
A Hierarchy of Interactions between Pathogenic Virus and Vertebrate Host


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
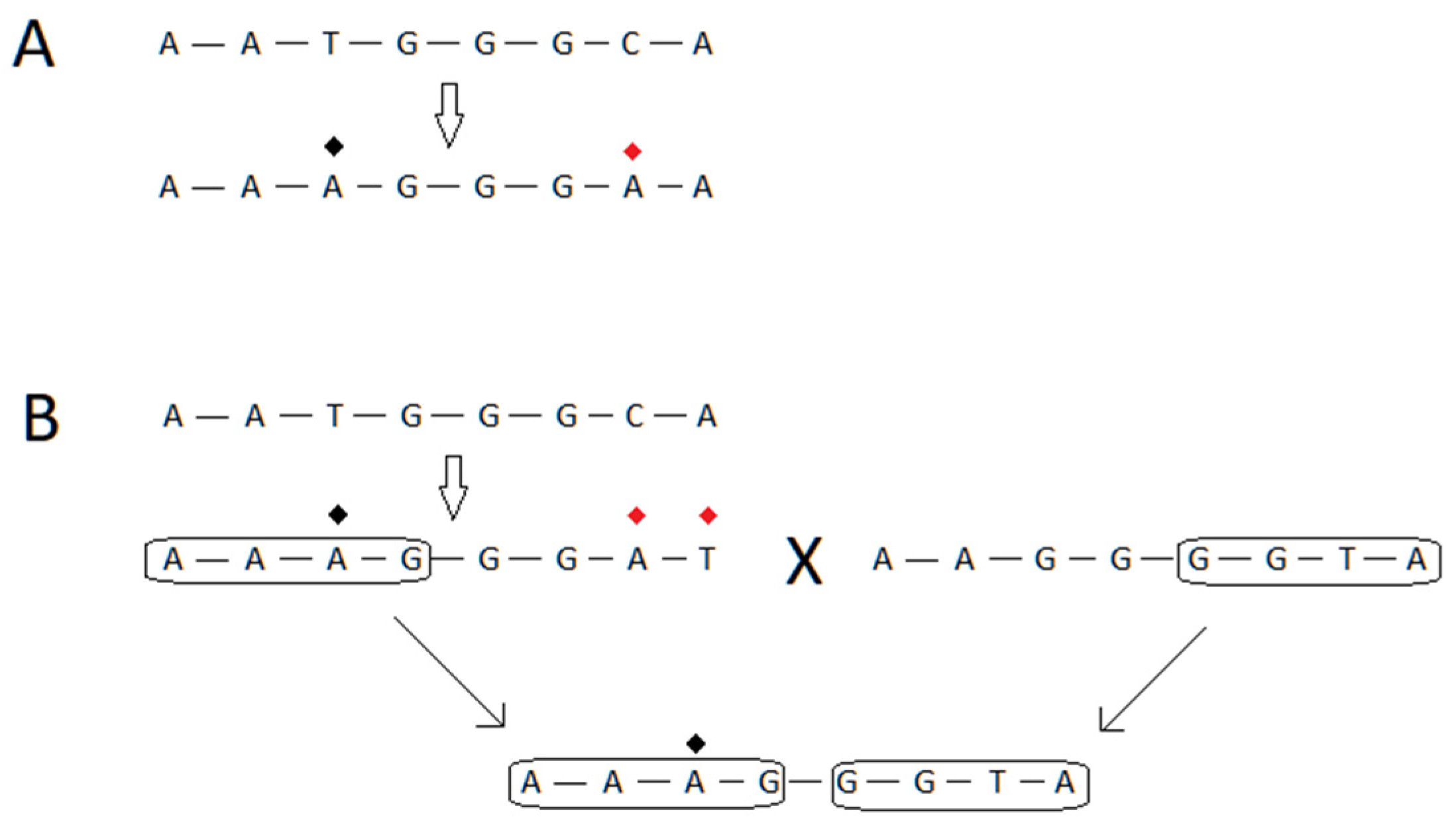{kind=link}
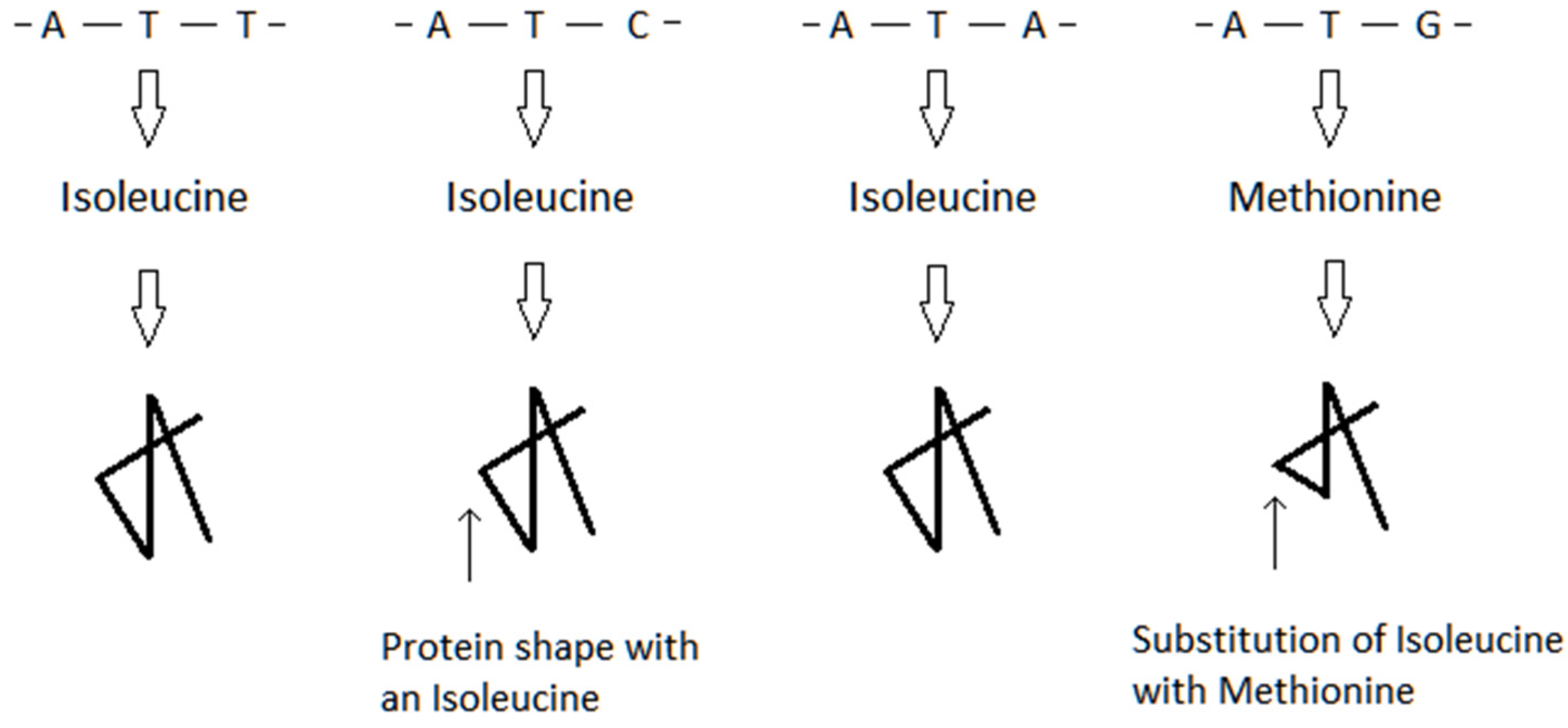{kind=link}
{kind=link}
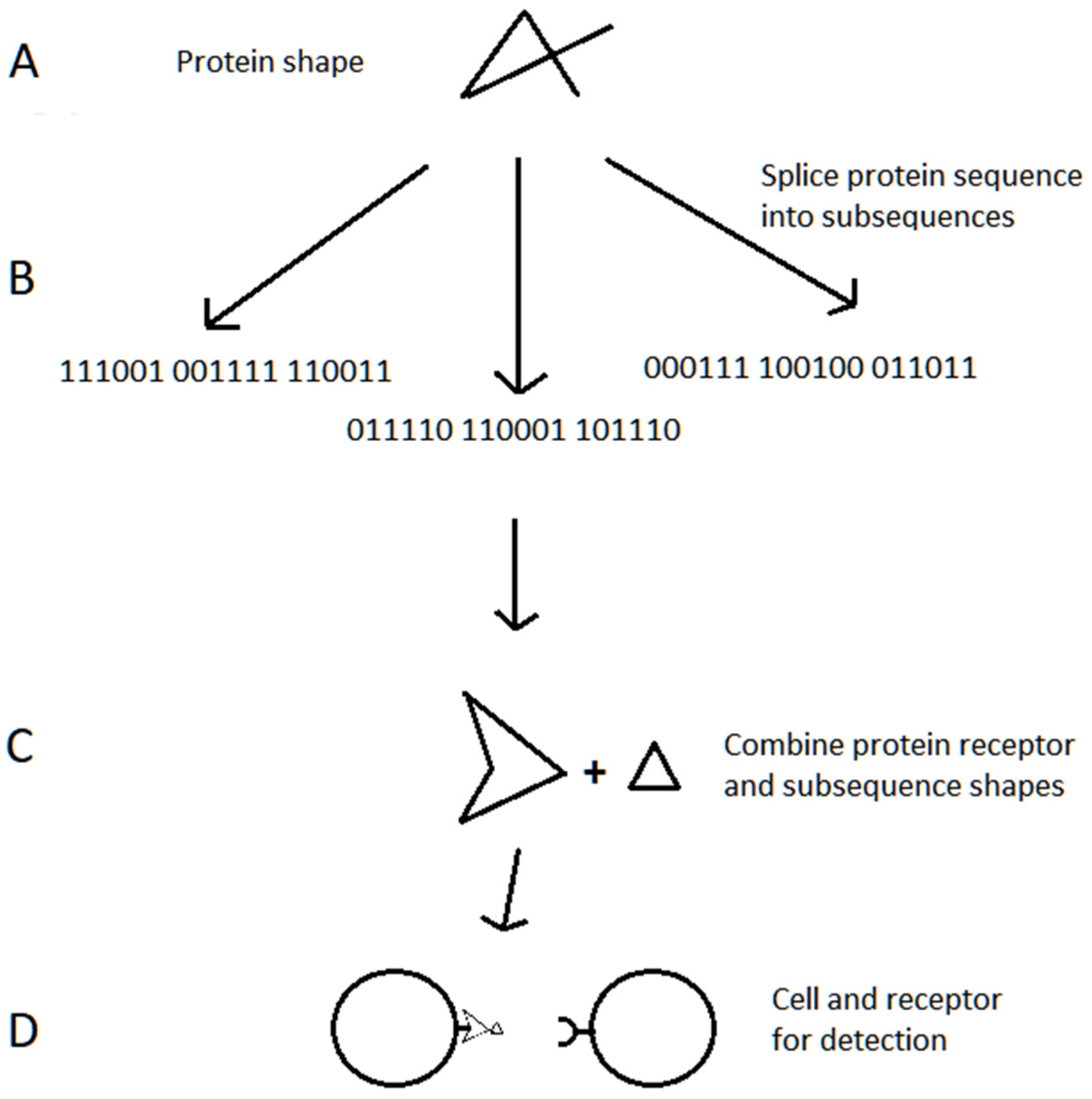{kind=link}
Abstract
1. Introduction
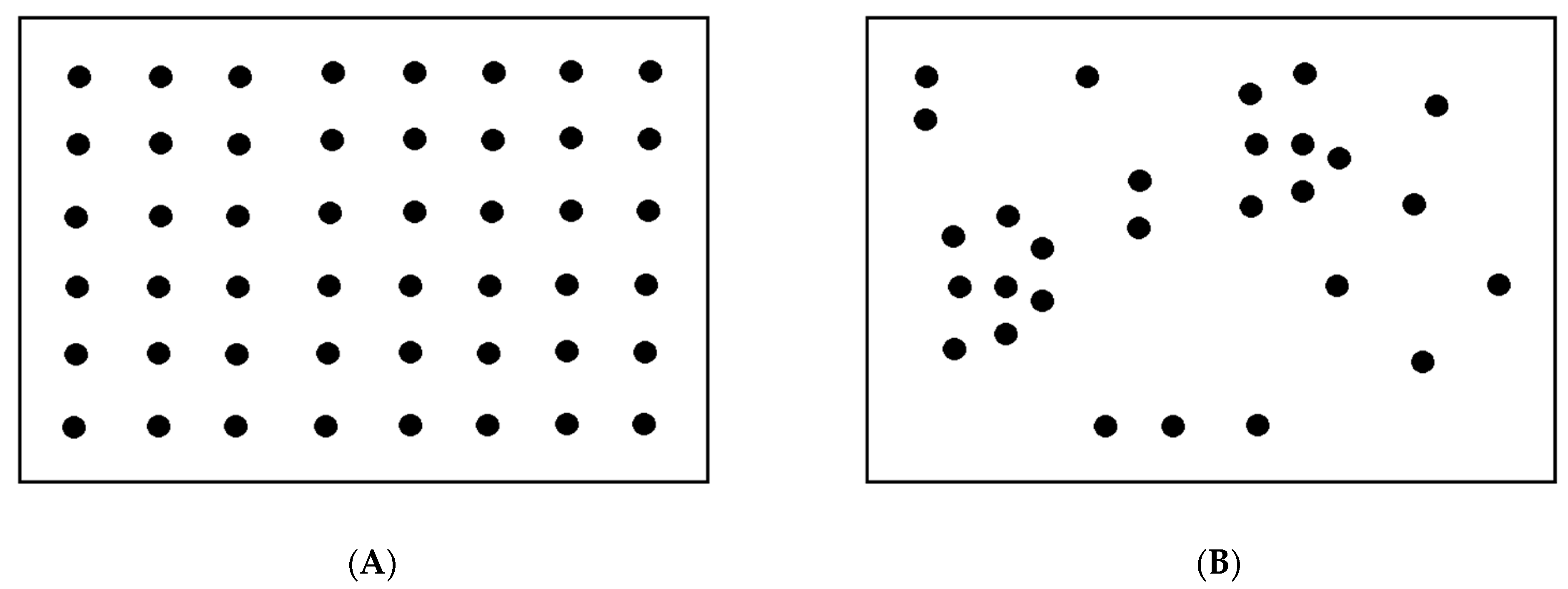
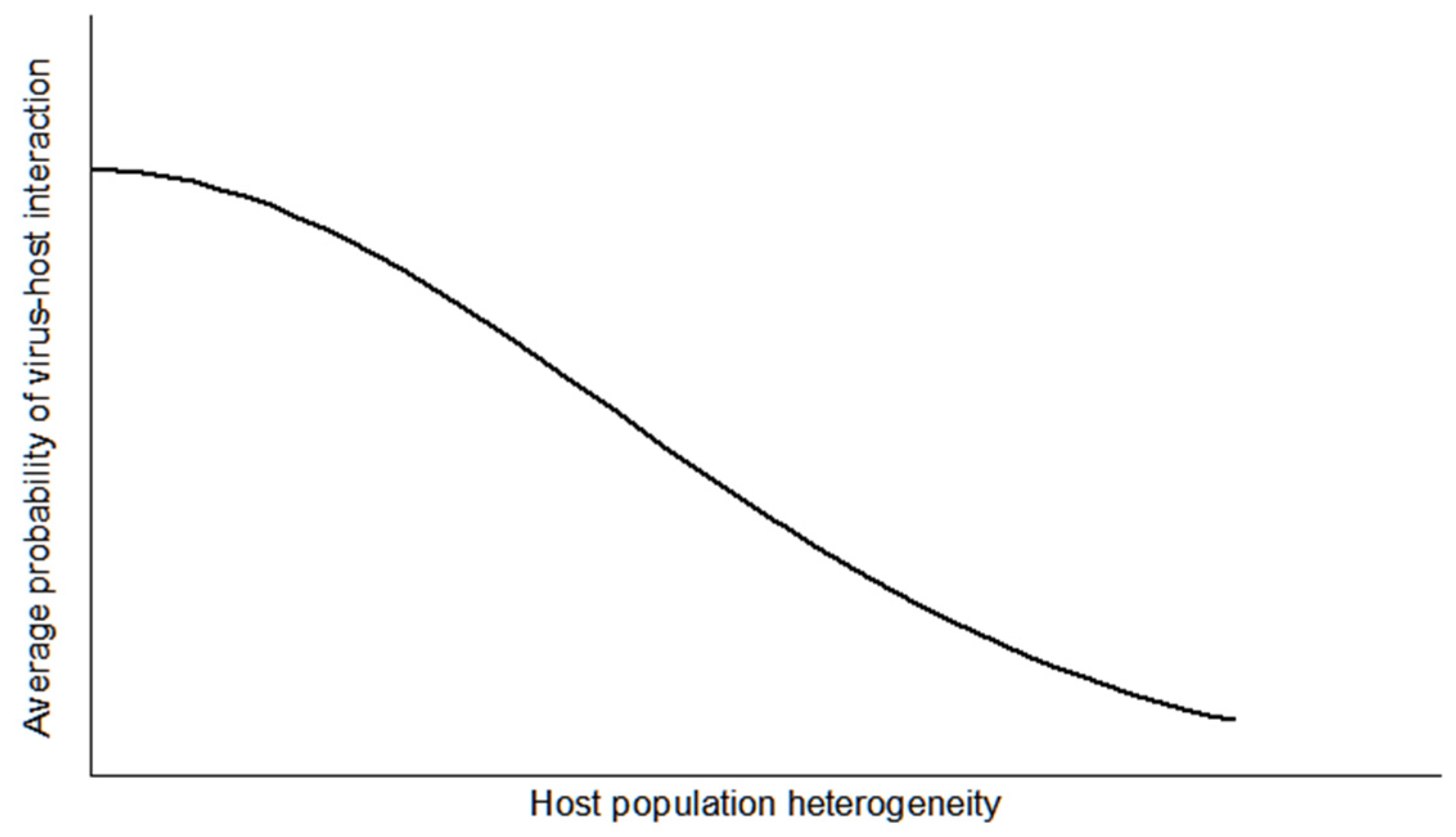
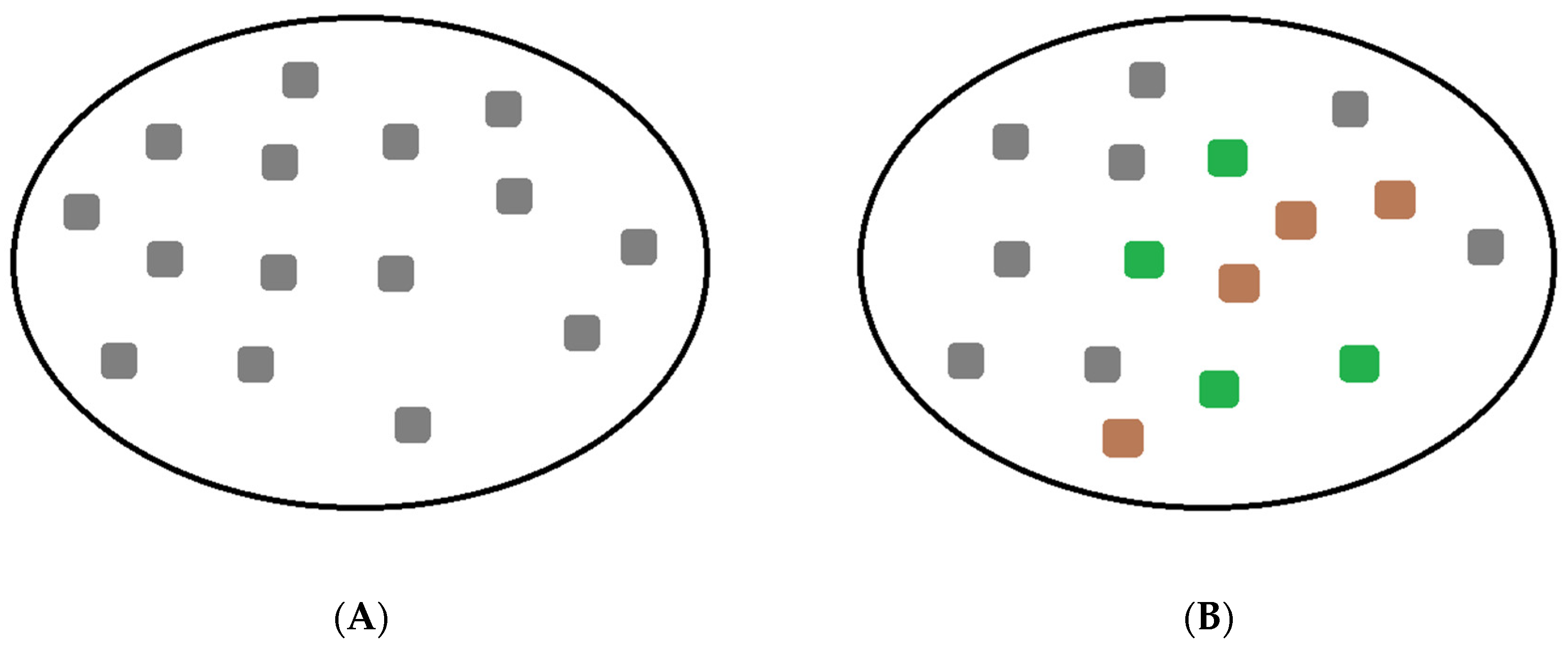
2. Population-Based Approach to the Virus–Host Interaction
3. Model of Virus–Host Interactions at the Population Level
3.1. Description of the Model
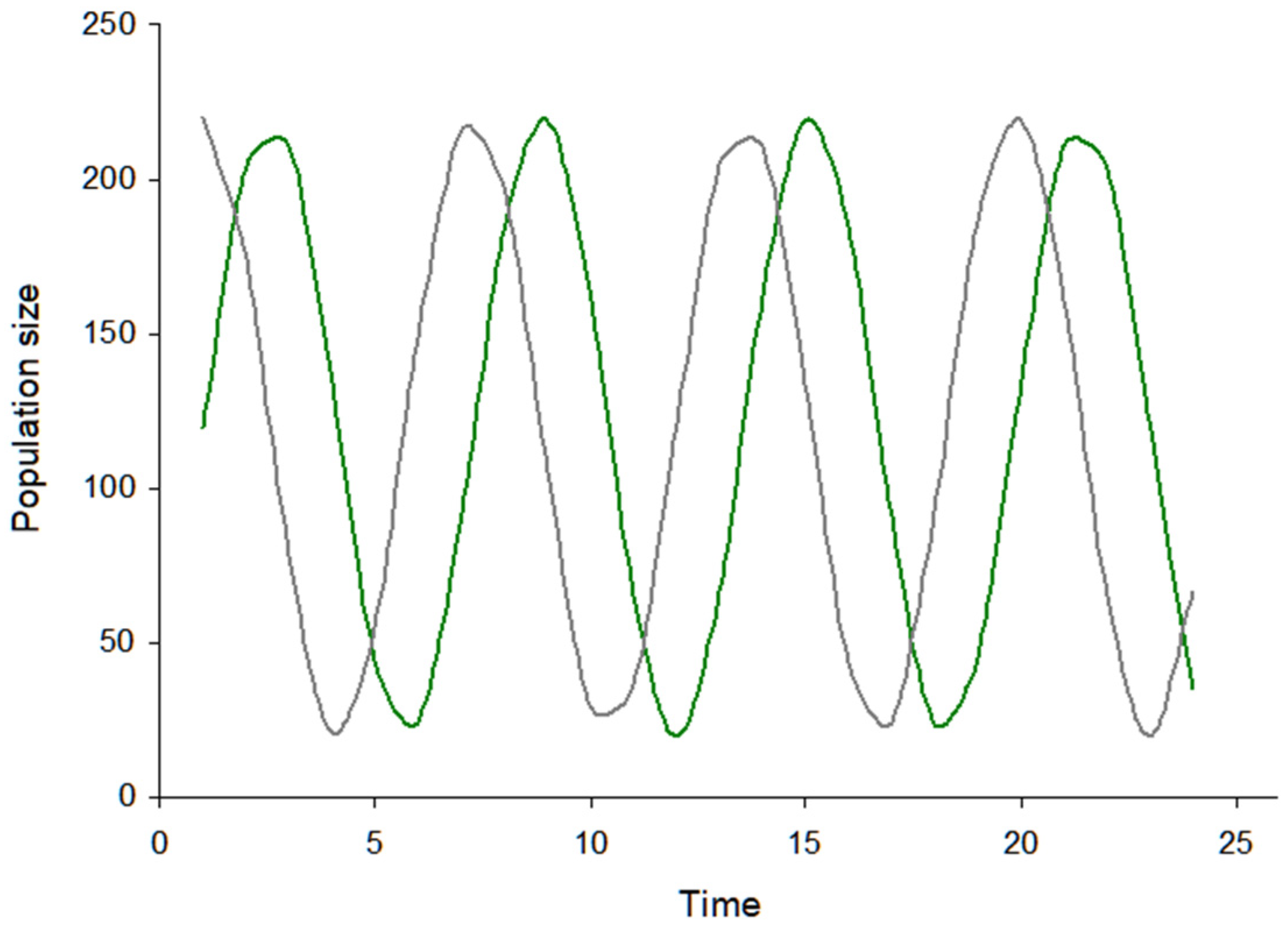
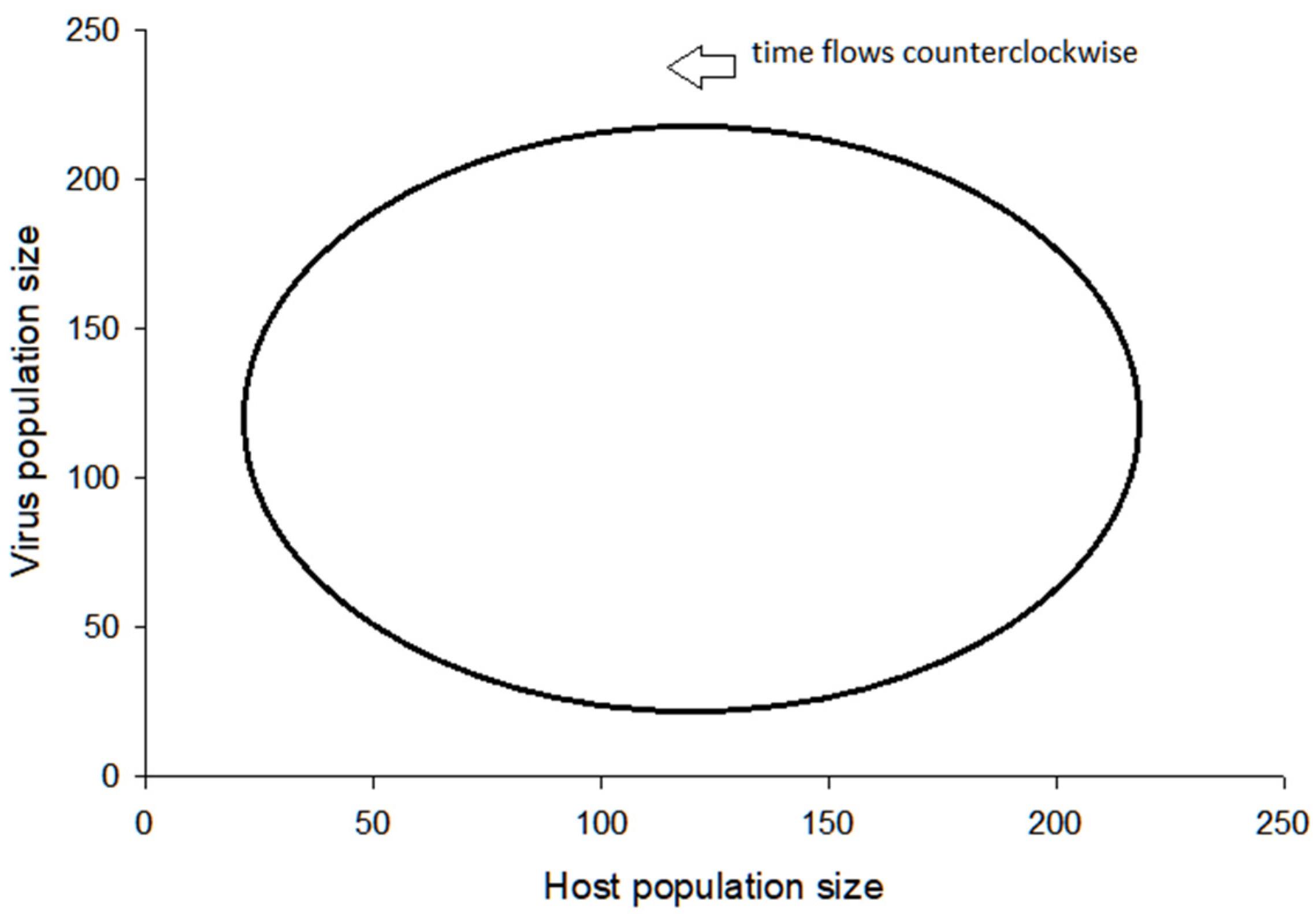
3.2. Visualization of the Virus–Host Model
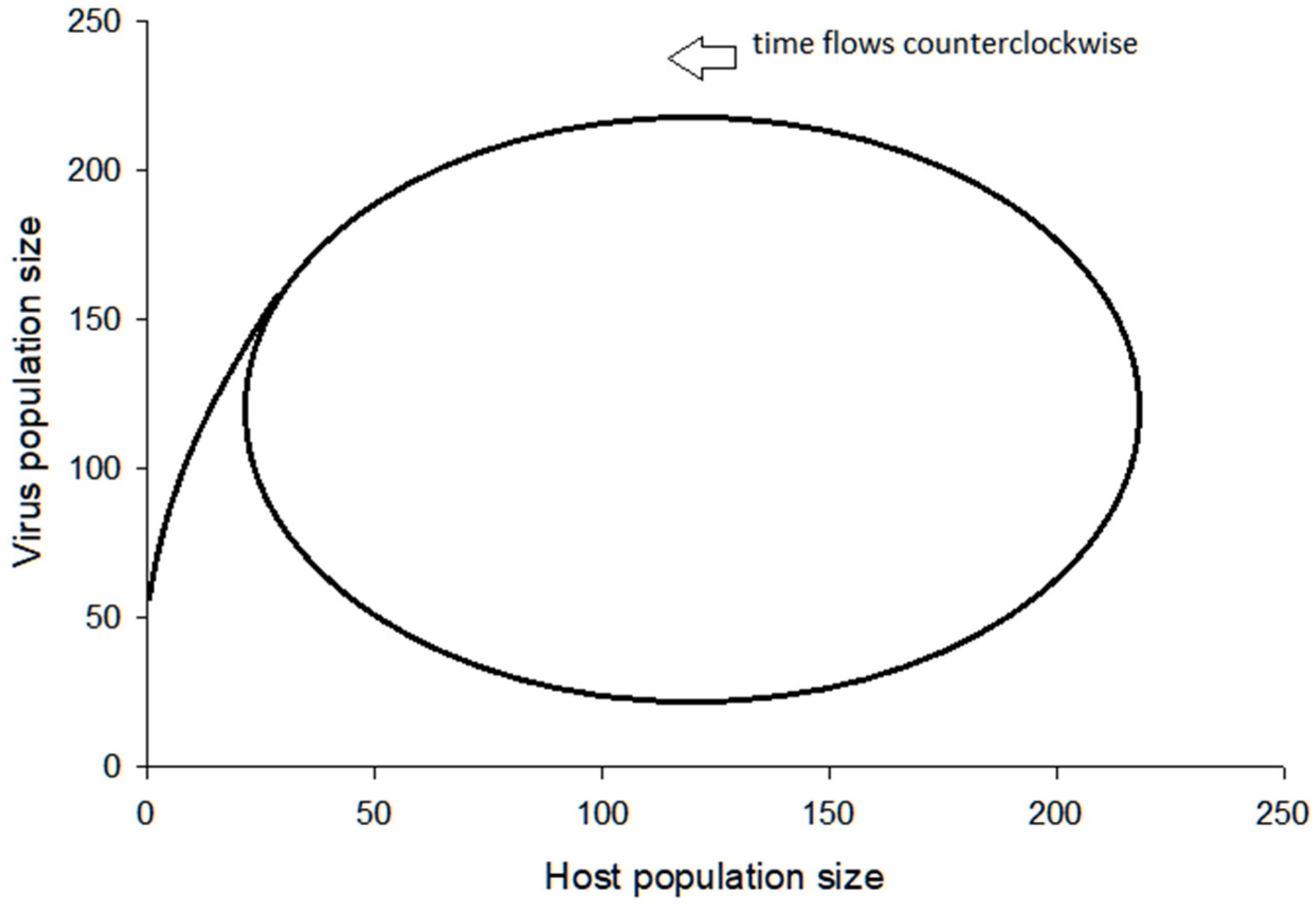
3.3. Further Comments on Virus–Host System Instability
4. Model of Virus–Host Interactions at the Genetic Level
5. Models for Generation of Protein Sequences
5.1. Overview of Deep Learning Methods
5.2. Deep Learning Models of Protein Structure
6. A Deep Learning Model for Generating Protein Sequences
6.1. GPT-2 as a Model for Sequence Generation
