3). When
Oi is higher than
Hi and
Ti which are also closer to 0, the values of D
KL(
Oi|Ti) and D
KL(
Oi|Hi) must increase. K132 and K142 make good examples of this phenomenon, showing relatively tall peaks in
Figure 2c. Reversing profile order in Equation (4) has a small to medium effect on
O vs.
T and
O vs.
R and a strong effect on
O vs.
H due to the fact that
O and
T are less often closer to 0 than
H. Moreover, the
O profile of 1DIV:A looks reasonably similar to the
H profile. It is not far from reality, since by definition,
O highly depends on its source. The contribution of the nearest (
Hi±1) neighbors in the sequence to
Oi can reach up to 50% in a typical protein. However, is
O really closer to
R than to
H in this structure as
O|H and
Figure 2d are implying? D
JS can help alleviate this ambiguity by joining pairs of opposing D
KL views in a similar manner to how
RD binds the compared coefficients together. Coincidentally, they even have the same value range. In this sense, Jensen–Shannon “equalizes” Kullback–Leibler by raising peaks everywhere the compared profiles differ (e.g., for both N20-T40 and G107-L117 regions in
Figure 2e), thus marking the residues exhibiting discrepancy while maintaining a balance of importance between the excess and deficiency of hydrophobicity. It resulted in the following pairs of values of Equation (7):
O|TJS = 0.166 =
T|OJS,
O|RJS = 0.055 =
R|OJS, and
O|HJS = 0.047 =
H|OJS. Calling upon Equation (5) reveals the final hydrophobic core status in 1DIV:A in the sense of both measures of entropy:
T-O-RKL = 0.785,
T-O-RJS = 0.749,
T-O-HKL = 0.641,
T-O-HJS = 0.778,
H-O-RKL = 0.672, and
H-O-RJS = 0.460.
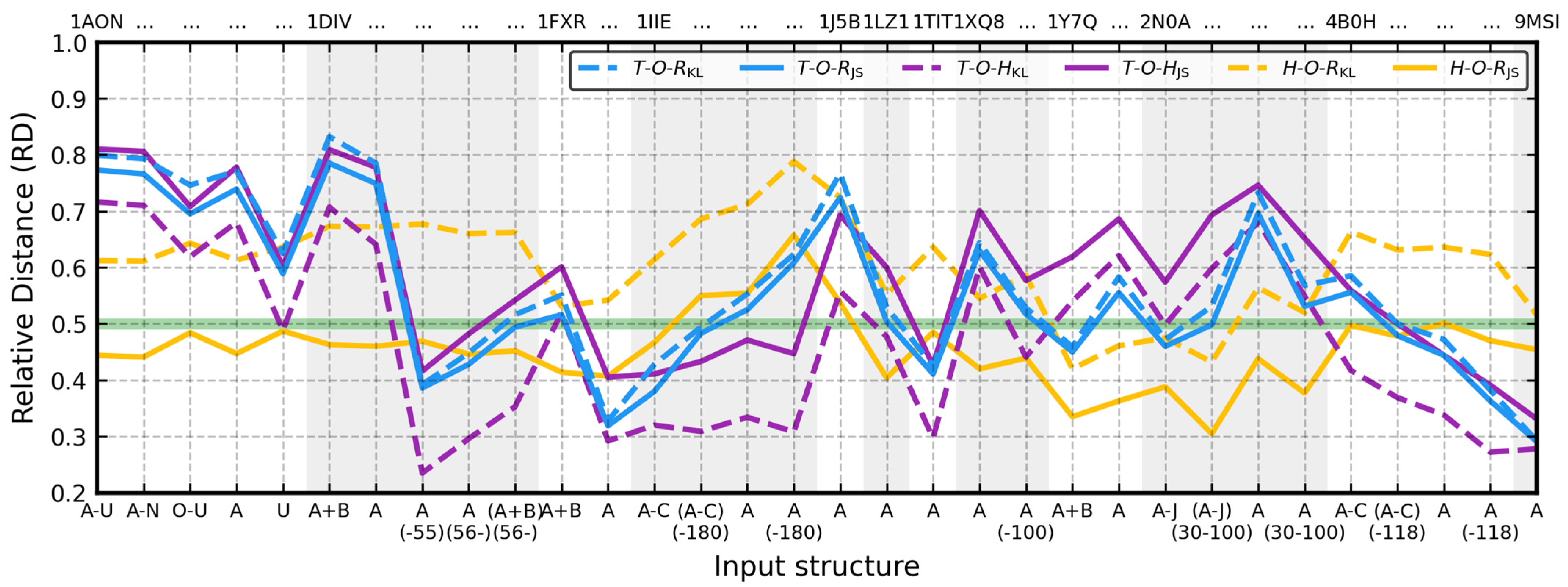
Experimental data for all proteins from the database can be found in
Table S1 and are plotted in
Figure 3. Each of the 32 inputs from
Table 2 was individually subject to the FOD model’s calculation and the status of its hydrophobic core was measured with D
KL and D
JS. Results in the form of
Figure 2 are available in
Supplement File S3.
