
Molecular Symmetry of Permethylated β-Cyclodextrins upon Complexation



Abstract
1. Introduction
2. Materials and Methods
2.1. X-ray Crystallography
2.1.1. Crystallization
2.1.2. Data Collection, Structure Solution and Refinement
2.2. Molecular Dynamics (MD) Study
2.2.1. System Preparation
2.2.2. MD Simulations
2.2.3. Free Energy Prediction
3. Results
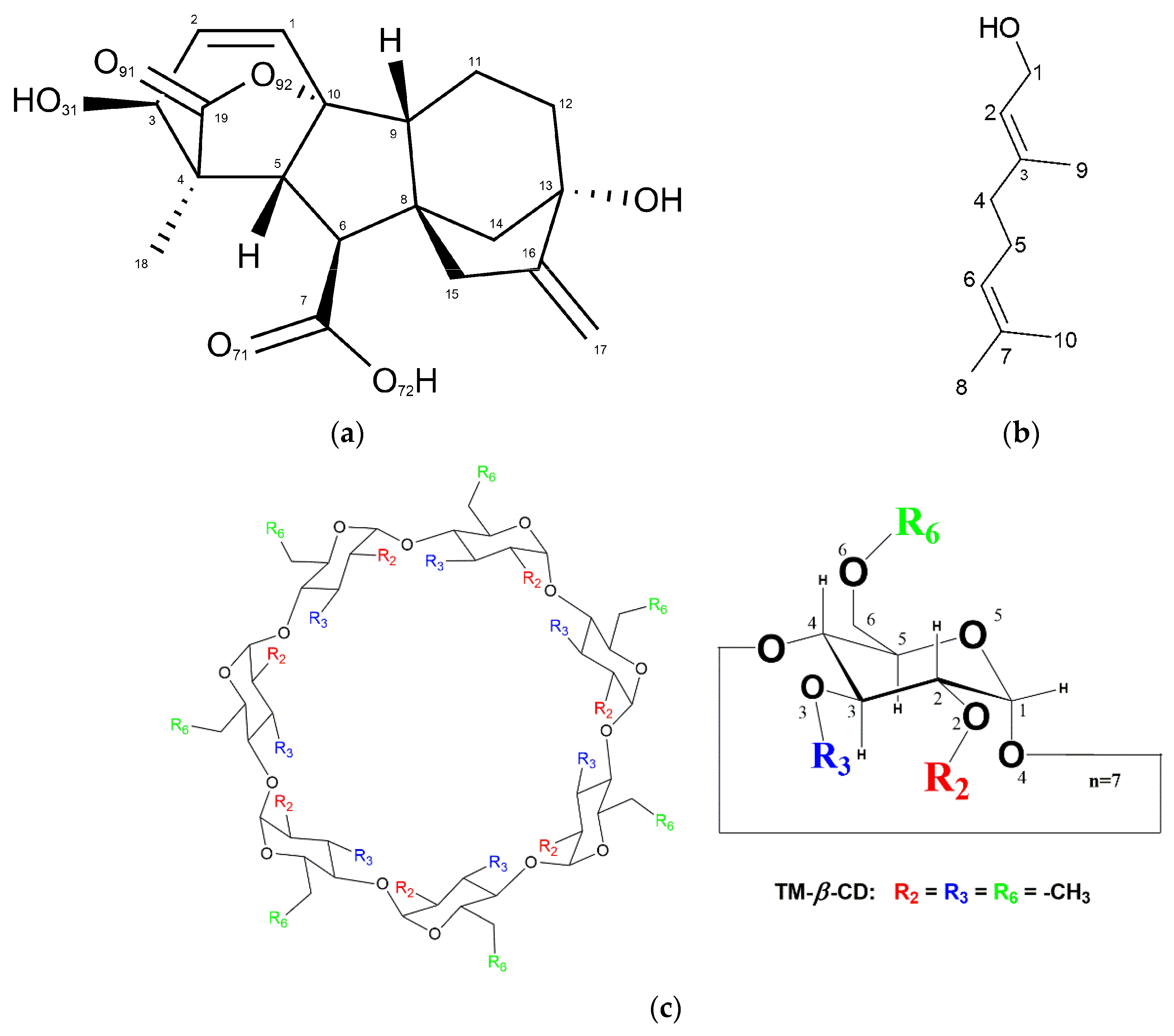
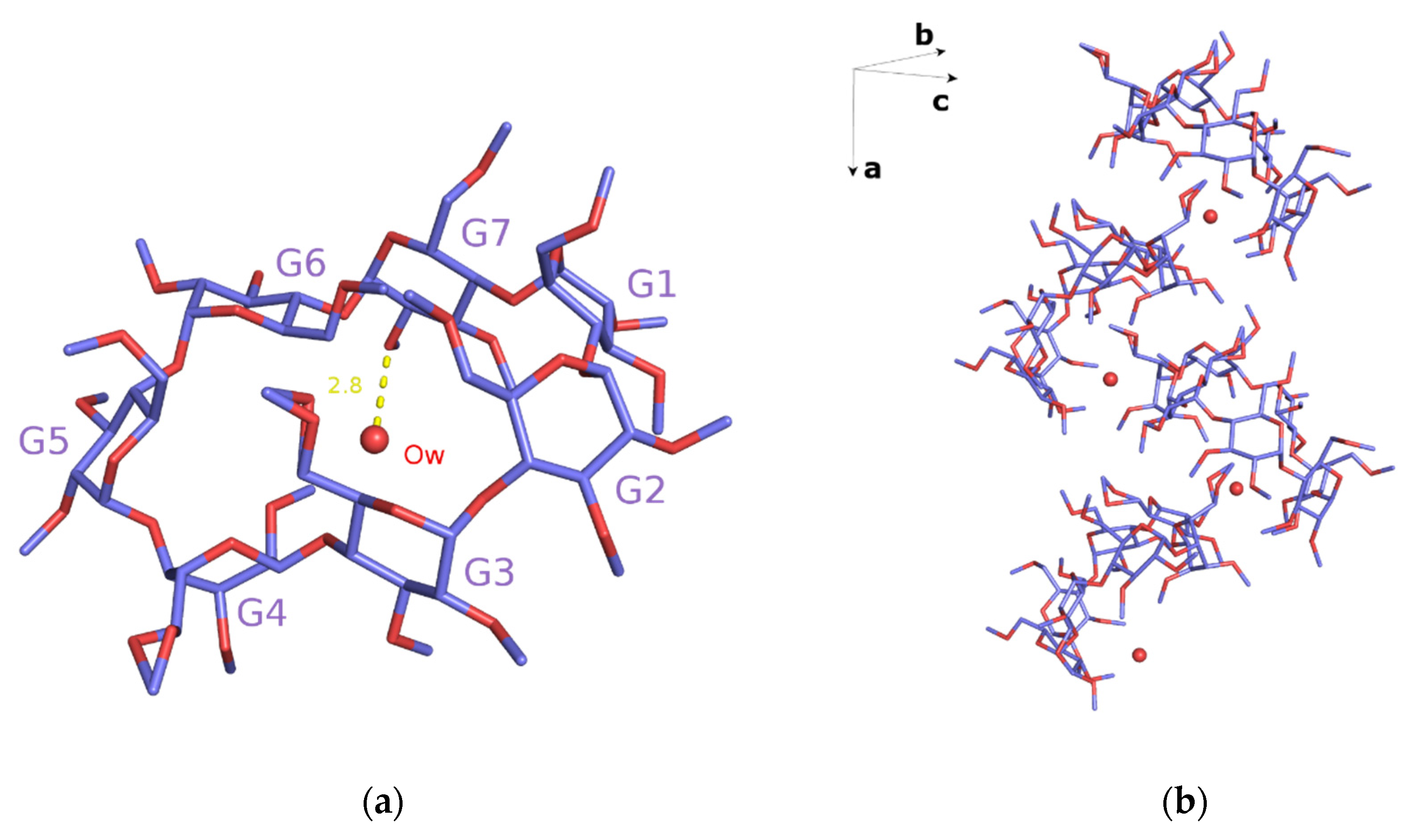
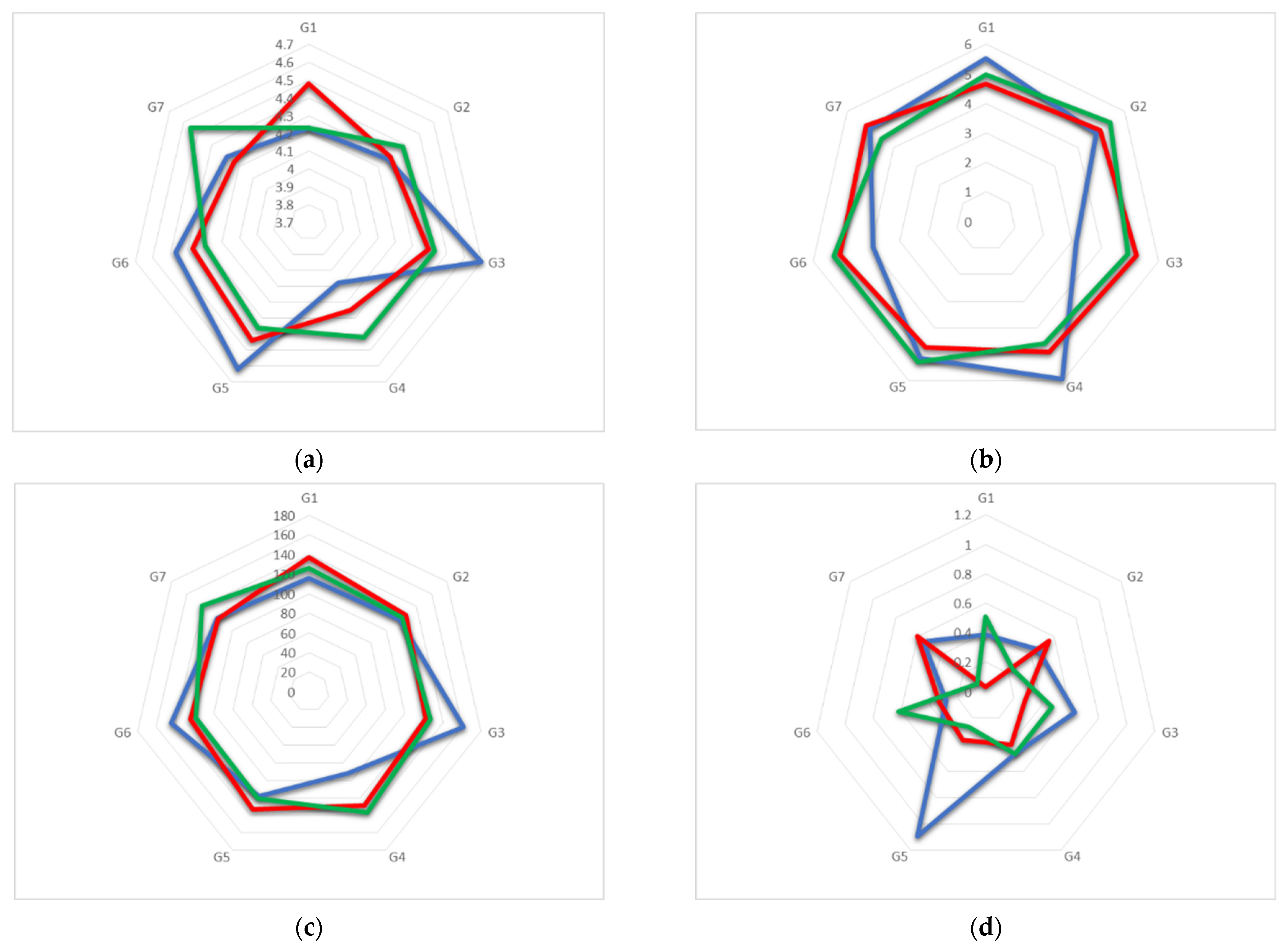
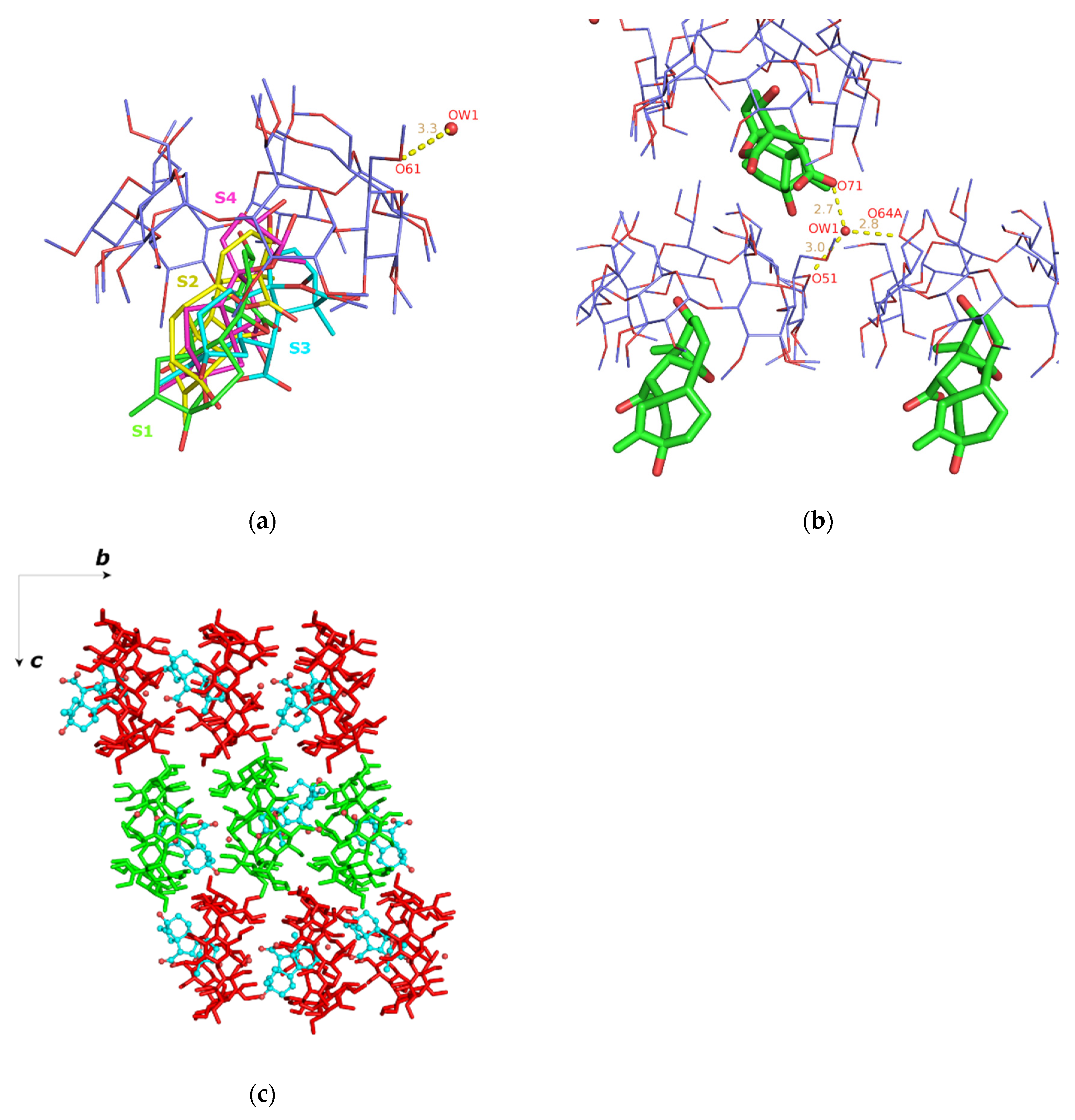
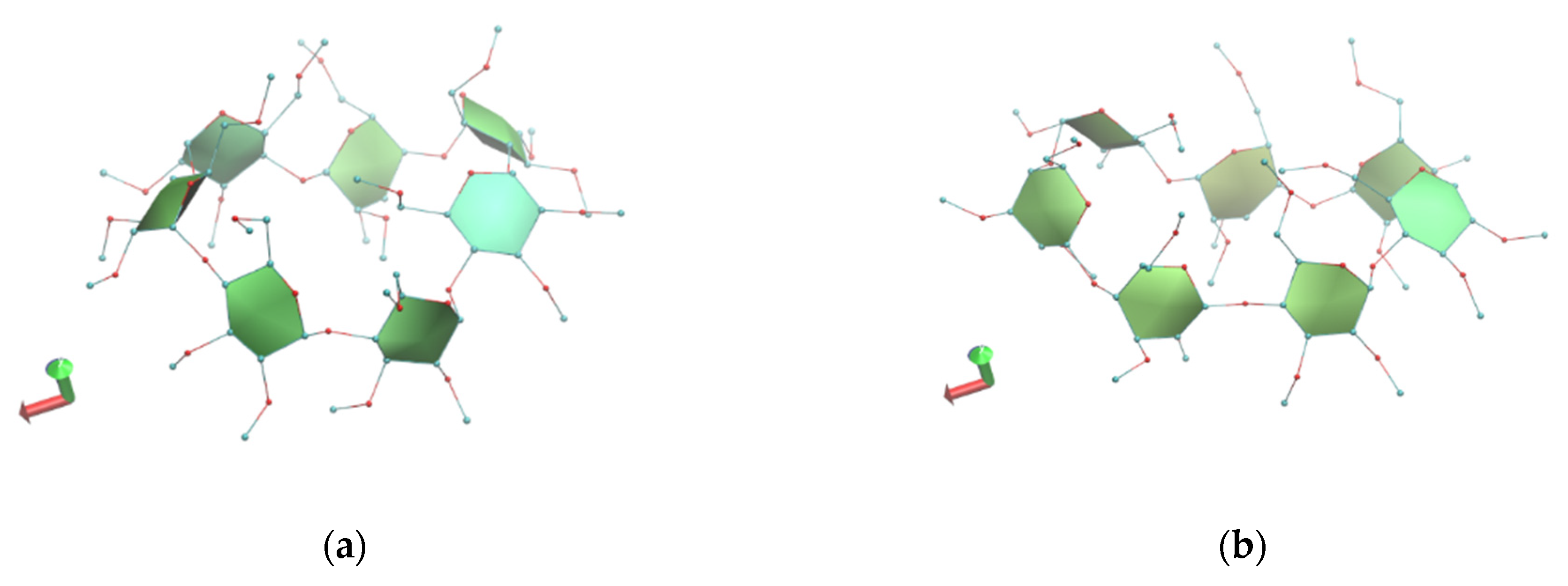
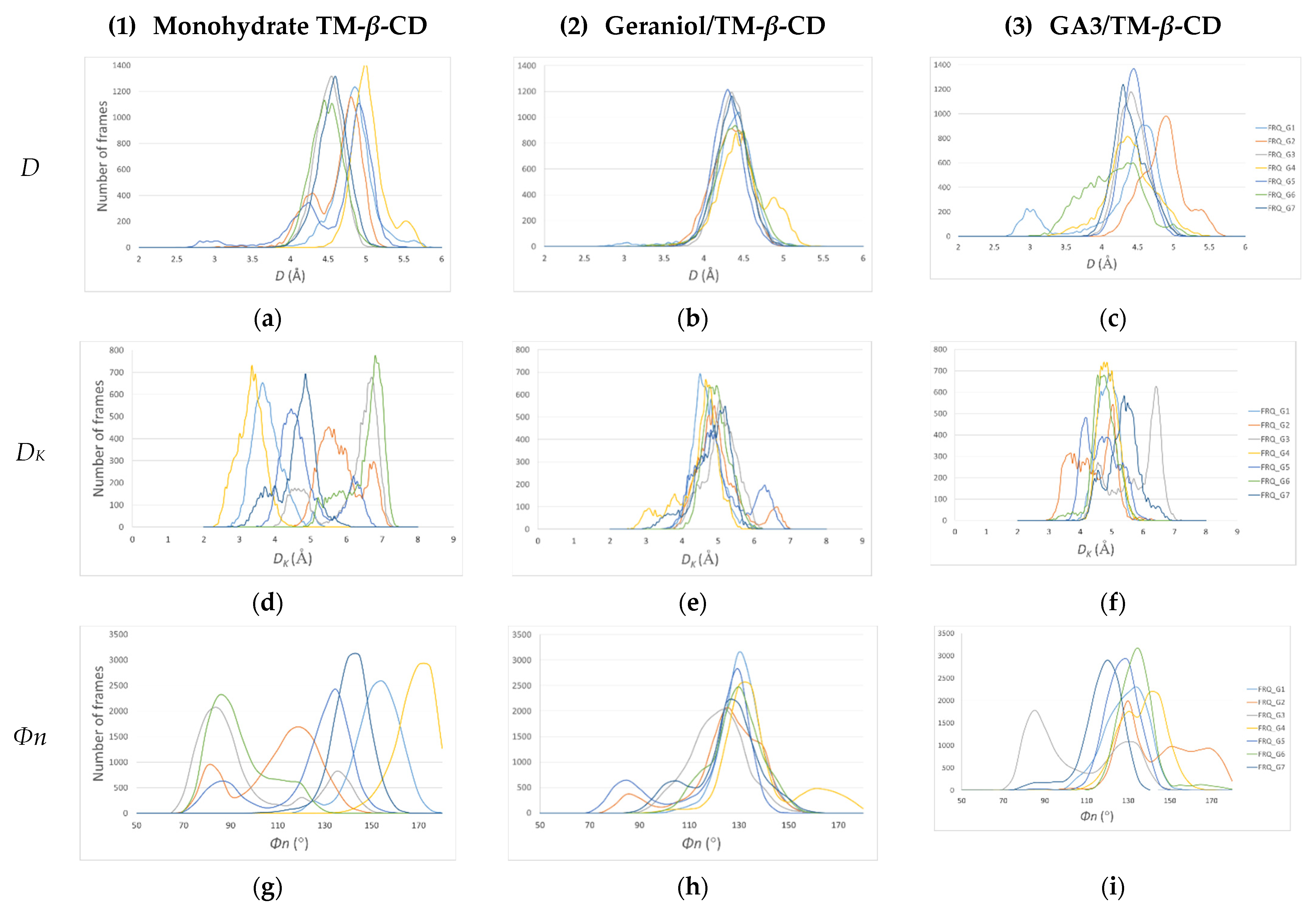
3.1. X-ray Analysis
Crystal Structures
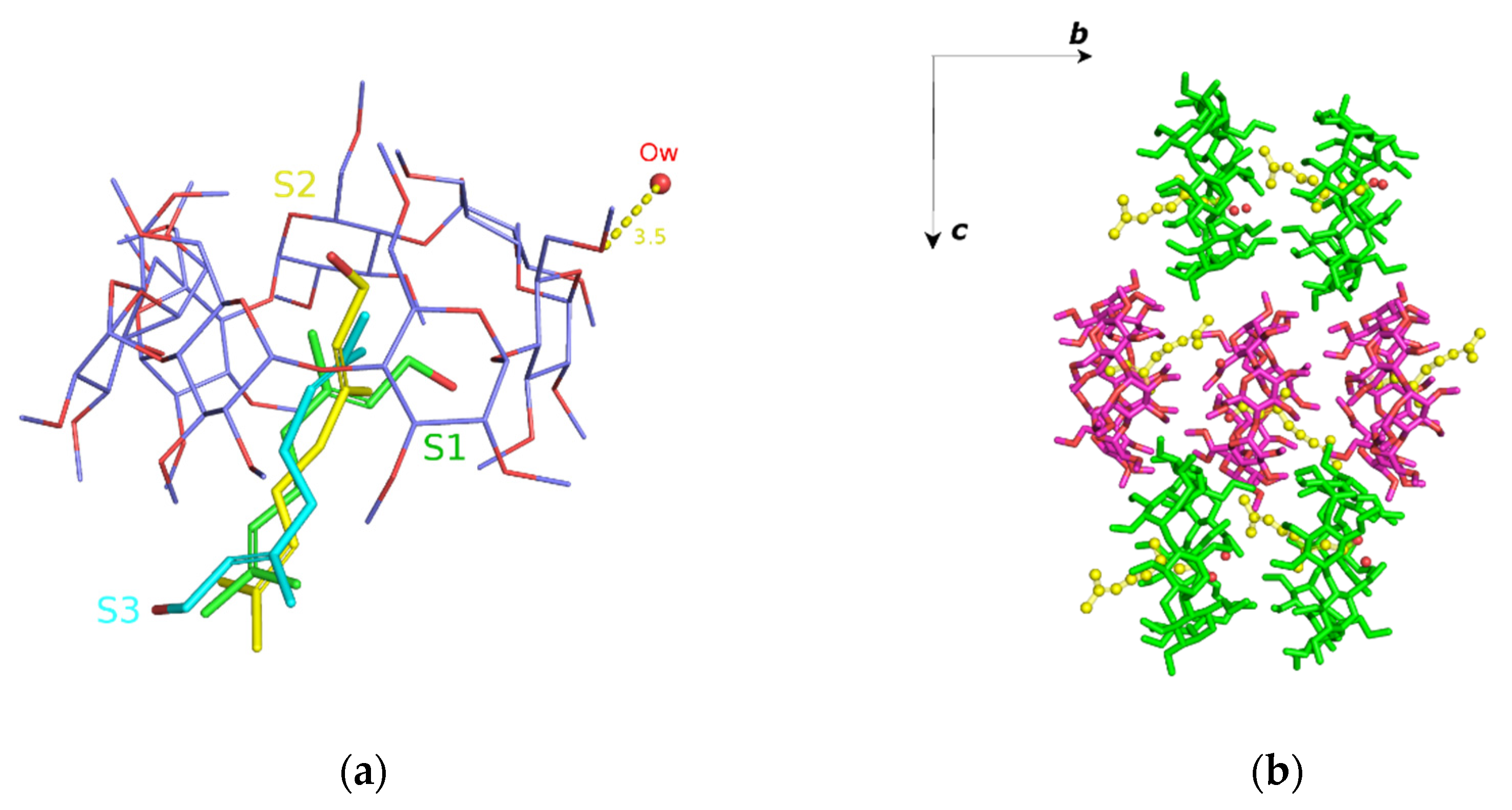
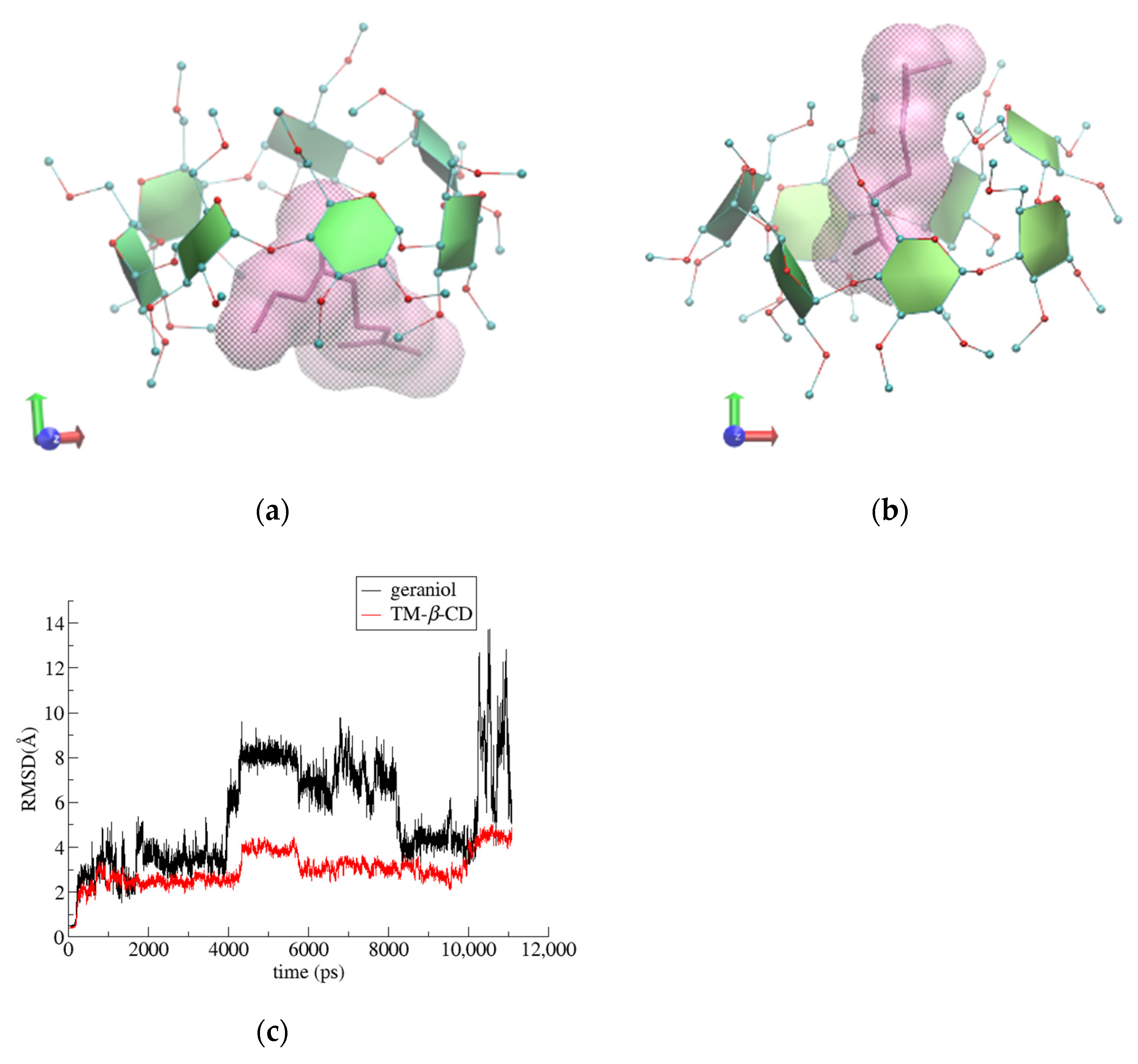
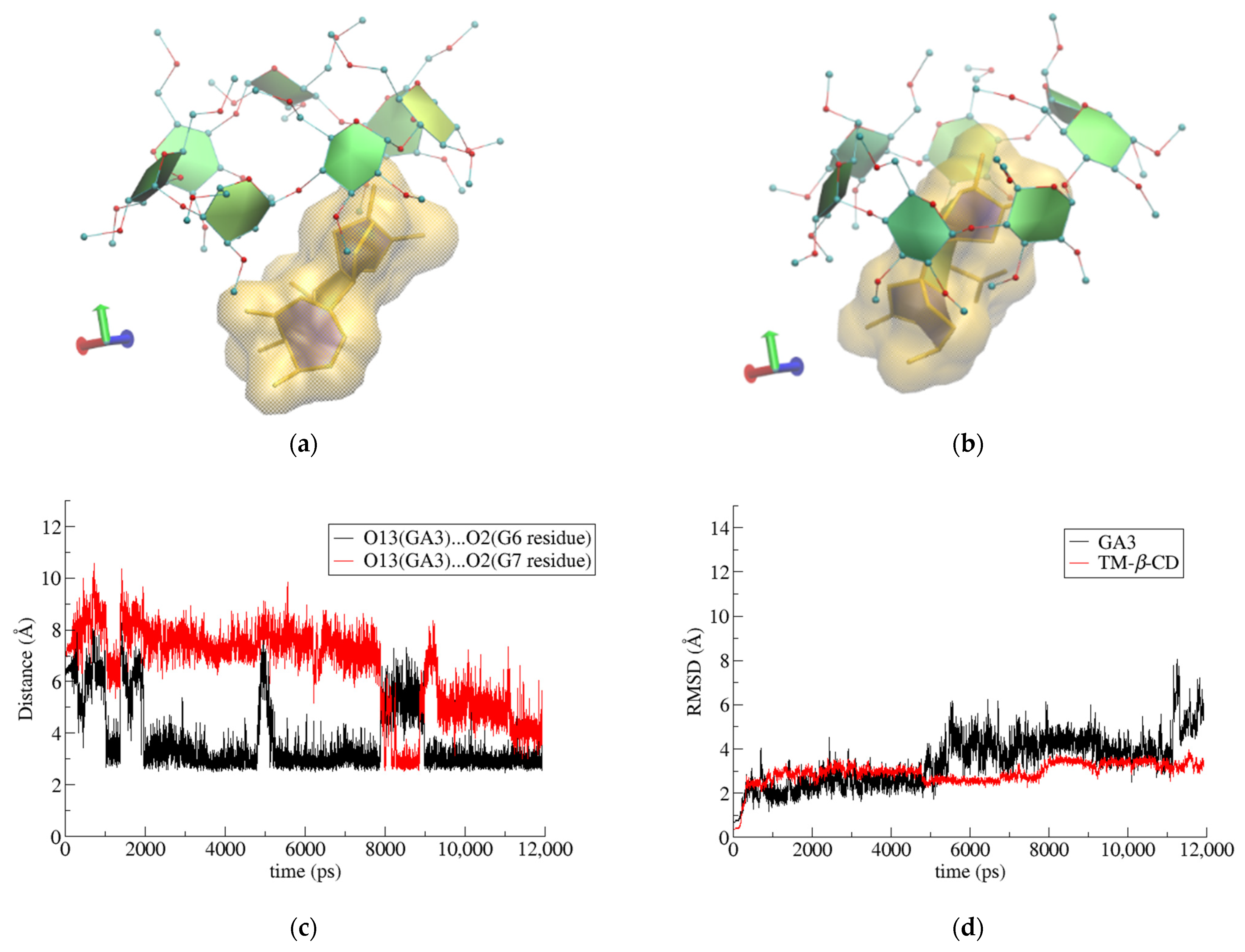
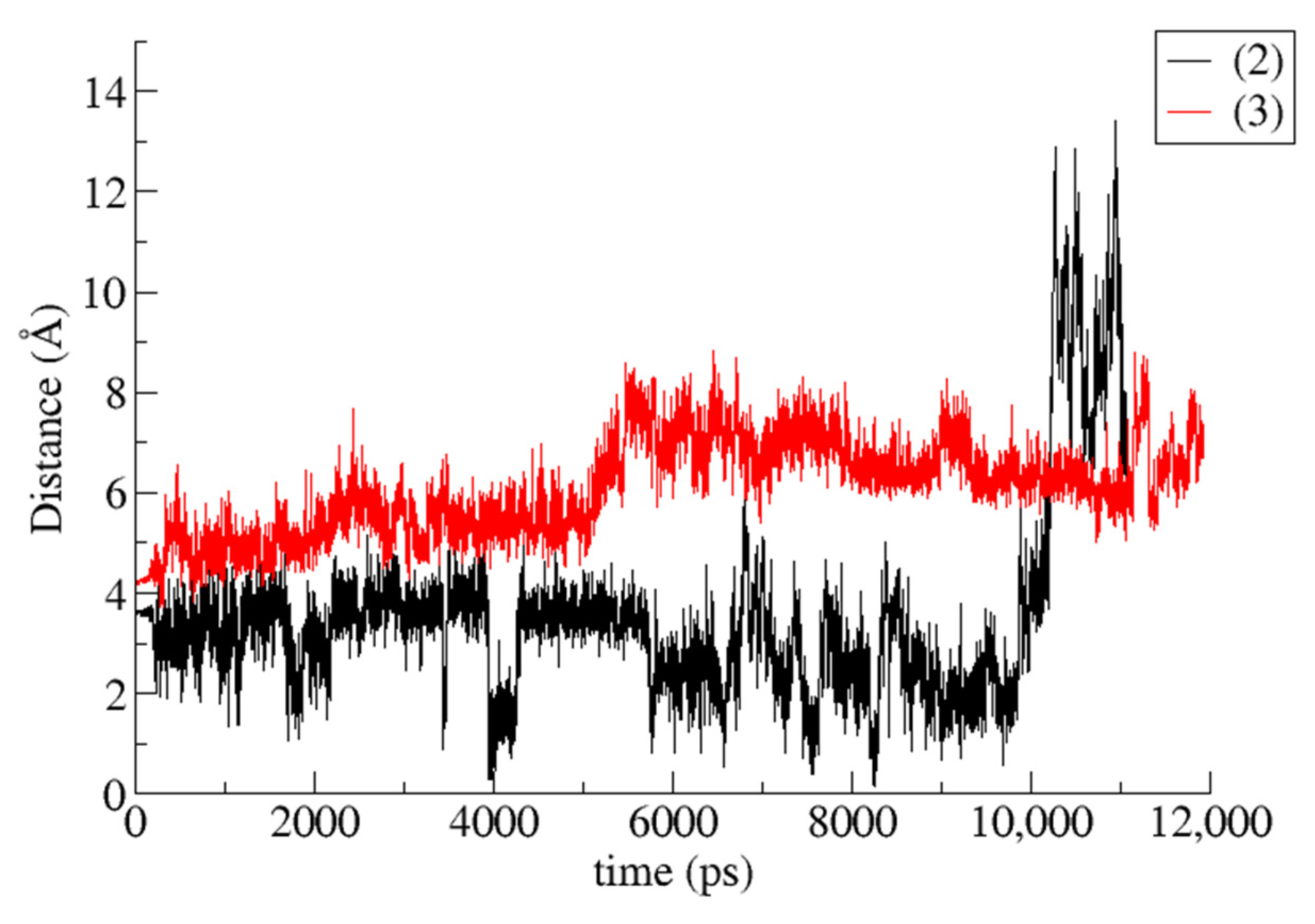
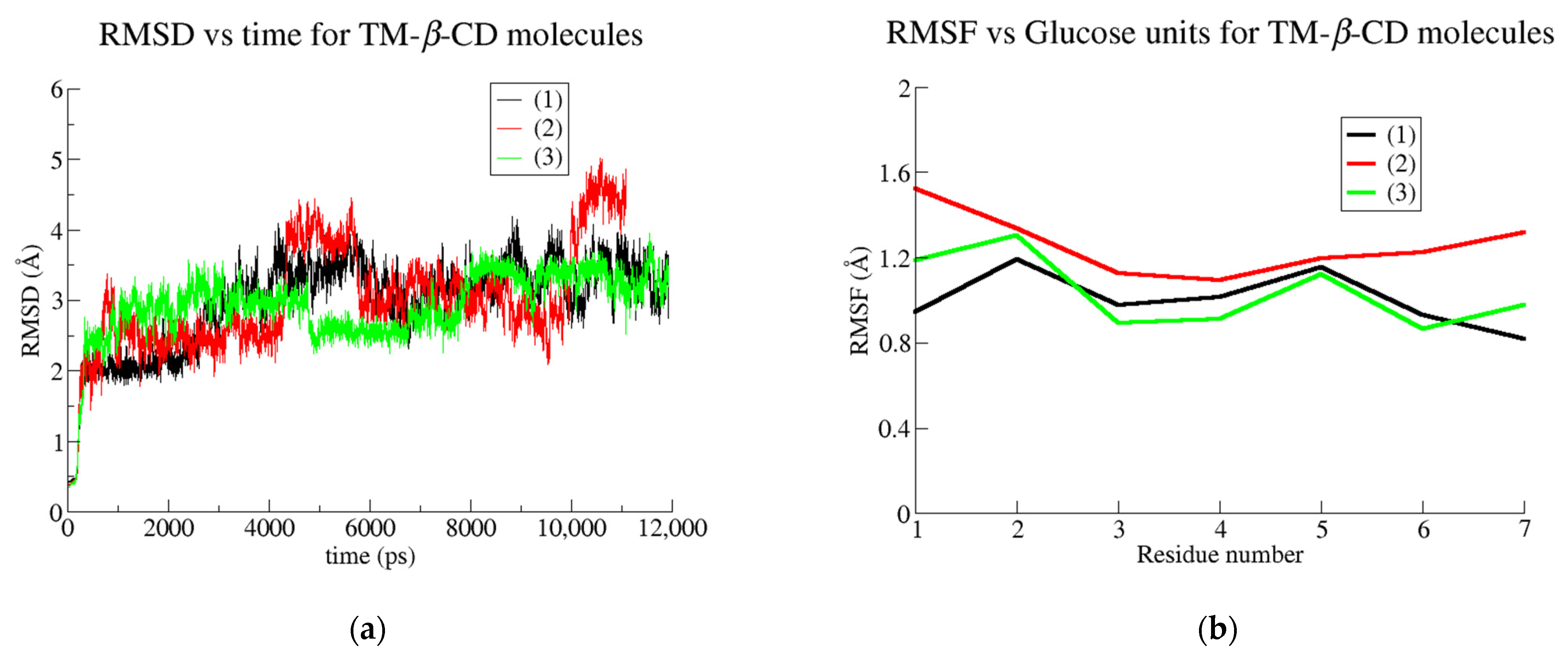
3.2. Trajectory Analysis
3.3. Binding Affinity Calculations
3.4. Comparison
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poulson, B.G.; Alsulami, Q.A.; Sharfalddin, A.; El Agammy, E.F.; Mouffouk, F.; Emwas, A.-H.; Jaremko, L.; Jaremko, M. Cyclodextrins: Structural, Chemical, and Physical Properties, and Applications. Polysaccharides 2022, 3, 1–31. [Google Scholar] [CrossRef]
- Fourmentin, S.; Crini, G.; Lichtfouse, E. Cyclodextrin Fundamentals, Reactivity and Analysis; Environmental Chemistry for a Sustainable World; Springer: Cham, Switzerland, 2018; ISBN 978-3-030-09418-8. [Google Scholar]
- Liu, Z.; Ye, L.; Xi, J.; Wang, J.; Feng, Z. Cyclodextrin Polymers: Structure, Synthesis, and Use as Drug Carriers. Prog. Polym. Sci. 2021, 118, 101408. [Google Scholar] [CrossRef]
- Bruns, C.J. Exploring and Exploiting the Symmetry-Breaking Effect of Cyclodextrins in Mechanomolecules. Symmetry 2019, 11, 1249. [Google Scholar] [CrossRef]