6.2. Large Data Collections for Training Deep Learning Models
6.3. Approaches for Tokenization of Protein Sequence Data
6.4. A Procedure for Generating Protein Sequences
7. Discussion
Funding
Conflicts of Interest
References
- Campillo-Balderas, J.A.; Lazcano, A.; Becerra, A. Viral genome size distribution does not correlate with the antiquity of the host lineages. Front. Ecol. Evol. 2015, 3, 143. [Google Scholar] [CrossRef]
- Sun, S.; Rao, V.B.; Rossmann, M.G. Genome packaging in viruses. Curr. Opin. Struct. Biol. 2010, 20, 114–120. [Google Scholar] [CrossRef]
- Chirico, N.; Vianelli, A.; Belshaw, R. Why genes overlap in viruses. Proc. R. Soc. B Biol. Sci. 2010, 277, 3809–3817. [Google Scholar] [CrossRef]
- Nasir, A.; Romero-Severson, E.; Claverie, J.M. Investigating the Concept and Origin of Viruses. Trends Microbiol. 2020, 28, 959–967. [Google Scholar] [CrossRef]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.F.; Pyle, J.D.; Yurkovetskiy, L.; Bosso, M.; Park, D.J.; Babadi, M.; MacInnis, B.L.; et al. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science 2021, 376, 1327–1332. [Google Scholar] [CrossRef]