Among the 32 test inputs, 11 had
T-O-RKL < 0.5, 18 had
T-O-HKL < 0.5, and only 4 had
H-O-RKL < 0.5. Switching to D
JS caused a mean change of
T-O-R in the database by 0.025 (
T-O-RKL−
T-O-RJS, σ = 0.013). Accordance with the model remained the same in 29 cases. The three that became accordant (
T-O-RJS < 0.5) were 1DIV:(A+B)(56-), 2N0A:(A-J)(30-100), and 4B0H:(A-C)(-118). In this group, 1DIV:(A+B)(56-) and 4B0H:(A-C)(-118) were initially closest to 0.5 (4B0H:(A-C)(-118) even had
T-O-RKL =
T-O-HKL = 0.500), so it is not surprising. However, the accordance of the central part of 2N0A (
T-O-RJS = 0.498) is not a favorable outcome. We correct it in
Section 3.2.
O|TKL and
T|OKL were similar in each input, with an average ratio of 1.011 (σ = 0.105). Lower
T-O-RJS was caused by
O|RKL being, on average, 0.83 of
R|OKL (σ = 0.05). The same conclusion can be arrived at by noticing that the mean
O|TKL /
O|TJS and
T|OKL /
O|TJS ratios are 4.28 (σ = 0.305) and 4.252 (σ = 0.249), but the mean
O|RKL /
O|RJS and
R|OKL /
O|RJS ratios are 3.799 (σ = 0.078) and 4.587 (σ = 0.197). When
O|T decreases and
O|R increases, Equation (5) mandates that
T-O-R must decrease toward accordance with the
T profile, here by a maximum of 0.051 in 1AON:(O-U) and a minimum of 0.001 in 9MSI. The correlation coefficient between
T-O-RJS and
T-O-R delta was measured in the entire test suite at 0.58.
O|HKL was nearly the double of H|OKL in the database (μ = 1.935, σ = 0.394), owing to average values of O|HKL / O|HJS and H|OKL / O|HJS ratios equal to 7.135 (σ = 1.078) and 3.726 (σ = 0.210), respectively. Joining them in O|HJS and putting them against O|TJS resulted in an increase in T-O-HJS in every case (μ = 0.113, σ = 0.033). It may seem to be a lot, but O switched to H’s side of the RD scale (T-O-HJS ≥ 0.5) only seven times, in 1AON:U, 1DIV:(A+B)(56-), 1LZ1, 1XQ8:A(-100), 2N0A:(A-J), 4B0H:(A-C), and 4B0H:(A-C)(-118). 1XQ8:A(-100) evened the classification of its hydrophobic core with 1XQ8 and unveiled K21 and K80 which face the center of the drop as highly discordant. Omitting them causes its core to gain stability (T-O-RKL = 0.497, T-O-RJS = 0.481). The highest RD change (0.189) was observed this time in 1DIV:(A+B)(56-) with the lowest shift once again reported by 9MSI (0.053). It can be attributed to the high values of the H profile in this structure (away from 0—it has only a single lysine, K61), resulting in comparable O|HKL and H|OKL (0.118 vs. 0.126). The other two pairs of its DKL coefficients also exhibit similarities. It makes sense for a type III antifreeze molecule.