- Lipkowitz, K.B. Symmetry Breaking in Cyclodextrins: A Molecular Mechanics Investigation. J. Org. Chem. 1991, 56, 6357–6367. [Google Scholar] [CrossRef]
- Saenger, W. Cyclodextrin Inclusion Compounds in Research and Industry. Angew. Chem. Int. Ed. Engl. 1980, 19, 344–362. [Google Scholar] [CrossRef]
- Dodziuk, H. Molecules with Holes–Cyclodextrins. In Cyclodextrins and Their Complexes; John Wiley & Sons, Ltd.: New York, NY, USA, 2006; pp. 1–30. ISBN 978-3-527-60898-0. [Google Scholar]
- Wang, J.; Li, S.; Ye, L.; Zhang, A.-Y.; Feng, Z.-G. Formation of a Polypseudorotaxane via Self-Assembly of γ-Cyclodextrin with Poly(N-Isopropylacrylamide). Macromol. Rapid Commun. 2012, 33, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Kida, T.; Akashi, M. Recognition of Stereoregularity of Poly(Methacrylic Acid)s with γ-Cyclodextrin. Macromolecules 2011, 44, 3723–3729. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, S.; Lin, Y.; Zhou, J.; Pang, L.; Nie, X.; Zhou, B.; Tang, W. Engineering Cyclodextrin Clicked Chiral Stationary Phase for High-Efficiency Enantiomer Separation. Sci. Rep. 2015, 5, 11523. [Google Scholar] [CrossRef]
- Betzel, C.; Saenger, W.; Hingerty, B.E.; Brown, G.M. Topography of Cyclodextrin Inclusion Complexes, Part 20. Circular and Flip-Flop Hydrogen Bonding in.Beta.-Cyclodextrin Undecahydrate: A Neutron Diffraction Study. J. Am. Chem. Soc. 1984, 106, 7545–7557. [Google Scholar] [CrossRef]
- Caira, M.R.; Bourne, S.A.; Mzondo, B. Encapsulation of the Antioxidant R-(+)-α-Lipoic Acid in Permethylated α- and β-Cyclodextrins: Thermal and X-Ray Structural Characterization of the 1:1 Inclusion Complexes. Molecules 2017, 22, 866. [Google Scholar] [CrossRef]