- Hamilton, W.D.; Axelrod, R.; Tanese, R. Sexual reproduction as an adaptation to resist parasites (A Review). Proc. Natl. Acad. Sci. USA 1990, 87, 3566–3573. [Google Scholar] [CrossRef]
- Agrawal, A.; Lively, C.M. Infection genetics: Gene-for-gene versus matching-alleles models and all points in between. Evol. Ecol. Res. 2002, 4, 91–107. [Google Scholar]
- Anderson, R.M.; May, R.M. Coevolution of hosts and parasites. Parasitology 1982, 85, 411–426. [Google Scholar] [CrossRef]
- Lotka, A.J. Analytical note on certain rhythmic relations in organic systems. Proc. Natl. Acad. Sci. USA 1920, 6, 410–415. [Google Scholar] [CrossRef]
- Lotka, A.J. Contribution to the mathematical theory of capture: I. Conditions for capture. Proc. Natl. Acad. Sci. USA 1932, 18, 172–178. [Google Scholar] [CrossRef]
- Volterra, V. Fluctuations in the abundance of a species considered mathematically. Nature 1926, 118, 558–560. [Google Scholar] [CrossRef]
- Volterra, V. Variazioni e fluttuazioni del numero d’individui in specie animali conviventi. Mem. Della R. Accad. Naz. Dei Lincei 1926, 2, 31–113. [Google Scholar]