The four inputs that were closer to
H than to
R in terms of D
KL (
H-O-RKL < 0.5) were 1Y7Q:(A+B), 1Y7Q:A, 2N0A:(A-J), and 2N0A:(A-J)(30-100). D
JS caused this observation to nearly reverse.
H-O-RJS was reduced in all database structures by 0.149 (
σ = 0.038) on average, resulting in all but five test cases—1IIE:(A-C)(-180), 1IIE:A, 1IIE:A(-180), 1J5B, and 4B0H:A—flipping their accordance from
R to
H (
H-O-RJS < 0.5). The highest and lowest
RD deltas were again found in 1DIV:(A+B)(56-) and 9MSI, respectively (0.21 and 0.062). It is a natural consequence of the findings from previous paragraphs; when
O|R increases while
O|H decreases,
H-O-R must go down. This coefficient complements the “
RD triangle” in which
T,
H, and
R are vertexes and
T-O-R,
T-O-H, and
H-O-R are edges of length 1 on which
O can slide. An interesting relation between this trio can be seen in
Figure 3;
H-O-R is below 0.5 when
T-O-R is below
T-O-H and vice versa with both D
KL and D
JS. In fact,
H-O-R can be approximated as 0.5+(
T-O-R)-(
T-O-H). Correlation coefficients between these two values were measured in the database at 0.987 for D
KL and 0.996 for D
JS. In some cases, their absolute difference was less than 1 × 10
−4 (1FXR, 1LZ1, 2N0A, 4B0H), while elsewhere it reached up to 0.017~0.048 with D
KL (1AON, 1DIV, 1IIE) and up to 0.014~0.019 with D
JS (1AON, 1DIV).
While unused by the baseline FOD model, H-O-R can nonetheless measure by how much T-O-R and T-O-H differ and how strongly the intrinsic hydrophobicity of residues (H) is affected by their local spatial neighborhood in the structure (expressed by O) without consideration for the surrounding water (i.e., it is unaffected by the drop size).
3.2. Symmetrization in Bounding in Drop Ellipsoid (FOD-PCA)
To calculate its T profile, the input protein must be bound inside an axis-aligned ellipsoid (drop) which represents this protein’s relationship with the surrounding aqueous environment. The first part of this procedure involves the translation of effective atoms to the origin and their rotation in alignment with the axes of the coordinate system in a way that maximizes their variance in all dimensions. The second step determines the size of the drop—the lengths of its radii constituting the basis for standard deviation parameters passed to Equation (2). O distribution, which is based solely on the hydrophobicity scale and pairwise hydrophobic interactions between residues, is naturally invariant to the spatial orientation of the input as a whole.
The baseline FOD algorithm for axis alignment and drop radii selection has the following description: after the translation of the mean position of all effective atoms to the origin, their most distant pair in 3D is found, and the whole set is rotated so that line connecting that pair becomes parallel to the X-axis. Next, effective atoms are orthogonally projected onto the YZ plane and rotated again, this time around the X-axis. The line connecting their most distant pair found in this plane must then become parallel to the Y-axis. The alignment step is complete. Both searches can be significantly accelerated by noticing that their solutions must belong to the convex hulls of the input data.
Absolute positions of effective atoms located furthest away from the origin in each dimension, all three extended by 9 Å, mark the final lengths of the radii of the drop. The three sigma rule allows the 3D Gauss function to reach nearly 0 at its surface and beyond, while the 9 Å radii extension counteracts domination over the whole profile by residues located near the origin, stretching it, and lowering its values. Drop size has a strong impact on theoretical hydrophobicity. The effects of its alterations (e.g., growing or shrinking) are more noticeable in small molecules than in large complexes.
The baseline approach works reasonably well in typical cases, particularly when the largest spread of effective atoms is sufficiently approximated by the line going through their most distant pair. However, sometimes it produces highly suboptimal alignments. It happens when structural outliers (e.g., disordered chain termini) disturb that balance. They are normally withdrawn a priori from the input structure by hand, for instance, by trimming 4B0H:A to 4B0H:A(-118). It is done because complete immersion in the solvent prevents those fragments from contributing much to the stability of the hydrophobic core in the main (globular) part of the molecule. Unfortunately, their manual removal is not feasible during bulk calculation when hundreds of structures pass through the algorithm. Any procedure of their detection must also consider the fact that in non-globular proteins, all or nearly all residues exhibit the same, low effective atom density (e.g., in 1XQ8).