- Papaioannou, A.; Christoforides, E.; Bethanis, K. Inclusion Complexes of Naringenin in Dimethylated and Permethylated β-Cyclodextrins: Crystal Structures and Molecular Dynamics Studies. Crystals 2019, 10, 10. [Google Scholar] [CrossRef]
- Bethanis, K.; Christoforides, E.; Tsorteki, F.; Fourtaka, K.; Mentzafos, D. Structural Studies of the Inclusion Compounds of α-Naphthaleneacetic Acid in Heptakis(2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis(2,3,6-Tri-O-Methyl)-β-Cyclodextrin by X-Ray Crystallography and Molecular Dynamics. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 157–171. [Google Scholar] [CrossRef]
- Hedden, P.; Sponsel, V. A Century of Gibberellin Research. J. Plant Growth Regul. 2015, 34, 740–760. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C. Gibberellin Signaling in Plants—The Extended Version. Front. Plant Sci. 2012, 2, 107. [Google Scholar] [CrossRef]
- Camara, M.C.; Vandenberghe, L.P.S.; Rodrigues, C.; de Oliveira, J.; Faulds, C.; Bertrand, E.; Soccol, C.R. Current Advances in Gibberellic Acid (GA3) Production, Patented Technologies and Potential Applications. Planta 2018, 248, 1049–1062. [Google Scholar] [CrossRef]
- Gao, X.-T.; Wu, M.-H.; Sun, D.; Li, H.-Q.; Chen, W.-K.; Yang, H.-Y.; Liu, F.-Q.; Wang, Q.-C.; Wang, Y.-Y.; Wang, J.; et al. Effects of Gibberellic Acid (GA3) Application before Anthesis on Rachis Elongation and Berry Quality and Aroma and Flavour Compounds in Vitis Vinifera L. “Cabernet Franc” and “Cabernet Sauvignon” Grapes. J. Sci. Food Agric. 2020, 100, 3729–3740. [Google Scholar] [CrossRef]
- Gao, S.; Chu, C. Gibberellin Metabolism and Signaling: Targets for Improving Agronomic Performance of Crops. Plant Cell Physiol. 2020, 61, 1902–1911. [Google Scholar] [CrossRef]