- Kingsland, S.; Alfred, J. Lotka and the origins of theoretical population ecology. Proc. Natl. Acad. Sci. USA 2015, 112, 9493–9495. [Google Scholar] [CrossRef] [PubMed]
- Anisiu, M.C. Lotka, Volterra and their model. Didact. Math. 2014, 32, 9–17. [Google Scholar]
- Huffaker, C. Experimental studies on predation: Dispersion factors and predator-prey oscillations. Hilgardia 1958, 27, 343–383. [Google Scholar] [CrossRef]
- Simonsen, K.L.; Churchill, G.A.; Aquadro, C.F. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics 1995, 141, 413–429. [Google Scholar] [CrossRef]
- Kimura, M. The Neutral Theory of Molecular Evolution. Sci. Am. 1979, 241, 98–129. [Google Scholar] [CrossRef]
- Freeland, S.J.; Hurst, L.D. The Genetic Code Is One in a Million. J. Mol. Evol. 1998, 47, 238–248. [Google Scholar] [CrossRef]
- Hie, B.; Zhong, E.D.; Berger, B.; Bryson, B. Learning the language of viral evolution and escape. Science 2021, 371, 284–288. [Google Scholar] [CrossRef]
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: NLP, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, K.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Marcu, S.-B.; Tabirca, S.; Tangney, M. An Overview of Alphafold’s Breakthrough. Front. Artif. Intell. 2022, 5, 875587. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, S.A. Form and Function in Organisms. Am. Zool. 1988, 28, 671–680. [Google Scholar] [CrossRef]
- Klein, J.; Figueroa, F. Evolution of the major histocompatibility complex. Crit. Rev. Immunol. 1986, 6, 295–386. [Google Scholar] [CrossRef]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef]
- Germain, R.N. MHC-dependent antigen processing and peptide presentation: Providing ligands for T lymphocyte activation. Cell 1994, 76, 287–299. [Google Scholar] [CrossRef]
- Friedman, R. A Perspective on Information Optimality in a Neural Circuit and Other Biological Systems. Signals 2022, 3, 410–427. [Google Scholar] [CrossRef]
- Garstka, M.A.; Fish, A.; Celie, P.H.; Joosten, R.P.; Janssen, G.M.; Berlin, I.; Hoppes, R.; Stadnik, M.; Janssen, L.; Ovaa, H.; et al. The first step of peptide selection in antigen presentation by MHC class I molecules. Proc. Natl. Acad. Sci. USA 2015, 112, 1505–1510. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst. 2020, 11, 42–48. [Google Scholar] [CrossRef]