The presence of outlier fragments has yet another, even more, severe consequence. They cause the drop to artificially inflate, misrepresenting the status of the hydrophobic core of the protein due to the overly stretched
T profile. 1IIE, 1Y7Q, 2N0A, and 4B0H are examples of such structures in the database. On the other hand, a lack of outliers does not guarantee a lack of suboptimal alignment. It can still occur when most distant pairs of effective atoms coincide with the diameter of the structure more than with their largest spread. Again, it happens because the baseline alignment process relies only on that pair while everything else is ignored. The GroEL molecule from 1AON is a perfect example of this issue. Results of its baseline ellipsoid bounding are shown in
Figure 4a–c. To correct the ≈45° rotation in the XY plane, a manual selection of the longest axis of the drop is required, for instance, by averaging two sets of effective atoms located at opposite sides of the molecule (e.g., residues with the same number from different chains, honoring axial symmetry). It works but requires the user’s active participation. Likewise,
Figure 4d–f illustrates how disordered outliers in 2N0A (outside the central A30-L100 region) skew its alignment with both a wrong angle and an overly spacious drop. It is more appropriate to leave some residues out of the drop—like the aforementioned highly exposed outliers—than grow it too large to keep them all inside (unless, of course, one wishes to encompass the structure exactly like it is). All distributions of the FOD model allow that.
Sometimes it happens that two or more chains with an identical sequence and nearly identical tertiary structure report conflicting coefficients from opposite halves of the RD scale. Even if they stay on the same side, the different status of individual residues may perturb their classification via hydrophobicity density maps. 1Y7Q exemplifies such RD disparity in the database. Monomers in its NMR conformer 1 that was used in this experiment exhibit T-O-RJS of 0.555 (chain A) and 0.534 (chain B) owing to the short bent N-terminal region in the second structure (G35-D41). Their RMSD is only 0.6 Å.
Ellipsoid bounding of 1AON:(A-N) and 2N0A:(A-J) presented in
Figure 4g–l is not encumbered by the difficulties mentioned above. Their structures are aligned in accordance with the model’s and user’s expectations. Axial symmetry of GroEL is properly discovered, while outlying segments of amyloid fibril do not interfere with finding the largest spread of effective atoms in the A30-L100 region. They are partially pushed out of the drop, providing a tighter, more accurate fit to the central part of the molecule (as it turns out, also in terms of
RD). These results were obtained via an alternative alignment method based on principal component analysis and a modified drop size selection scheme. They are the focus of this part of the paper (see
Section 2.3 for a general description of PCA).
Unlike the baseline alignment algorithm which relies on the longest distance between effective atoms, the PCA-based approach considers them all, delivering optimal translation vector and rotation matrix which position the input data set at origin and rotate it in a way that truly (rather than approximately) maximizes variance in each dimension. Put differently, a diagonal 3 × 3 covariance matrix is produced. 1AON:(A-N) illustrates this very well. On
Figure 4a–c,
σ2(
x) = 1548.7,
σ2(
y) = 1401.6,
σ2(
z) = 1234.3, cov(
x,
y) = 247.3, cov(
x,
z) = −75.7, and cov(
y,
z) = −52.4, while on
Figure 4g–i,
σ2(
x) = 1749.5,
σ2(
y) = 1220.8,
σ2(
z) = 1214.2, and the rest of matrix is zero. As a beneficial side effect, taking all effective atoms into account allows PCA to automatically (i.e., without the user’s intervention) discover symmetry in the molecule, even in the highly disordered 2N0A:(A-J) (
Figure 4j–l). This is exactly what the theoretical hydrophobicity of the FOD model expects to be done with its effective atoms. Aligning them with PCA is also faster than the baseline approach between 6 and 9 times (≈7 on average,
σ ≈ 0.4). The speed-up magnitude naturally depends on the computer environment, but it omits an additional and more complicated algorithm (convex hull) and a subsequent search among pairs of elements of its output.
At this moment, effective atoms are aligned with axes of the coordinate system. Now it is time to choose the right size of the drop. The baseline algorithm relies for that on the longest distance to the origin in each dimension, which in turn relies on only up to three effective atoms, making it subject to potential drop radii length overestimation, especially along the Y- and Z-axes. Since it can be problematic for working with small structures, we would like to alleviate this problem or at least diminish its severity.