- Caira, M.R.; Griffith, V.J.; Nassimbeni, L.R.; van Oudtshoorn, B. Unusual 1C4 Conformation of a Methylglucose Residue in Crystalline Permethyl-[Small Beta]-Cyclodextrin Monohydrate. J. Chem. Soc. Perkin Trans. 2 1994, 2071–2072. [Google Scholar] [CrossRef]
- Steiner, T.; Saenger, W. Closure of the Cavity in Permethylated Cyclodextrins through Glucose Inversion, Flipping, and Kinking. Angew. Chem. Int. Ed. 1998, 37, 3404–3407. [Google Scholar] [CrossRef]
- Christoforides, E.; Fourtaka, K.; Andreou, A.; Bethanis, K. X-ray Crystallography and Molecular Dynamics Studies of the Inclusion Complexes of Geraniol in β-Cyclodextrin, Heptakis (2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis (2,3,6-Tri-O-Methyl)-β-Cyclodextrin. J. Mol. Struct. 2020, 1202, 127350. [Google Scholar] [CrossRef]
- Sheldrick G, M. SAINT, Version 8.37; Bruker AXS Inc.: Madison, WI, USA, 2013. [Google Scholar]
- Sheldrick, G.M. SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. XPREP; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. Experimental Phasing with It SHELXC/It D/It E: Combining Chain Tracing with Density Modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A Tool for High-Throughput Crystallography of Protein–Ligand Complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Cezard, C.; Trivelli, X.; Aubry, F.; Djedaini-Pilard, F.; Dupradeau, F.-Y. Molecular Dynamics Studies of Native and Substituted Cyclodextrins in Different Media: 1. Charge Derivation and Force Field Performances. Phys. Chem. Chem. Phys. 2011, 13, 15103–15121. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Fourtaka, K.; Christoforides, E.; Mentzafos, D.; Bethanis, K. Crystal Structures and Molecular Dynamics Studies of the Inclusion Compounds of β-Citronellol in β-Cyclodextrin, Heptakis(2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis(2,3,6-Tri-O-Methyl)-β-Cyclodextrin. J. Mol. Struct. 2018, 1161, 1–8. [Google Scholar] [CrossRef]