- Montemurro, A.; Schuster, V.; Povlsen, H.R.; Bentzen, A.K.; Jurtz, V.; Chronister, W.D.; Crinklaw, A.; Hadrup, S.R.; Winther, O.; Peters, B.; et al. NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCR and sequence data. Commun. Biol. 2021, 4, 1060. [Google Scholar] [CrossRef]
- Beattie, C.; Koppe, T.; Duenez-Guzman, E.A.; Leibo, J.Z. DeepMind Lab2D. arXiv 2020, arXiv:2011.07027. [Google Scholar]
- Silver, D.; Singh, S.; Precup, D.; Sutton, R.S. Reward is enough. Artif. Intell. 2021, 299, 103535. [Google Scholar] [CrossRef]
- Radford, A.; Wu, J.; Child, R.; Luan, D.; Amodei, D.; Sutskever, I. Language Models are Unsupervised Multitask Learners. Available online: openai.com/blog/better-language-models;github.com/openai/gpt-2 (accessed on 5 September 2022).
- Ferruz, N.; Schmidt, S.; Hocker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention Is All You Need. In Proceedings of the 31st Conference on Neural Information Processing System, Long Beach, CA, USA, 4–9 December 2017; pp. 5998–6008. [Google Scholar]
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C.; Moi, A.; Cistac, P.; Rault, T.; Louf, R.; Funtowicz, M.; et al. Transformers: State-of-the-Art Natural Language Processing. In Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations, Online, 16–20 November 2020; pp. 38–45. [Google Scholar]
- Bisong, E. Google Colaboratory. In Building Machine Learning and Deep Learning Models on Google Cloud Platform; Apress: Berkeley, CA, USA, 2019. [Google Scholar]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Bai, H.; Shi, P.; Lin, J.; Tan, L.; Xiong, K.; Gao, W.; Liu, J.; Li, M. Semantics of the Unwritten: The Effect of End of Paragraph and Sequence Tokens on Text Generation with GPT2. arXiv 2020, arXiv:2004.02251. [Google Scholar]
- Gage, P. New Algorithm for Data Compression. C Users J. 1994, 12, 23–38. [Google Scholar]
- Generative Model for Protein Sequences. Available online: github.com/bob-friedman/protein-sequence-generation (accessed on 4 September 2022).
- Madani, A.; McCann, B.; Naik, N.; Keskar, N.S.; Anand, N.; Eguchi, R.R.; Huang, P.-S.; Socher, R. ProGen: Language Modeling for Protein Generation. arXiv 2020, arXiv:2004.03497. [Google Scholar]