PCA only provides radii direction vectors (principal components) of the ellipsoid fit to the input data, but not their lengths. It is left to the user to decide at which size this ellipsoid separates the interior model from exterior outliers. In the FOD model, it must encompass the molecule in a manner that approximates its assumed globular shape, not too large and without carving too much into the structure. If the spread of effective atoms is utilizable to achieve their optimal axis alignment, it can be used to determine drop size as well. Drop radii are sought on behalf of theoretical distribution which models the distribution of residues via a multivariate normal distribution. Hence, their lengths can be determined from standard deviations of effective atoms in each dimension (
σx,
σy,
σz) and the value from the χ
2 distribution with three degrees of freedom at confidence level
P [
59]. The point [
x, y, z] belongs then to the surface of an axis-aligned ellipsoid when:
Because [μx, μy μz] = [0, 0, 0], to obtain the radius of this ellipsoid in the X dimension, y and z need to be set to 0, resulting in x = σx√s and so on for y and z.
We realized, however, that the above approach may, in turn, overestimate
x after the standard deviation along the X-axis is maximized, while too small value of
P can produce too small drops. Hence, we decided to combine its anti-outlier properties with the baseline method. After the PCA-based alignment, the modified drop radii were set in this experiment to an average of baseline radii and the results of Equation (10) with
P = 0.75 (
s ≈ 4.108, √
s ≈ 2.027, very close to 2
σ) and then extended by 9 Å. Whether this protocol can be further optimized is something to discover in another paper. Drops shown as red ellipses in
Figure 4g–l were obtained by adhering to it. They are nearly identical to baseline radii (the orange ellipses) for 1AON:(A-N), but visibly shorter for 2N0A:(A-J).
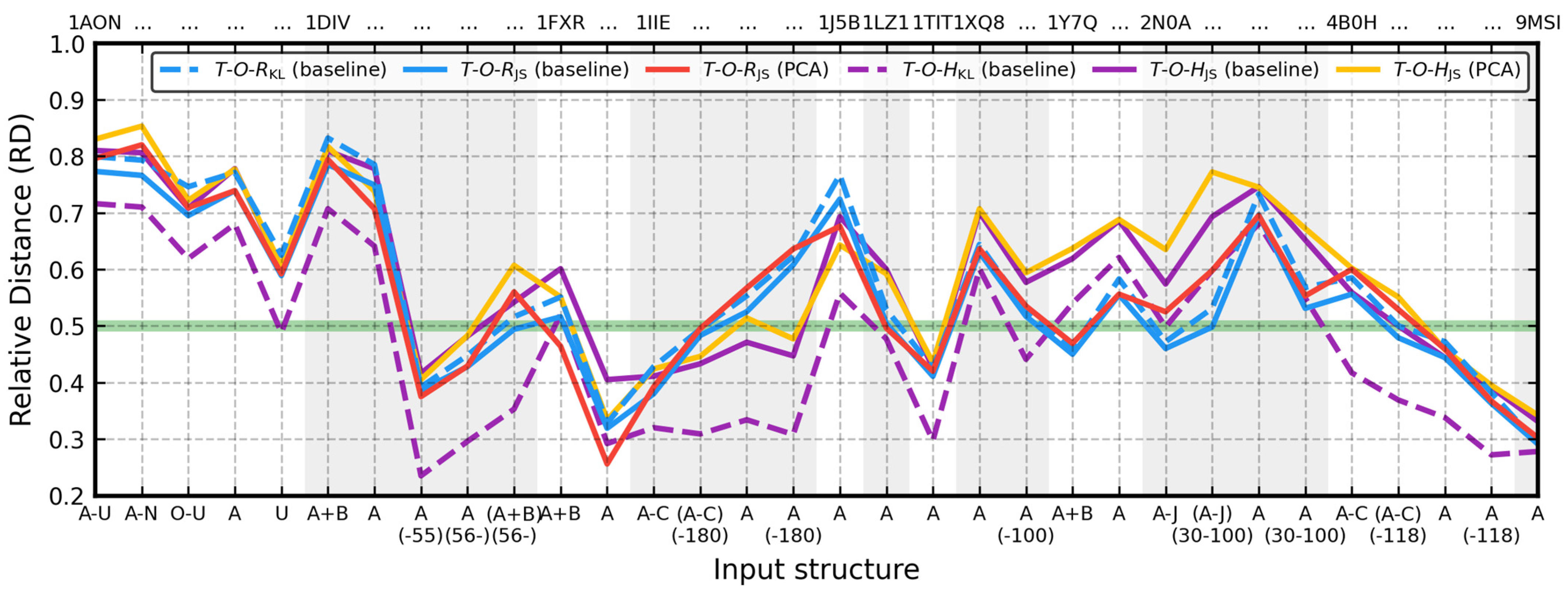
Experimental data for all proteins from the database can be found in
Tables S2 and S3 and are plotted in
Figure 5. Each of the 32 inputs from
Table 2 was individually subject to the FOD model’s calculation following a separate baseline and modified bounding in the drop ellipsoid after which the status of its hydrophobic core was measured with D
KL and D
JS. Additionally, we checked the post-alignment standard deviations of the effective atoms and the sizes of the drops (radii length and volume coefficient
V =
rx·
ry·
rz/1000). The comparison of those drops in a form similar to
Figure 4 is available in
Supplement File S4.