- Fourtaka, K.; Christoforides, E.; Tzamalis, P.; Bethanis, K. Inclusion of Citral Isomers in Native and Methylated Cyclodextrins: Structural Insights by X-Ray Crystallography and Molecular Dynamics Simulation Analysis. J. Mol. Struct. 2021, 1234, 130169. [Google Scholar] [CrossRef]
- Cruickshank, D.L.; Rougier, N.M.; Maurel, V.J.; de Rossi, R.H.; Buján, E.I.; Bourne, S.A.; Caira, M.R. Permethylated β-Cyclodextrin/Pesticide Complexes: X-Ray Structures and Thermogravimetric Assessment of Kinetic Parameters for Complex Dissociation. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 47–56. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Shi, J.; Guo, D.-S.; Ding, F.; Liu, Y. Unique Regioselective Binding of Permethylated β-Cyclodextrin with Azobenzene Derivatives. Eur. J. Org. Chem. 2009, 2009, 923–931. [Google Scholar] [CrossRef]
- Triantafyllopoulou, V.; Tsorteki, F.; Mentzafos, D.; Bethanis, K. Inclusion Compounds of Plant Growth Regulators in Cyclodextrins, Part VII: Study of the Crystal Structures of 2-Naphthylacetic Acid Encapsulated in β-Cyclodextrin and Heptakis(2,3,6-Tri-O-Methyl)-β-Cyclodextrin Complexes by X-Ray Crystallography. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 303–310. [Google Scholar] [CrossRef][Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (1) Monohydrate TM-β-CD | (2) Geraniol/ TM-β-CD 1 | (3) GA3/ TM-β-CD | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | C63H112O35·H2O | C63H112O35· C10 H18O·0.5(H2O) | C62H112O35·C19 H22O6·H2O |
| Mr | 1445.52 | 1591.76 | 1791.88 |