- Wu, K.; Yost, K.E.; Daniel, B.; Belk, J.A.; Xia, Y.; Egawa, T.; Satpathy, A.; Chang, H.Y.; Zou, J. TCR-BERT: Learning the grammar of T-cell receptors for flexible antigen-xbinding analyses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Park, M.; Seo, S.W.; Park, E.; Kim, J. EpiBERTope: A sequence-based pre-trained BERT model improves linear and structural epitope prediction by learning long-distance protein interactions effectively. bioRxiv 2022. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedman, R. A Hierarchy of Interactions between Pathogenic Virus and Vertebrate Host. Symmetry 2022, 14, 2274. https://doi.org/10.3390/sym14112274
Friedman R. A Hierarchy of Interactions between Pathogenic Virus and Vertebrate Host. Symmetry. 2022; 14(11):2274. https://doi.org/10.3390/sym14112274
Chicago/Turabian StyleFriedman, Robert. 2022. "A Hierarchy of Interactions between Pathogenic Virus and Vertebrate Host" Symmetry 14, no. 11: 2274. https://doi.org/10.3390/sym14112274
APA StyleFriedman, R. (2022). A Hierarchy of Interactions between Pathogenic Virus and Vertebrate Host. Symmetry, 14(11), 2274. https://doi.org/10.3390/sym14112274