As expected, PCA-based alignment produced a diagonal effective atoms covariance matrix for all test cases. In comparison to the baseline approach, it maximized their standard deviation along the X-axis and decreased it along the Y- and Z-axes, reaching, on average, 102.7% (σ = 3.2%), 99% (σ = 5.1%), and 92.7% (σ = 7.1%) respectively. It resulted in slightly smaller drops, confirmed by these average modified/baseline ratios: rx/rx = 94.9% (σ = 5.7%), ry/ry = 91% (σ = 8.6%), rz/rz = 91.1% (σ = 6.9%), and V/V = 79.1% (σ = 12%). When 1IIE:(A-C), 1Y7Q:(A+B), 2N0A:(A-J), and 4B0H:A (i.e., inputs with significant outliers) are discounted, these ratios change to 95.7% (σ = 5.4%), 92.4% (σ = 8%), 92% (σ = 6.7%), and 81.4% (σ = 10.5%). The only protein in which the V coefficient was increased, although just by 3%, was 1J5B. The rx/rx ratio exceeded 1 five times and was exceeded the most in 1DIV:A (1.07) and 1J5B (1.08). Conversely, drops of even longer 1X8Q and 1XQ8:(-100) remained akin to their baseline counterparts. The same happened to globular inputs: 1AON:(O-U), 1AON:U, the domains of 1DIV:A, 1IIE:A(-180), 1TIT, 4B0H:A(-118), and 9MSI. Automated alignment correction to properly handle the structural symmetry and a drop shrink resulting from it was observed in 1AON:(A-U), 1AON:(A-N), 1AON:A, 1DIV(A+B), 1DIV:A, 1DIV:(A+B)(56-), 1FXR:(A+B), 1IIE:(A-C), 1IIE:(A-C)(-180), 1Y7Q:(A+B), 2N0A:(A-J), and 2N0A:(A-J)(30-100).
Unstructured outlier chain segments were managed by the modified FOD algorithm in all four test cases in which they were prominently exposed to the solvent: 1IIE:(A-C), 1Y7Q:(A+B), 2N0A:(A-J), and 4B0H:A. By managed, we mean counteracting their negative influence by reducing size of the drop toward a correct approximation of the dense part of the structure—the assumed location of the hydrophobic core. We do not consider the S181-K192 region of 1IIE:A as an outlier because of the short length of this chain (75 aa) and its loose tertiary structure. In this sense, it is closer to 1XQ8 and 2N0A:A which lack any tertiary structure. Thus, terminal regions in those inputs (and in 1XQ8:A(-100)) should not be classified as different from the rest in terms of effective atom density. Our modification recognizes that and does not needlessly shrink their drops. On the other hand, its use resulted in the following
V/V ratios (modified / baseline): 058 for 1IIE:(A-C), 0.73 for 1Y7Q:(A+B), 0.53 for 2N0A:(A-J), and 0.66 for 4B0H:A. However, 1AON:(A-N) obtained 0.63, and 2N0A:(A-J)(30-100) obtained 0.47 because of the correction of their alignment. This gave us, however, an idea as to how we can detect and report the possible presence of outliers in the input. Effective atoms should be first aligned with the PCA-powered approach and then the
V/V ratio calculated for drops with radii lengths chosen using modified and baseline methods (e.g., red vs. orange on
Figure 4g–l). This changes the above numbers to 0.73 for 1IIE:(A-C), 0.75 for 1Y7Q:(A+B), 0.6 for 2N0A:(A-J), 0.79 for 4B0H:A, 1.01 for 1AON:(A-N), 0.82 for 2N0A:(A-J)(30-100), and 0.84 for 1IIE:A.