| Crystal system, space group | Orthorhombic, P212121 | Orthorhombic, P212121 | Orthorhombic, P212121 |
| Temperature (K) | 293 | 100 | 100 |
| a, b, c (Å) | 14.847(2), 19.391(5), 26.556(5) | 14.903(6), 20.888(1), 27.686(8) | 14.7487(8), 22.0113(13), 27.6009(15) |
| V (Å3) | 7645(3) | 8618(4) | 8960.3(9) |
| Z | 4 | 4 | 4 |
| Radiation type | Cu Ka λ = 1.54 Å | Synchrotron, λ = 0.81 Å | Cu Ka λ = 1.54 Å |
| μ (mm−1) | 0.87 | 0.13 | 0.87 |
| Crystal size (mm3) | 0.5 × 0.4 × 0.3 | 0.3 × 0.2 × 0.1 | 0.4 × 0.2 × 0.05 |
| Data collection | |||
| Diffractometer | Bruker APEX-II | - | Bruker APEX-II |
| Absorption correction | Multi-scan SADABS2014/5—Bruker AXS area detector scaling and absorption correction | Multi-scan SADABS 2014/5 | Multi-scan SADABS2014/5—Bruker AXS area detector scaling and absorption correction |
| Tmin, Tmax | 0.525, 0.752 | 0.684, 0.746 | 0.648, 0.75 |
| No. of measured, independent and observed [I > 2s(I)] reflections | 71,972, 11,002, 10,232 | 87,735, 11,978, 10,712 | 75,037, 9134, 6687 |
| Rint | 0.041 | 0.06 | 0.070 |
| θmax (°) | 59.2 | 26.7 | 50.6 |
| (sin θ/λ)max (Å−1) | 0.557 | 0.550 | 0.501 |
| Refinement | |||
| R[F2 > 2s(F2)], wR(F2), S | 0.048, 0.135, 1.03 | 0.071, 0.187, 1.06 | 0.098, 0.279, 1.06 |
| No. of parameters | 891 | 1100 | 1172 |
| No. of restraints | - | 148 | 414 |
| ∆ρmax, ∆ρmin (e Å−3) | 0.39, −0.30 | 0.57, −0.45 | 0.40, −0.43 |
| Absolute structure parameter | 0.03(3) | −0.14(14) | 0.12(6) |
| A. Glucose Residues of Monohydrate TM-β-CD (1) 1 | DK (Å) | D (Å) | d (Å) | Φn (°) | τ (°) | t (°) | C |
|---|---|---|---|---|---|---|---|
| G1 | 5.52(3) | 4.233(4) | −0.395(2) | 116.11(8) | +20.74(14) | −77.3(4) | gg |
| G2 | 4.785(3) | 4.273(4) | −0.459(2) | 118.05(9) | +37.73(17) | 64.8(5) | gt |
| G3 | 3.414(3) | 4.703(4) | 0.629(3) | 161.28(10) | +24.62(18) | 60.9(8) 92.9(8) | Gt gt |
| G4 | 5.940(3) | 4.082(4) | 0.472(2) | 92.27(8) | −24.4(2) | 67.1(7) 128.0(9) | Gt tg |
| G5 | 5.127(3) | 4.619(4) | −1.089(2) | 118.62(8) | +57.10(15) | −82.4(4) | gg |
| G6 | 3.931(3) | 4.466(4) | 0.285(2) | 144.64(8) | +72.98(16) | −64.0(0) | gg |
| G7 | 5.041(2) | 4.287(4) | 0.553(19) | 120.14(8) | −4.71(13) | −78.3(3) | gg |
| B. Glucose Residues of TM-β-CD in (2) 2 | DK(Å) | D(Å) | d(Å) | Φn (°) | τ (°) | t(°) | C |
| G1 | 4.66(4) | 4.48(5) | 0.030(3) | 136.99(12) | +43.3(2) | 85.3(7) | gt |
| G2 | 4.95(4) | 4.29(5) | 0.556(3) | 125.71(12) | −13.9(2) | −68.7(6) | gg |
| G3 | 5.22(4) | 4.39(5) | −0.278(3) | 122.25(12) | +15.55(2) | 75.2(6) | gt |
| G4 | 4.92(4) | 4.25(7) | −0.399(3) | 128.87(13) | +29.0(3) | −74.0(6) | gg |
| G5 | 4.73(6) | 4.44(7) | 0.368(4) | 132.78(16) | +36.1(3) | 72.1(9) −65.3(2) | Gt gg |
| G6 | 5.09(4) | 4.37(6) | 0.332(3) | 124.80(12) | −16.9(4) | −81.5(9) −90.2(2) | Gg gg |
| G7 | 5.21(4) | 4.24(5) | −0.609(3) | 119.39(11) | +38.8(2) | −74.9(5) | gg |
| C. Glucose Residues of TM-β-CD in (3) 3 | DK(Å) | D(Å) | d(Å) | Φn (°) | τ (°) | t(°) | C |
| G1 | 4.983(8) | 4.229(12) | 0.513(7) | 125.9(4) | −11.5(6) | −69.1(13) | gg |
| G2 | 5.369(8) | 4.376(11) | −0.241(7) | 120.8(3) | +13.9(4) | −60.1(15) 89.0(2) 65.0(3) | gg gt gt |
| G3 | 4.932(8) | 4.432(14) | −0.467(7) | 127.1(3) | +32.0(6) | −74.9(11) | gg |
| G4 | 4.581(11) | 4.421(14) | 0.468(8) | 136.7(3) | +39.0(6) | −71.8(18) 83.0(2) | gg gt |
| G5 | 5.287(8) | 4.358(12) | 0.266(7) | 120.8(3) | −10.5 (6) | −79.1(10) | gg |
| G6 | 5.286(8) | 4.294(13) | −0.621(7) | 118.6(3) | +37.0(6) | −62.1(2) 51.2(14) | gg gt |