The average
RD difference between baseline (D
KL) and modified (D
JS) bounding was measured at 0.013 (
σ = 0.038) for
T-O-R, −0.125 (
σ = 0.048) for
T-O-H, and −0.149 (
σ = 0.038) for
H-O-R.
H-O-R is the same as in
Section 3.1 as only the
T profile was altered here. Omitting 1IIE:(A-C), 1Y7Q:(A+B), 2N0A:(A-J), and 4B0H:A causes these values to change to 0.015 (
σ = 0.038) for
T-O-R, −0.126 (
σ = 0.05) for
T-O-H, and −0.155 (
σ = 0.036) for
H-O-R. With D
KL replaced by D
JS, baseline and alternative approaches differ on average by −0.013 (
σ = 0.035) in terms of
T-O-R and −0.011 (
σ = 0.033) in terms of
T-O-H. Without structures with outliers, both
RD variants report a mean change of ≈−0.01 (
σ ≈ 0.035).
Jensen–Shannon entropy caused three inputs to accord with the model in
Section 3.1 (
T-O-RJS < 0.5): 1DIV:(A+B)(56-), 2N0A:(A-J)(30-100), and 4B0H:(A-C)(-118). PCA-based alignment and alternative drop size selection took them back to the discordant side of the
RD scale. This is particularly beneficial with the amyloid which received higher
T-O-RJS (0.598 vs. 0.531) than before. 2N0A:(A-J)—the complete fibril—also “fixed” the baseline status of its hydrophobic core: to
T-O-RJS = 0.525 from
T-O-RKL = 0.472. This time it was 1FXR:(A+B) and 1LZ1 which switched to accordance with the model, suggesting that the Ferredoxin I complex of two highly stable monomers is actually stable (
T-O-RJS = 0.464)—a change of core status classification caused by an alignment correction mostly in the XZ plane. 1DIV:(A+B)(56-) reports a similar rotation fix, only in the XY and XZ planes and with the opposite effect on perception of its stability. 1FXR:A also became the most stable structure in the database (
T-O-RJS = 0.256), overtaking 9MSI (
T-O-RJS = 0.302).
The situation with
T-O-H did not change much. Only 1IIE:A joined the seven structures that moved closer to
H in
Section 3.1 (
T-O-HJS ≥ 0.5), confirming a stronger impact on this coefficient by the symmetric measure of entropy rather than by manipulation of the drop. More interesting is the fact that
RD coefficients calculated for monomers of 1Y7Q were brought closer to each other.
T-O-RJS changed from 0.555 to 0.556 for chain A and from 0.534 to 0.554 for chain B, overcoming
RD discrepancy caused by the 1Y7Q:B(35-41) region.
T-O-HJS for chain A changed from 0.686 to 0.688 and from 0.641 to 0.659 for chain B, reducing the
RD difference by ≈35%.
T-O-RJS of 1Y7Q:(A+B) remained similar to value of this coefficient for its structure with outliers removed from it a priori (0.469 vs. 0.472). It is a very promising outcome. A similar phenomenon was observed for its
T-O-HJS, but to a lesser extent as expected (0.637 vs. 0.654). We also noticed that in the entire NMR ensemble of 1Y7Q (all 20 conformers), the standard deviation of
T-O-RJS was reduced by 9.5% for the dimer, by 4.8% for chain A, and by 16.7% for chain B. For
T-O-HJS, these ratios were measured at 16.7%, 12.5%, and 8.7%, respectively.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}