| G7 | 4.511(9) | 4.549(14) | 0.082(8) | 140.4(3) | +44.4(7) | 64.0(2) 58.0(30) | gt gt |
| Atom Name (Symmetry Operation) | Atom Name (Symmetry Operation) | Distance (Å) |
|---|---|---|
| Ow1(x, y, z) | O51(x, y, z) | 2.951(1) |
| Ow1(x, y, z) | O71(1 − x, −1/2 + y, 3/2 − z) 1 | 2.665(1) |
| Ow1(x, y, z) | O64A(1 + x, y, z) 2 | 2.838(2) |
| Geraniol/TM-β-CD | GA3/TM-β-CD | |
|---|---|---|
| ΔEvdW | −23.89 ± 2.80 | −23.13± 5.12 |
| ΔEele | −2.96 ± 1.74 | −7.25 ± 3.84 |
| ΔEGB | 13.56 ± 1.78 | 19.39 ± 3.23 |
| ΔEsurf | −3.22 ± 0.29 | −2.73 ± 0.36 |
| ΔGgas | −26.86 ± 3.59 | −30.38 ± 5.13 |
| ΔGsolv | 10.34 ± 1.67 | 16.66 ± 3.20 |
| ΔG(GB)1 | −16.52 ± 2.88 | −13.72 ± 3.94 |
| T·∆S | −17.22 ± 2.00 | −17.59 ± 1.50 |
| ΔGbind2 | +0.70 ± 3.50 | +3.87 ± 4.21 |
| RoG Value of TM-β-CD (Å) | |
|---|---|
| (1) | 6.26 ± 0.10 |
| (2) | 6.06 ± 0.14 |
| (3) | 6.27 ± 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bethanis, K.; Christoforides, E.; Andreou, A.; Eliopoulos, E. Molecular Symmetry of Permethylated β-Cyclodextrins upon Complexation. Symmetry 2022, 14, 2214. https://doi.org/10.3390/sym14102214
Bethanis K, Christoforides E, Andreou A, Eliopoulos E. Molecular Symmetry of Permethylated β-Cyclodextrins upon Complexation. Symmetry. 2022; 14(10):2214. https://doi.org/10.3390/sym14102214
Chicago/Turabian StyleBethanis, Kostas, Elias Christoforides, Athena Andreou, and Elias Eliopoulos. 2022. "Molecular Symmetry of Permethylated β-Cyclodextrins upon Complexation" Symmetry 14, no. 10: 2214. https://doi.org/10.3390/sym14102214
APA StyleBethanis, K., Christoforides, E., Andreou, A., & Eliopoulos, E. (2022). Molecular Symmetry of Permethylated β-Cyclodextrins upon Complexation. Symmetry, 14(10), 2214. https://doi.org/10.3390/sym14102214